
Autophagy and Alzheimer’s Disease: Mechanisms and Impact Beyond the Brain

 ,
,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Methodology
2. Overview of Autophagy
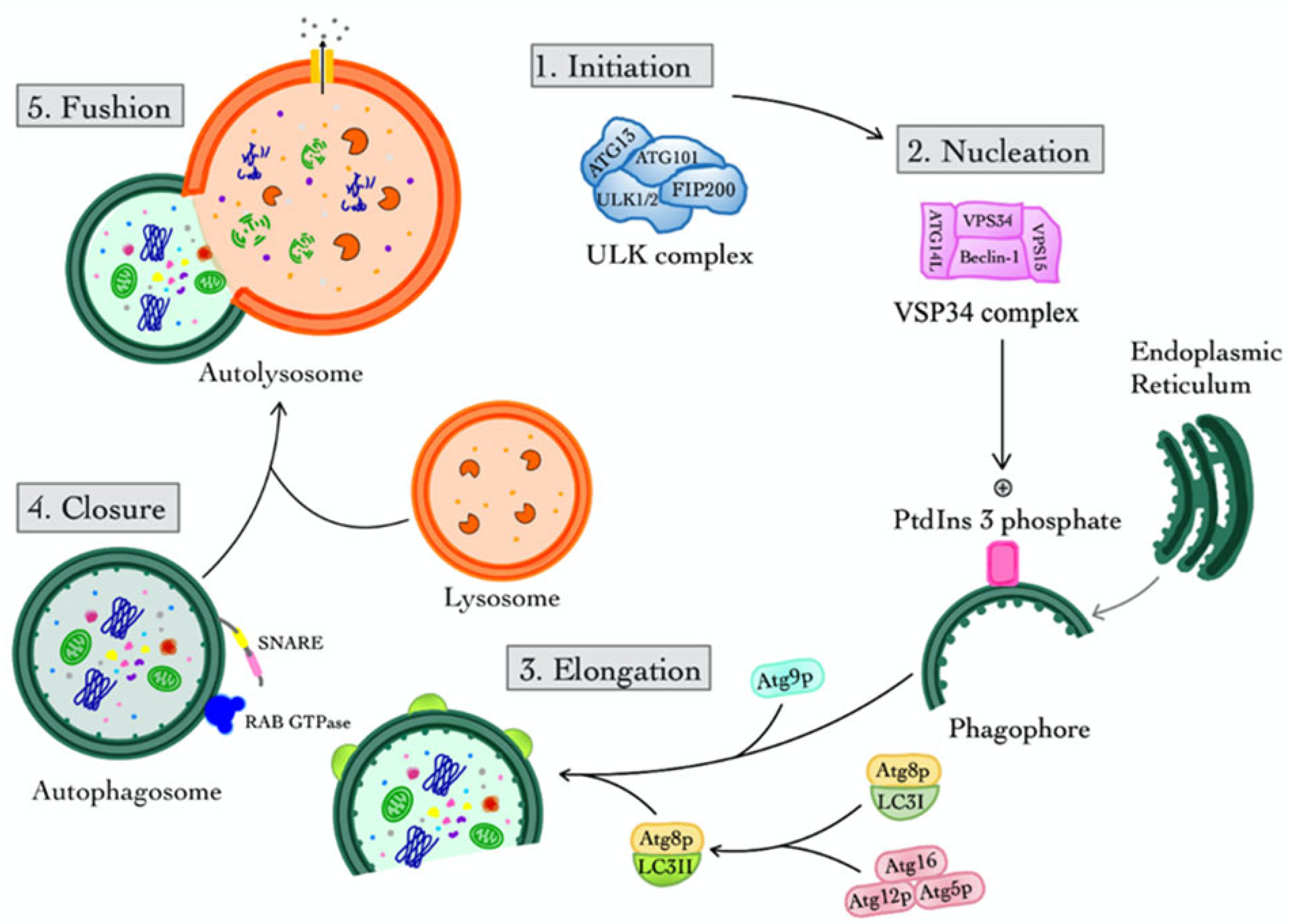
2.1. Macroautophagy
2.1.1. Initiation
2.1.2. Nucleation
2.1.3. Elongation
2.1.4. Closure
2.1.5. Fusion
2.2. Selective Features and Regulation of Macroautophagy
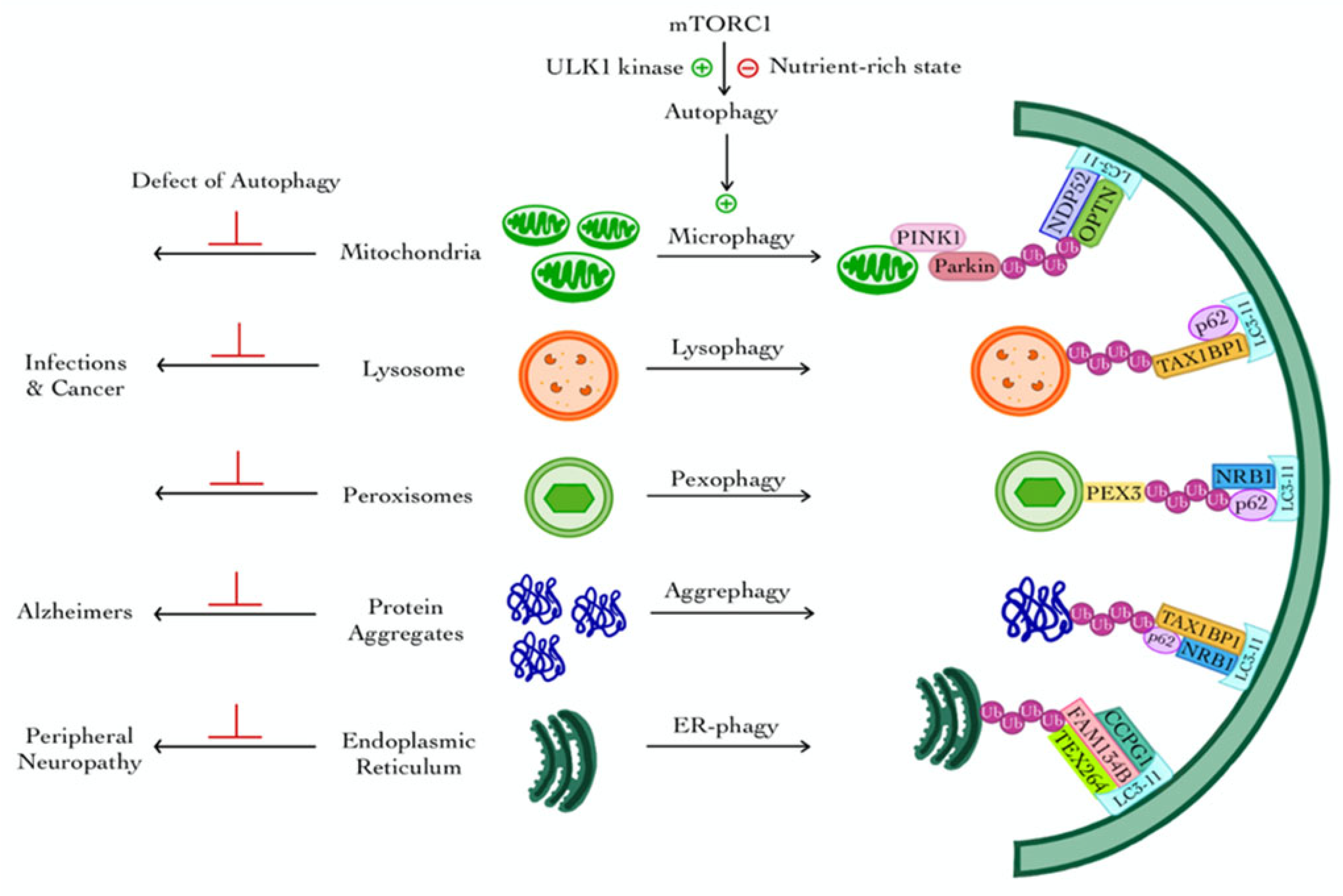
3. Selective Autophagy
3.1. Mitophagy
3.2. Lysophagy
3.3. Pexophagy
3.4. Aggrephagy
3.5. ER-Phagy (Reticulophagy)
4. Chaperone-Mediated Autophagy (CMA)
5. Microautophagy and Endosomal Microautophagy
6. Autophagy in AD and Organ-Specific Impacts
6.1. Autophagic Impairment in Protein Aggregate Clearance
6.2. Lysosomal Dysfunction and Genetic Regulators
6.3. Autophagic Flux
6.4. Axonal Transport and Autophagic Vacuole Accumulation
6.5. Autophagy–Lysosome Pathway Disruption: Mechanistic Insights
6.6. Aβ Secretion and Autophagy Crosstalk
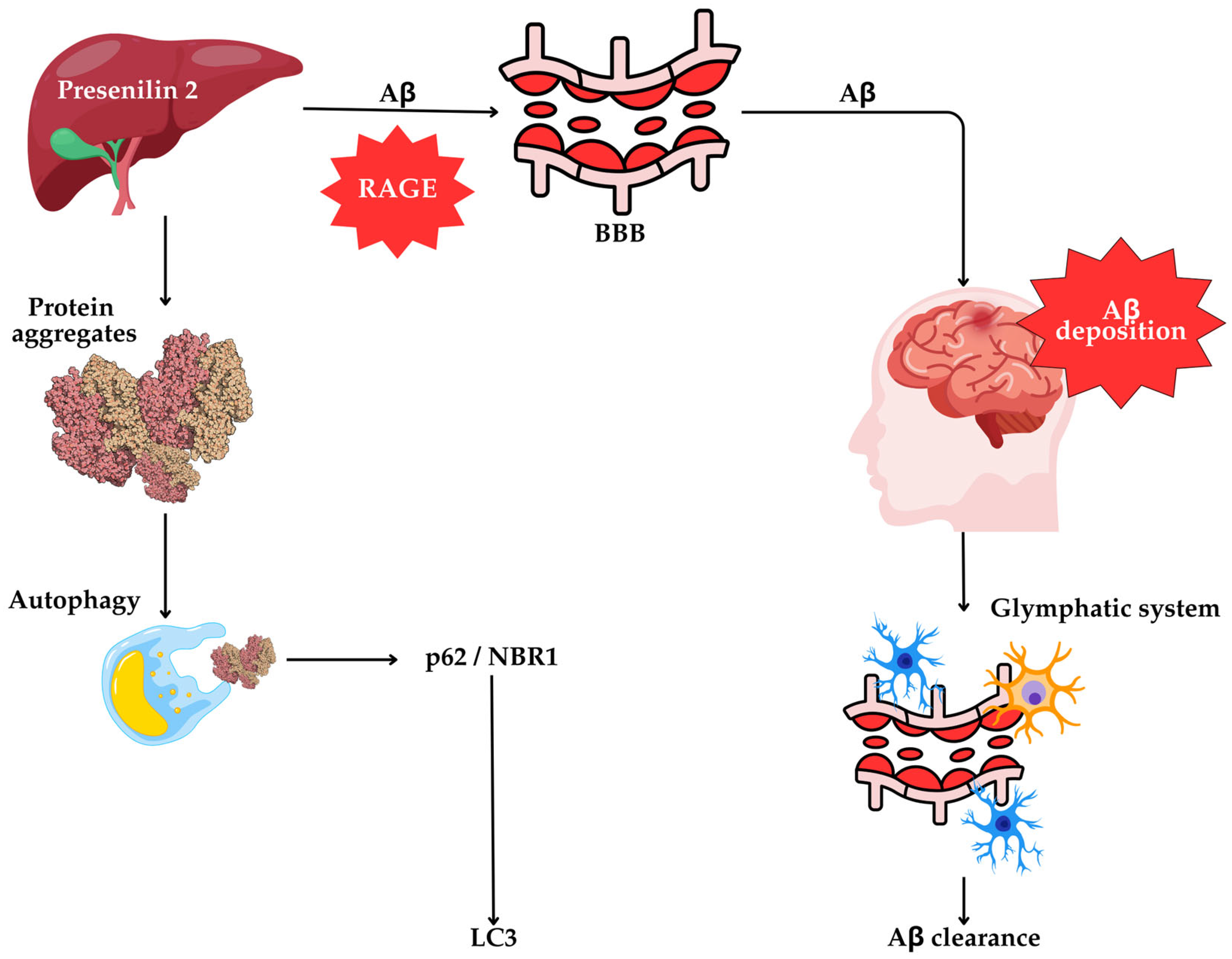
6.7. The Liver’s Role in Autophagy and AD Pathogenesis
6.8. Relative Contributions of Autophagic Pathways to AD Pathogenesis
7. Future Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA-ATPase VCP/p9 | ATP-driven chaperone valosin-containing protein |
| AD | Alzheimer’s disease |
| AMPK | AMP-activated protein kinase |
| ApoE4 | Apolipoprotein E4 |
| APP | Amyloid precursor protein |
| ATG | Autophagy-related genes |
| AVs | Autophagic vacuoles |
| Aβ | Amyloid-beta |
| BBB | Blood–brain barrier |
| BECN | Beclin |
| BNIP3L | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like |
| CCPG1 | Cell cycle progression gene 1 |
| CMA | Chaperone-mediated autophagy |
| DFCPI | Double FYVE-containing protein 1 |
| EF1α | Elongation factor-1α |
| ESCRT | Endosomal sorting complex required for transport |
| FAM134B | Family with Sequence Similarity 134 Member B |
| FIP200 | FAK family kinase-interacting protein of 200 kDa |
| GFAP | Glial fibrillary acidic protein |
| HIF-1α | Hypoxia-inducible factor-1α |
| HSC70 | Cytosolic chaperone heat shock cognate 71-kDa protein |
| JLK | UNC-51-like autophagy-activating kinase |
| LAMP-2A | Lysosome-associated membrane protein type 2A |
| LANDO | LC3-associated endocytosis |
| LAP | LC3-associated phagocytosis |
| LC3 | Microtubule-associated protein 1A/1B light chain 3 |
| LE/MVBs | Late endosomes/multivesicular bodies |
| LLOMe | L-leucyl-L-leucine methyl ester |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| NDP52 | Nuclear dot protein 52 kDa |
| NF-κB | Nuclear factor kappa B |
| NFAT | Nuclear factor of activated T-cells |
| NIX | NIP3-like protein X |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| OPTN | Optineurin |
| PARP | Poly(ADP-ribose) polymerase |
| PAS | Phagophore assembly site |
| PE | Phosphatidyl-ethanolamine |
| PEX3 | Peroxisomal biogenesis factor 3 |
| PINK1 | PTEN-induced kinase 1 |
| PS-1 or 2 | Presenilin-1 or 2 |
| PtdIns3P | Phosphatidylinositol 3-phosphate |
| RAGE | Receptor for advanced glycation end-products |
| RARα | Retinoic acid receptor alpha |
| SNARE | Soluble N-ethylmaleimide-sensitive-factor attachment protein receptor |
| SQSTM1 | Sequestosome 1 |
| STX17 | Syntaxin 17 |
| TAX1BP1 | Tax1 (Human T-cell Leukemia Virus Type I) binding protein 1 |
| TBK1 | Tank-binding kinase 1 |
| TPD52 | Tumor protein D52 |
| TEX264 | Testis expressed 264 |
| TFEB | Transcription factor EB |
| TLR | Toll-like receptors |
| TNKS1 | Tankyrase 1 |
| UPS | Ubiquitin-proteasome system |
| VHL | von Hippel–Lindau protein |
| VPS34 | Vacuolar protein sorting 34 |
| WIPI | WD repeat domain, phosphoinositide interacting |
References
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Vishnumukkala, T.; Gopalakrishna, P.K.; Jagadeesan, S.; Chiroma, S.M.; Mohd Nor, N.H.; Baharuldin, M.T.H.; Thomas, W.; Mohd Moklas, M.A. Herbal Medicine: A Promising Approach for the Treatment of Alzheimer’s Disease. Int. J. Ayurveda Pharma Res. 2024, 12, 142–150. [Google Scholar] [CrossRef]
- Mat Nuri, T.H.; Hong, Y.H.; Ming, L.C.; Mohd Joffry, S.; Othman, M.F.; Neoh, C.F. Knowledge on Alzheimer’s Disease among Public Hospitals and Health Clinics Pharmacists in the State of Selangor, Malaysia. Front. Pharmacol. 2017, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Qi, J.; Lin, S.; Liu, X.; Yin, P.; Wang, Z.; Tang, R.; Wang, J.; Huang, Q.; Li, J.; et al. The China Alzheimer Report 2022. Gen. Psychiatry 2022, 35, e100751. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Y.; Chen, Y.; Lobanov-Rostovsky, S.; Liu, Y.; Zeng, M.; Bandosz, P.; Xu, D.R.; Wang, X.; Liu, Y.; et al. Projection for Dementia Burden in China to 2050: A Macro-Simulation Study by Scenarios of Dementia Incidence Trends. Lancet Reg. Health West. Pac. 2024, 50, 101158. [Google Scholar] [CrossRef]
- Nakahori, N.; Sekine, M.; Yamada, M.; Tatsuse, T.; Kido, H.; Suzuki, M. Future Projections of the Prevalence of Dementia in Japan: Results from the Toyama Dementia Survey. BMC Geriatr. 2021, 21, 602. [Google Scholar] [CrossRef]
- Tsering, W.; Prokop, S. Neuritic Plaques—Gateways to Understanding Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 2808–2821. [Google Scholar] [CrossRef]
- Azargoonjahromi, A. The Duality of Amyloid-β: Its Role in Normal and Alzheimer’s Disease States. Mol. Brain 2024, 17, 44. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in Neurodegenerative Diseases: Pathogenesis and Therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, Y.; Huang, Z.; Jing, F.; Qiao, P.; Zou, Q.; Li, J.; Cai, Z. Impaired Autophagy in Amyloid-Beta Pathology: A Traditional Review of Recent Alzheimer’s Research. J. Biomed. Res. 2022, 37, 30–46. [Google Scholar] [CrossRef]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-Specific Autophagy in Inflammatory Diseases: A Potential Therapeutic Target Underlying the Quality Control of Multiple Organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Harnet, M.M. From Christian de Duve to Yoshinori Ohsumi: More to Autophagy Than Just Dining at Home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Kroemer, G. Autophagy in Metabolism and Quality Control: Opposing, Complementary or Interlinked Functions? Autophagy 2022, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Riebisch, A.K.; Mühlen, S.; Beer, Y.Y.; Schmitz, I. Autophagy—A Story of Bacteria Interfering with the Host Cell Degradation Machinery. Pathogens 2021, 10, 110. [Google Scholar] [CrossRef]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Ruga, S.; Zito, M.C.; et al. From Metabolic Syndrome to Neurological Diseases: Role of Autophagy. Front. Cell Dev. Biol. 2021, 9, 651021. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-Mediated Autophagy: A Unique Way to Enter the Lysosome World. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in Mammalian Cells: Revisiting a 40-Year-Old Conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-Deficient Mutants and Conditions for Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-Mediated Induction of Autophagy via an Apg1 Protein Kinase Complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.Y.; Li, L.; Zhu, X.M.; Lu, J.P.; Liu, X.H.; Lin, F.C. The Crucial Role of the Regulatory Mechanism of the Atg1/ULK1 Complex in Fungi. Front. Microbiol. 2022, 13, 1019543. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 Affects Autophagosome Fusion and Links Chaperone-Mediated Autophagy to Macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Kamada, Y.; Suzuki, K.; Kuboshima, N.; Akimatsu, H.; Ota, S.; Ohsumi, M.; Ohsumi, Y. Characterization of a Novel Autophagy-Specific Gene, ATG29. Biochem. Biophys. Res. Commun. 2005, 338, 1884–1889. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two Distinct Vps34 Phosphatidylinositol 3-Kinase Complexes Function in Autophagy and Carboxypeptidase Y Sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M. The Different Autophagy Degradation Pathways and Neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P–RAB11A. Dev. Cell 2018, 45, 114–131.e8. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-Dependent Movement of Autophagosomes Mediates Efficient Encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Kristensen, A.R.; Schandorff, S.; Høyer-Hansen, M.; Nielsen, M.O.; Jäättelä, M.; Dengjel, J.; Andersen, J.S. Ordered Organelle Degradation During Starvation-Induced Autophagy. Mol. Cell. Proteomics 2008, 7, 2419–2428. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation During Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions Between Autophagy Receptors and Ubiquitin-Like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Li, Z.; Chen, X. Ubiquitination in the Regulation of Autophagy. Acta Biochim. Biophys. Sin. 2023, 55, 1348–1357. [Google Scholar] [CrossRef]
- Marshall, R.S.; Hua, Z.; Mali, S.; McLoughlin, F.; Vierstra, R.D. ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell 2019, 177, 766–781. [Google Scholar] [CrossRef]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329. [Google Scholar] [CrossRef]
- Turco, E.; Fracchiolla, D.; Martens, S. Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 2019, 431, 822–836. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Li, J. PINK1/Parkin-Mediated Mitophagy in Neurodegenerative Diseases. Ageing Res. Rev. 2022, 73, 101817. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Morita, E.; Itoh, T.; Tanaka, A.; Nakaoka, M.; Osada, Y.; Umemoto, T.; Saitoh, T.; Nakatogawa, H.; Kobayashi, S.; et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J. Cell Biol. 2013, 203, 115–128. [Google Scholar] [CrossRef]
- Bussi, C.; Peralta Ramos, J.M.; Arroyo, D.S.; Gallea, J.I.; Ronchi, P.; Kolovou, A.; Wang, J.M.; Florey, O.; Celej, M.S.; Schwab, Y.; et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J. Cell Sci. 2018, 131, jcs226241. [Google Scholar] [CrossRef]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef]
- Nuttall, J.M.; Motley, A.M.; Hettema, E.H. Deficiency of the exportomer components Pex1, Pex6, and Pex15 causes enhanced pexophagy in Saccharomyces cerevisiae. Autophagy 2014, 10, 835–845. [Google Scholar] [CrossRef]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Shah, H.V.; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol. Cell 2020, 80, 779–795.e10. [Google Scholar] [CrossRef]
- Cui, Z.; Napolitano, G.; de Araujo, M.E.G.; Esposito, A.; Monfregola, J.; Huber, L.A.; Ballabio, A.; Hurley, J.H. Structure of the lysosomal mTORC1-TFEB-Rag-Ragulator megacomplex. Nature 2023, 614, 572–579. [Google Scholar] [CrossRef]
- Malampati, S.; Song, J.-X.; Tong, B.C.-K.; Nalluri, A.; Yang, C.-B.; Wang, Z.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.; et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells 2020, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Eapen, V.V.; Swarup, S.; Hoyer, M.J.; Paulo, J.A.; Harper, J.W. Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. ELife 2021, 10, e72328. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef]
- Karabiyik, C.; Vicinanza, M.; Son, S.M.; Rubinsztein, D.C. Glucose starvation induces autophagy via ULK1-mediated activation of PIKfyve in an AMPK-dependent manner. Dev. Cell 2021, 56, 1961–1975. [Google Scholar] [CrossRef]
- Vicinanza, M.; Korolchuk, V.I.; Ashkenazi, A.; Puri, C.; Menzies, F.M.; Clarke, J.H.; Rubinsztein, D.C. PI(5)P regulates autophagosome biogenesis. Mol. Cell 2015, 57, 219–234. [Google Scholar] [CrossRef]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif- promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef]
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef]
- Li, X.; Han, H.; Zhou, M.-T.; Yang, B.; Ta, A.P.; Li, N.; Chen, J.; Wang, W. Proteomic Analysis of the Human Tankyrase Protein Interaction Network Reveals Its Role in Pexophagy. Cell Rep. 2017, 20, 737–749. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Honda, S.; Torii, S.; Shimizu, K.; Katoh, K.; Miyake, K.; Miyake, N.; Fujikake, N.; Sakurai, H.T.; Arakawa, S.; et al. Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration. Nat. Commun. 2020, 11, 5311. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Honda, S.; Arakawa, S.; Yamaguchi, H.; Torii, S.; Sakurai, H.T.; Tsujioka, M.; Murohashi, M.; Shimizu, S. Association between Atg5-independent alternative autophagy and neurodegenerative diseases. J. Mol. Biol. 2020, 432, 2622–2632. [Google Scholar] [CrossRef]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The roles of ubiquitin in mediating autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef]
- Sun-Wang, J.L.; Ivanova, S.; Zorzano, A. The dialogue between the ubiquitin-proteasome system and autophagy: Implications in ageing. Ageing Res. Rev. 2020, 64, 101203. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of Regulators of Chaperone-Mediated Autophagy. Mol. Cell 2010, 39, 535–547. [Google Scholar] [CrossRef]
- Endicott, S.J.; Ziemba, Z.J.; Beckmann, L.J.; Boynton, D.N.; Miller, R.A. Inhibition of Class I PI3K Enhances Chaperone-Mediated Autophagy. J. Cell Biol. 2020, 219, e202001031. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical Modulation of Chaperone-Mediated Autophagy by Retinoic Acid Derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S. Microautophagy—Distinct Molecular Mechanisms Handle Cargoes of Many Sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.J.; Cuervo, A.M. Assessment of Mammalian Endosomal Microautophagy. Methods Cell Biol. 2021, 164, 167–185. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-Wide Analysis of Chaperone-Mediated Autophagy Targeting Motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.A.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation Induces Rapid Degradation of Selective Autophagy Receptors by Endosomal Microautophagy. J. Cell Biol. 2018, 217, 3640–3655. [Google Scholar] [CrossRef]
- Suzuki, K.; Terry, R.D. Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol. 1967, 8, 276–284. [Google Scholar] [CrossRef]
- Ruan, Z.; Pathak, D.; Kalavai, S.V.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s Disease Brain-Derived Extracellular Vesicles Spread Tau Pathology in Interneurons. Brain 2021, 144, 288–309. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Song, Y.Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty Autolysosome Acidification in Alzheimer’s Disease Mouse Models Induces Autophagic Build-Up of Aβ in Neurons, Yielding Senile Plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal Cell Death Mechanisms in Alzheimer’s Disease: An Insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Zhang, J.; Zhang, X.; Yang, G. Molecular Mechanism of Autophagy: Its Role in the Therapy of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 720–739. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; An, P.; Li, H.; Zhou, Z.; Sun, Y.; Wang, J.; Ma, L.; Lu, B. Tau Accumulation via Reduced Autophagy Mediates GGGGCC Repeat Expansion-Induced Neurodegeneration in Drosophila Model of ALS. Neurosci. Bull. 2020, 36, 1414–1428. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sealey, M.; Ruiz, E.; Pegasiou, C.M.; Brookes, K.; Green, S.; Crisford, A.; Duque-Vasquez, M.; Luckett, E.; Robertson, R.; et al. Age-Related Changes in Tau and Autophagy in Human Brain in the Absence of Neurodegeneration. PLoS ONE 2023, 18, e0262792. [Google Scholar] [CrossRef]
- Deng, Z.; Dong, Y.; Zhou, X.; Lu, J.H.; Yue, Z. Pharmacological Modulation of Autophagy for Alzheimer’s Disease Therapy: Opportunities and Obstacles. Acta Pharm. Sin. B 2022, 12, 1688–1706. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome Dysfunction as a Cause of Neurodegenerative Diseases: Lessons from Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu. Rev. Pathol. 2022, 17, 73–99. [Google Scholar] [CrossRef]
- Pires, M.; Rego, A.C. Apoe4 and Alzheimer’s Disease Pathogenesis—Mitochondrial Deregulation and Targeted Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 778. [Google Scholar] [CrossRef]
- Journal of Healthcare Engineering. Retracted: Inhibiting Autophagy Pathway of PI3K/AKT/mTOR Promotes Apoptosis in SK-N-SH Cell Model of Alzheimer’s Disease. J. Healthc. Eng. 2023, 2023, 9780432. [Google Scholar] [CrossRef] [PubMed]
- Suelves, N.; Saleki, S.; Ibrahim, T.; Palomares, D.; Moonen, S.; Koper, M.J.; Vrancx, C.; Vadukul, D.M.; Papadopoulos, N.; Viceconte, N.; et al. Senescence-Related Impairment of Autophagy Induces Toxic Intraneuronal Amyloid-β Accumulation in a Mouse Model of Amyloid Pathology. Acta Neuropathol. Commun. 2023, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Reed, E.G.; Keller-Norrell, P.R. Minding the Gap: Exploring Neuroinflammatory and Microglial Sex Differences in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 17377. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamilanban, T.; Alsayari, A.; Ramachawolran, G.; Wong, L.S.; Sekar, M.; Gan, S.H.; Subramaniyan, V.; Chinni, S.V.; Izzati Mat Rani, N.N.; et al. Trilateral Association of Autophagy, mTOR and Alzheimer’s Disease: Potential Pathway in the Development for Alzheimer’s Disease Therapy. Front. Pharmacol. 2022, 13, 1094351. [Google Scholar] [CrossRef]
- Krishnan, S.; Shrestha, Y.; Jayatunga, D.P.W.; Rea, S.; Martins, R.; Bharadwaj, P. Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6739. [Google Scholar] [CrossRef]
- Feng, Q.; Luo, Y.; Zhang, X.N.; Yang, X.F.; Hong, X.Y.; Sun, D.S.; Li, X.C.; Hu, Y.; Li, X.G.; Zhang, J.F.; et al. MAPT/Tau Accumulation Represses Autophagy Flux by Disrupting IST1-Regulated ESCRT-III Complex Formation: A Vicious Cycle in Alzheimer Neurodegeneration. Autophagy 2020, 16, 641–658. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, J.; Zhu, Y.; Wang, L.; Jiang, X.; Liu, B.; He, G. Targeting Autophagy and Beyond: Deconvoluting the Complexity of Beclin-1 from Biological Function to Cancer Therapy. Acta Pharm. Sin. B 2023, 13, 4688–4714. [Google Scholar] [CrossRef]
- He, Z.; Yang, Y.; Xing, Z.; Zuo, Z.; Wang, R.; Gu, H.; Qi, F.; Yao, Z. Intraperitoneal Injection of IFN-γ Restores Microglial Autophagy, Promotes Amyloid-β Clearance and Improves Cognition in APP/PS1 Mice. Cell Death Dis. 2020, 11, 440. [Google Scholar] [CrossRef]
- Deng, M.; Huang, L.; Zhong, X. β-Asarone Modulates Beclin 1, LC3 and p62 Expression to Attenuate Aβ40 and Aβ42 Levels in APP/PS1 Transgenic Mice with Alzheimer’s Disease. Mol. Med. Rep. 2020, 21, 2095–2102. [Google Scholar] [CrossRef]
- Lachance, V.; Wang, Q.; Sweet, E.; Choi, I.; Cai, C.Z.; Zhuang, X.X.; Zhang, Y.; Jiang, J.L.; Blitzer, R.D.; Bozdagi-Gunal, O.; et al. Autophagy Protein NRBF2 Has Reduced Expression in Alzheimer’s Brains and Modulates Memory and Amyloid-Beta Homeostasis in Mice. Mol. Neurodegener. 2019, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- McLeod, I.X.; Saxena, R.; Carico, Z.; He, Y.W. Class I PI3K Provide Lipid Substrate in T Cell Autophagy through Linked Activity of Inositol Phosphatases. Front. Cell Dev. Biol. 2021, 9, 709398. [Google Scholar] [CrossRef] [PubMed]
- Jalin, A.M.A.; Jin, R.; Wang, M.; Li, G. EPPS Treatment Attenuates Traumatic Brain Injury in Mice by Reducing Aβ Burden and Ameliorating Neuronal Autophagic Flux. Exp. Neurol. 2019, 314, 20–33. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, Regulation and Pathophysiological Implications of Autophagosome Maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Qin, Z.H.; Sheng, R. The Multiple Roles of Autophagy in Neural Function and Diseases. Neurosci. Bull. 2023, in press. [CrossRef]
- Kulkarni, V.V.; Stempel, M.H.; Anand, A.; Sidibe, D.K.; Maday, S. Retrograde Axonal Autophagy and Endocytic Pathways Are Parallel and Separate in Neurons. J. Neurosci. 2022, 42, 8524–8541. [Google Scholar] [CrossRef]
- Fahimi, A.; Noroozi, M.; Salehi, A. Enlargement of Early Endosomes and Traffic Jam in Basal Forebrain Cholinergic Neurons in Alzheimer’s Disease. Handb. Clin. Neurol. 2021, 179, 207–218. [Google Scholar] [CrossRef]
- Conway, O.; Akpinar, H.A.; Rogov, V.V.; Kirkin, V. Selective Autophagy Receptors in Neuronal Health and Disease. J. Mol. Biol. 2020, 432, 2483–2509. [Google Scholar] [CrossRef]
- Ganesan, D.; Cai, Q. Understanding Amphisomes. Biochem. J. 2021, 478, 1959–1976. [Google Scholar] [CrossRef]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Mol. Neurosci. 2020, 13, 586731. [Google Scholar] [CrossRef]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated Tau Inhibits Chaperone-Mediated Autophagy and Promotes Tau Pathology Propagation in Mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Ho, C.S.; Huang, Y.W.; Wu, T.Y.; Lee, T.H.; Huang, Z.D.; Wang, T.J.; Yang, S.J.; Chiang, M.F. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Long, Z.; Ding, Y.; Jiang, T.; Liu, J.; Li, Y.; Liu, Y.; Peng, X.; Wang, K.; Feng, M.; et al. Dihydroartemisinin Ameliorates Learning and Memory in Alzheimer’s Disease through Promoting Autophagosome–Lysosome Fusion and Autolysosomal Degradation for Aβ Clearance. Front. Aging Neurosci. 2020, 12, 47. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s Disease: Molecular Defects and Therapeutic Approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef]
- Kendall, R.L.; Holian, A. The Role of Lysosomal Ion Channels in Lysosome Dysfunction. Inhal. Toxicol. 2021, 33, 41–54. [Google Scholar] [CrossRef]
- Mustaly-Kalimi, S.; Gallegos, W.; Marr, R.A.; Gilman-Sachs, A.; Peterson, D.A.; Sekler, I.; Stutzmann, G.E. Protein Mishandling and Impaired Lysosomal Proteolysis Generated through Calcium Dysregulation in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2211999119. [Google Scholar] [CrossRef]
- Sose, P.M.; Doshi, G.M.; Kale, P.P. An Update on Autophagy as a Target in the Treatment of Alzheimer’s Disease. Curr. Drug Targets 2023, 24, 547–567. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Q.; Ni, Y.; Le, W. Autophagy and Alzheimer’s Disease. In Advances Experimental Medicine and Biology; Springer: Singapore, Singapore, 2020; Volume 1207, pp. 3–19. [Google Scholar] [CrossRef]
- Murariu, M.; Drochioiu, G. Biostructural theory of the living systems. BioSystems 2012, 109, 126–132. [Google Scholar] [CrossRef]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Han, X.; Tang, Y.; Zhang, Y.; Zhang, J.; Hu, Z.; Xu, W.; Xu, S.; Niu, Q. Impaired V-ATPase leads to increased lysosomal pH, results in disrupted lysosomal degradation and autophagic flux blockage, contributes to fluoride-induced developmental neurotoxicity. Ecotoxicol. Environ. Saf. 2022, 236, 113500. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the Bi-Directional Relationship between Autophagy and Alzheimer’s Disease. CNS Neurosci. Ther. 2020, 26, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.R.; Martins, R.N. Autophagy Modulates Aβ Accumulation and Formation of Aggregates in Yeast. Mol. Cell. Neurosci. 2020, 104, 103466. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Taylor-Robinson, S.D.; Fertleman, M.; Khan, M.; Neely, D. Is Alzheimer’s Disease a Liver Disease of the Brain? J. Alzheimers Dis. 2020, 75, 1–14. [Google Scholar] [CrossRef]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.Y.; Wang, Y.J. Peripheral Clearance of Brain-Derived Aβ in Alzheimer’s Disease: Pathophysiology and Therapeutic Perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. [Google Scholar] [CrossRef]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective Autophagy as a Therapeutic Target for Neurological Diseases. Cell. Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef]
- Le Guerroué, F.; Youle, R.J. Ubiquitin Signaling in Neurodegenerative Diseases: An Autophagy and Proteasome Perspective. Cell Death Differ. 2021, 28, 439–454. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Li, Q.; Xing, S.; Liu, Y.; Liu, Y.; Chen, Y.; Liu, W.; Feng, F.; Sun, H. p62/SQSTM1, a Central but Unexploited Target: Advances in Its Physiological/Pathogenic Functions and Small Molecular Modulators. J. Med. Chem. 2020, 63, 10135–10157. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.; Lin, M.; Rios-Colon, L.; Qi, Q.; Moore, J.T.; Kumar, D. Emerging Roles of Impaired Autophagy in Fatty Liver Disease and Hepatocellular Carcinoma. Int. J. Hepatol. 2021, 2021, 6675762. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Mo, S.T.; Chen, I.T.; Hsieh, F.-Y.; Hsieh, S.-L.; Zhang, J.; Lai, M.-Z. Caspase-8 Inactivation Drives Autophagy-Dependent Inflammasome Activation in Myeloid Cells. Sci. Adv. 2022, 8, eabn9912. [Google Scholar] [CrossRef] [PubMed]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H.-D. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.-X. Autophagy in Liver Diseases: A Review. Mol. Asp. Med. 2021, 82, 100973. [Google Scholar] [CrossRef]
- Gao, P.Y.; Ou, Y.N.; Wang, H.F.; Wang, Z.B.; Fu, Y.; He, X.Y.; Ma, Y.H.; Feng, J.F.; Cheng, W.; Tan, L.; et al. Associations of liver dysfunction with incident dementia, cognition, and brain structure: A prospective cohort study of 431 699 adults. J. Neurochem. 2024, 168, 26–38. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, W.; Hou, X.; Wu, J.; Wang, Y.; Fan, J.; Zhang, Z.; Yuan, Z.; Sun, C.; Lu, B.; et al. Metabolic-associated steatotic liver disease and risk of Alzheimer’s disease: A real-world retrospective cohort study. Front. Endocrinol. 2024, 15, 1451908. [Google Scholar] [CrossRef]
- Weinstein, G.; Schonmann, Y.; Yeshua, H.; Zelber-Sagi, S. The association between liver fibrosis score and incident dementia: A nationwide retrospective cohort study. Alzheimers Dement. 2024, 20, 5385–5397. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Silvey, S.G.; Rogal, S.; O’Leary, J.G.; Patton, H.; Morgan, T.R.; Kanagalingam, G.; Gentili, A.; Godschalk, M.; Patel, N. Undiagnosed Cirrhosis and Hepatic Encephalopathy in a National Cohort of Veterans With Dementia. JAMA Netw. Open 2024, 7, e2353965. [Google Scholar] [CrossRef]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. ELife 2017, 6, e21776. [Google Scholar] [CrossRef]
- Son, J.; Shim, J.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.-W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.-H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714. [Google Scholar] [CrossRef]
- Fote, G.M.; Geller, N.R.; Efstathiou, N.E.; Hendricks, N.; Vavvas, D.G.; Reidling, J.C.; Thompson, L.M.; Steffan, J.S. Isoform-dependent lysosomal degradation and internalization of apolipoprotein E requires autophagy proteins. J. Cell Sci. 2022, 135, jcs258687. [Google Scholar] [CrossRef]
- Valdor, R.; Martinez-Vicente, M. The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis. Biomedicines 2024, 12, 257. [Google Scholar] [CrossRef]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Angeletti, M.; Keller, J.N.; Eleuteri, A.M. The fine-tuning of proteolytic pathways in Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 3433–3451. [Google Scholar] [CrossRef]
- Limone, A.; Veneruso, I.; D’Argenio, V.; Sarnataro, D. Endosomal trafficking and related genetic underpinnings as a hub in Alzheimer’s disease. J. Cell. Physiol. 2022, 237, 3803–3815. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological Amyloid-Beta Clearance in the Periphery and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Walker, J.E.; Sue, L.I.; Dugger, B.N.; Beach, T.G.; Serrano, G.E. Impaired Hepatic Amyloid-Beta Degradation in Alzheimer’s Disease. PLoS ONE 2018, 13, e0203659. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus Promotes Autophagic Clearance of Amyloid-β and Provides Protective Effects in Cellular and Animal Models of Alzheimer’s Disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lin, H.W.K.; Zhang, Q.; Zong, X. Targeting Alzheimer’s Disease: The Critical Crosstalk between the Liver and Brain. Nutrients 2022, 14, 4298. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Marambaud, P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Kandimalla, R.; Kuruva, C.S. Protective effects of a natural product, curcumin, against amyloid β induced mitochondrial and synaptic toxicities in Alzheimer’s disease. J. Investig. Med. 2016, 64, 1220–1234. [Google Scholar] [CrossRef]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef]
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric restriction mimetics against age-associated disease: Targets, mechanisms, and therapeutic potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef]
- Jiang, Y.; Uhm, H.; Ip, F.C.; Ouyang, L.; Lo, R.M.N.; Cheng, E.Y.L.; Cao, X.; Tan, C.M.C.; Law, B.C.H.; Ortiz-Romero, P.; et al. A Blood-Based Multi-Pathway Biomarker Assay for Early Detection and Staging of Alzheimer’s Disease across Ethnic Groups. Alzheimers Dement. 2024, 20, 2000–2015. [Google Scholar] [CrossRef]
- Ren, J.; Sowers, J.R.; Zhang, Y. Metabolic Stress, Autophagy, and Cardiovascular Aging: From Pathophysiology to Therapeutics. Trends Endocrinol. Metab. 2018, 29, 699–711. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hein, Z.M.; Vishnumukkala, T.; Karikalan, B.; Alkatiri, A.; Hussan, F.; Jagadeesan, S.; Kamaruzzaman, M.A.; Che Ramli, M.D.; Che Mohd Nassir, C.M.N.; Gopalakrishna, P.K. Autophagy and Alzheimer’s Disease: Mechanisms and Impact Beyond the Brain. Cells 2025, 14, 911. https://doi.org/10.3390/cells14120911
Hein ZM, Vishnumukkala T, Karikalan B, Alkatiri A, Hussan F, Jagadeesan S, Kamaruzzaman MA, Che Ramli MD, Che Mohd Nassir CMN, Gopalakrishna PK. Autophagy and Alzheimer’s Disease: Mechanisms and Impact Beyond the Brain. Cells. 2025; 14(12):911. https://doi.org/10.3390/cells14120911
Chicago/Turabian StyleHein, Zaw Myo, Thirupathirao Vishnumukkala, Barani Karikalan, Aisyah Alkatiri, Farida Hussan, Saravanan Jagadeesan, Mohd Amir Kamaruzzaman, Muhammad Danial Che Ramli, Che Mohd Nasril Che Mohd Nassir, and Prarthana Kalerammana Gopalakrishna. 2025. "Autophagy and Alzheimer’s Disease: Mechanisms and Impact Beyond the Brain" Cells 14, no. 12: 911. https://doi.org/10.3390/cells14120911
APA StyleHein, Z. M., Vishnumukkala, T., Karikalan, B., Alkatiri, A., Hussan, F., Jagadeesan, S., Kamaruzzaman, M. A., Che Ramli, M. D., Che Mohd Nassir, C. M. N., & Gopalakrishna, P. K. (2025). Autophagy and Alzheimer’s Disease: Mechanisms and Impact Beyond the Brain. Cells, 14(12), 911. https://doi.org/10.3390/cells14120911