
Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy

 ,
,  , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Collection
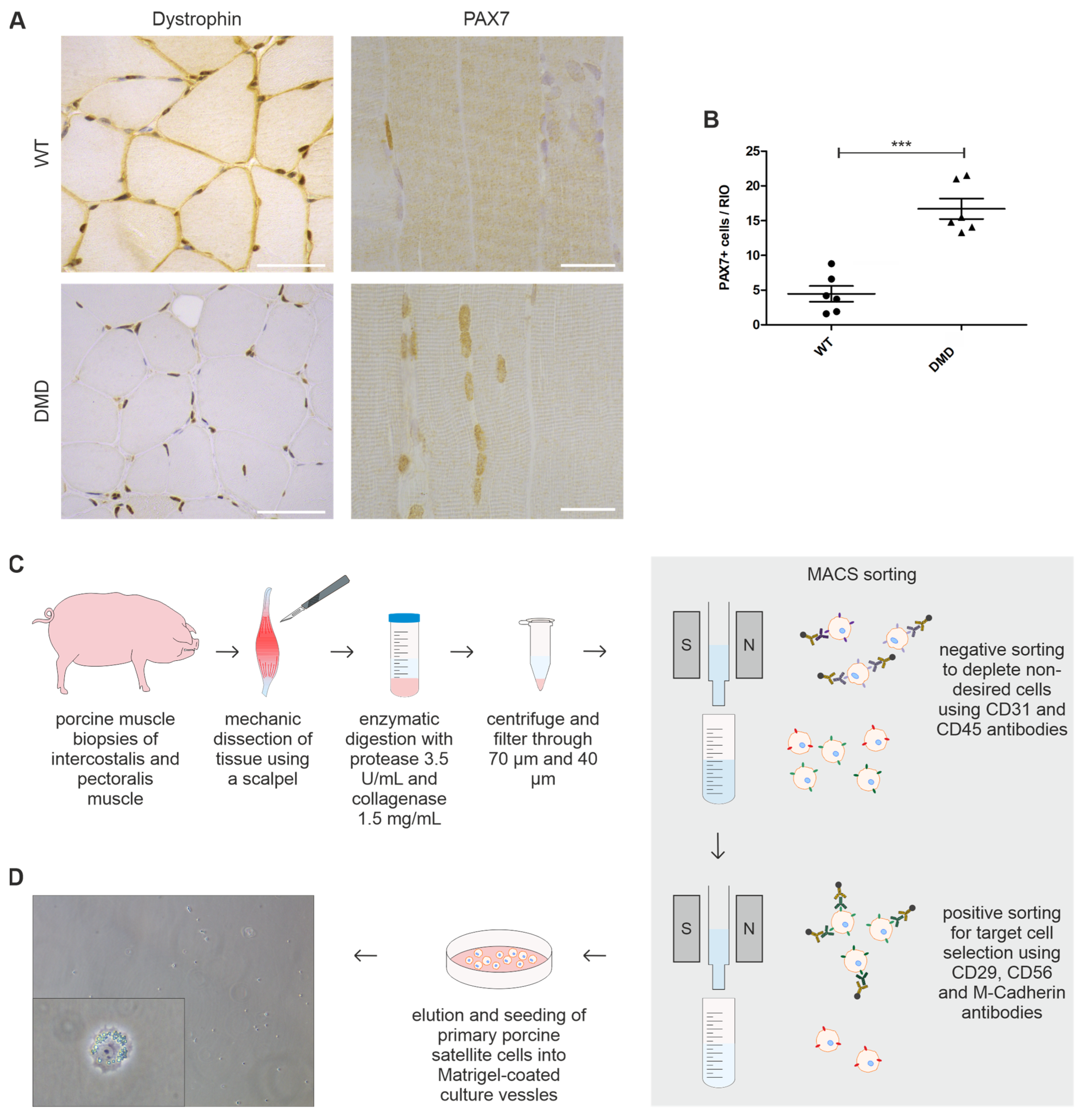
2.2. Isolation of Primary Porcine Satellite Cells
2.2.1. Dissociation of Porcine Muscle Tissue
2.2.2. Magnetically Activated Cell Sorting
2.3. Primary Cell Culture
2.4. Traction Force Microscopy
2.4.1. Silicone Substrate Preparation
2.4.2. Traction Force Measurements and Data Analysis
2.5. Immunohistochemistry
2.6. Immunofluorescence
2.7. Immunoblotting
2.8. RNA Isolation from Cell Lines, Library Preparation, Sequencing, and Data Analysis
2.9. Sample Preparation for LC-MS/MS and Proteome Data Analysis
2.10. Multi-Omics Factor Analysis
2.11. Statistical Analysis
3. Results
3.1. Muscle Sections of DMD Animals Harbor Significantly Elevated Levels of PAX7+ SCs
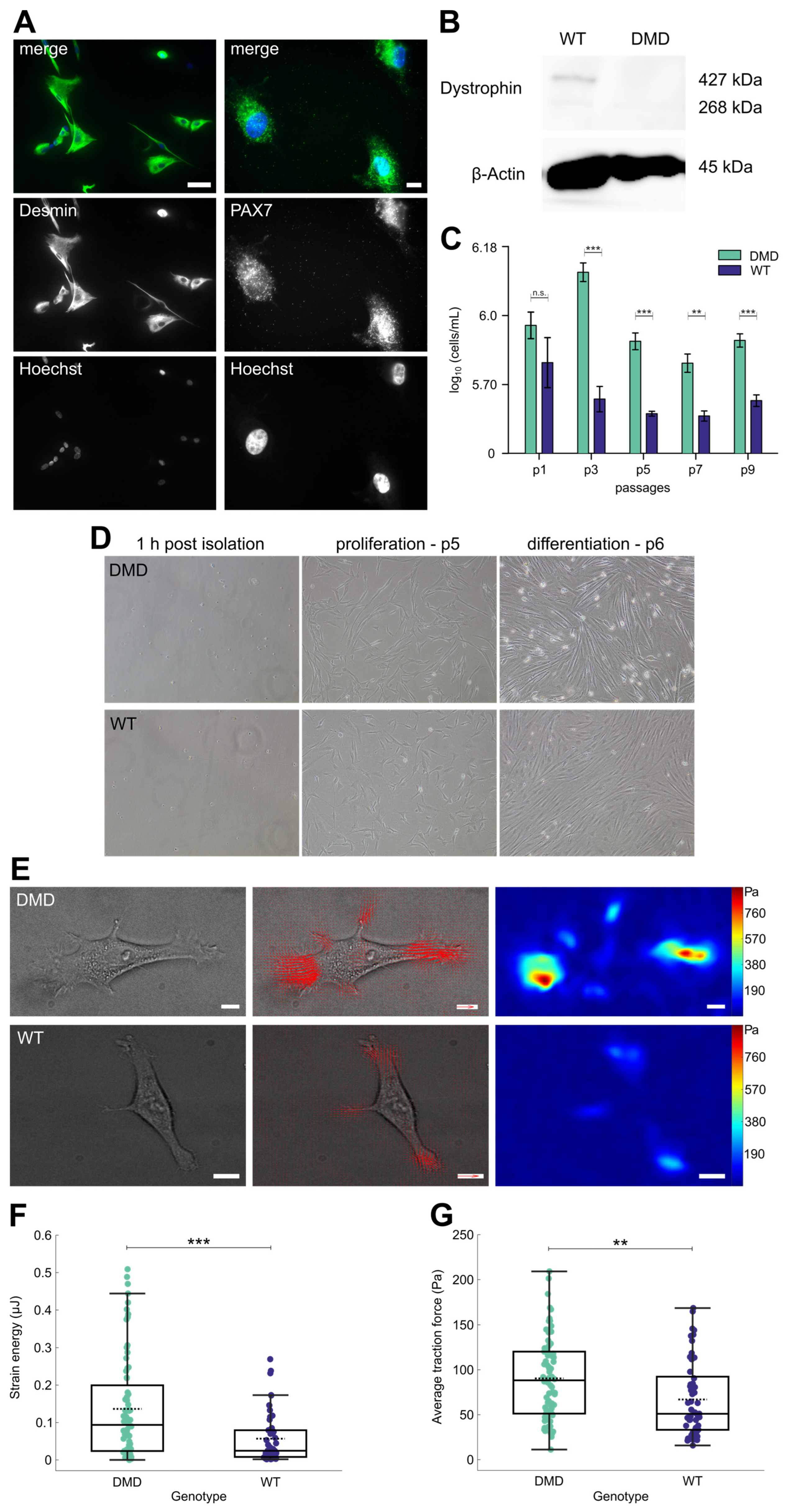
3.2. Isolated PAX7+ SCs from DMD Piglets Lack Dystrophin Expression
3.3. DMD SCs Morphology Under Proliferation and Differentiation Conditions
3.4. DMD SCs Exhibit Increased Force Generation
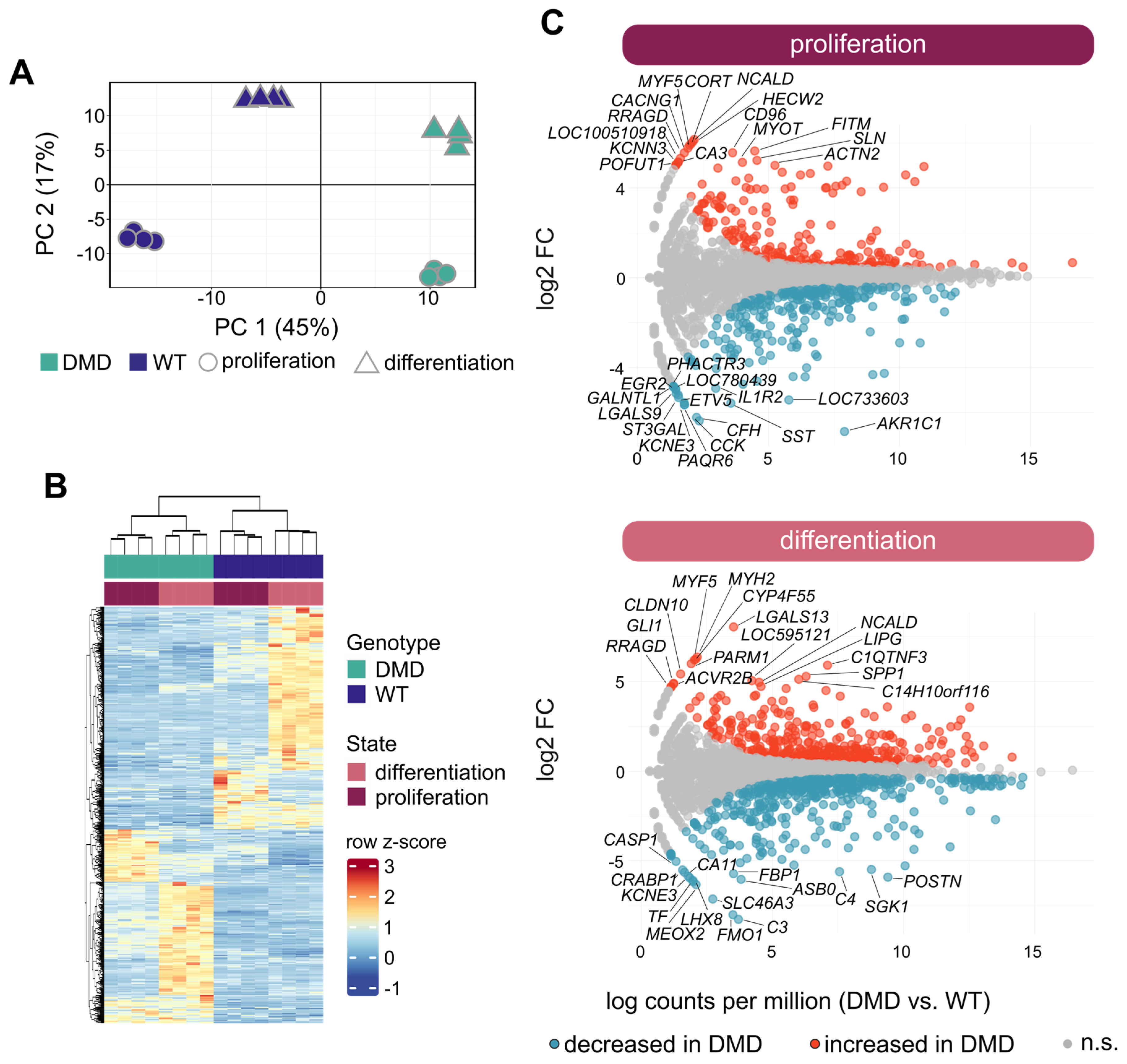
3.5. Overview of Transcriptome Profiles of DMD and WT SCs
3.6. Transcriptional Changes in DMD SCs Analyzed in Proliferative and Differentiated States
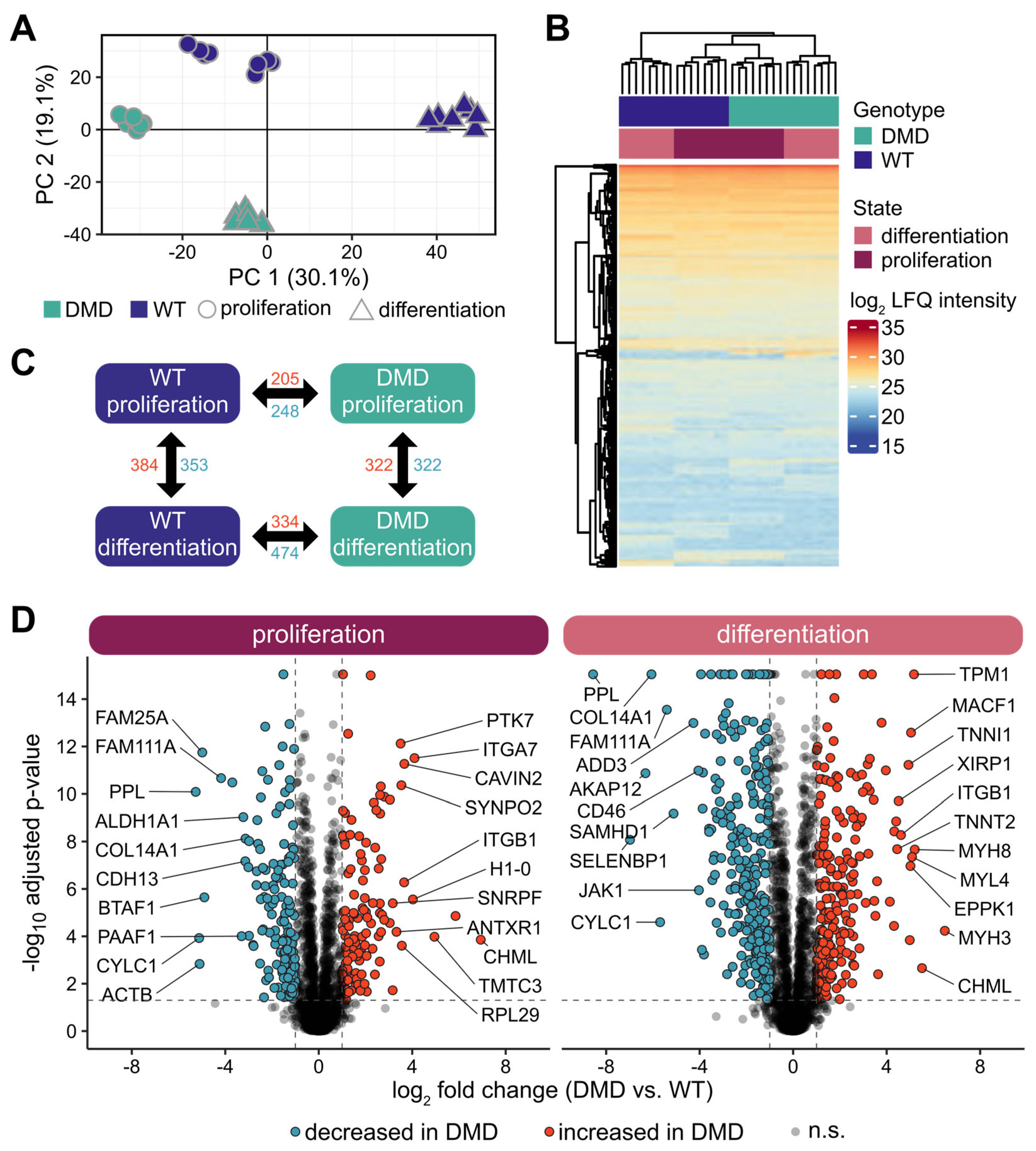
3.7. Overview of the Proteome Profiles of DMD and WT SCs
3.8. Changes in Protein Expression in DMD SCs Analyzed in Proliferative and Differentiated States
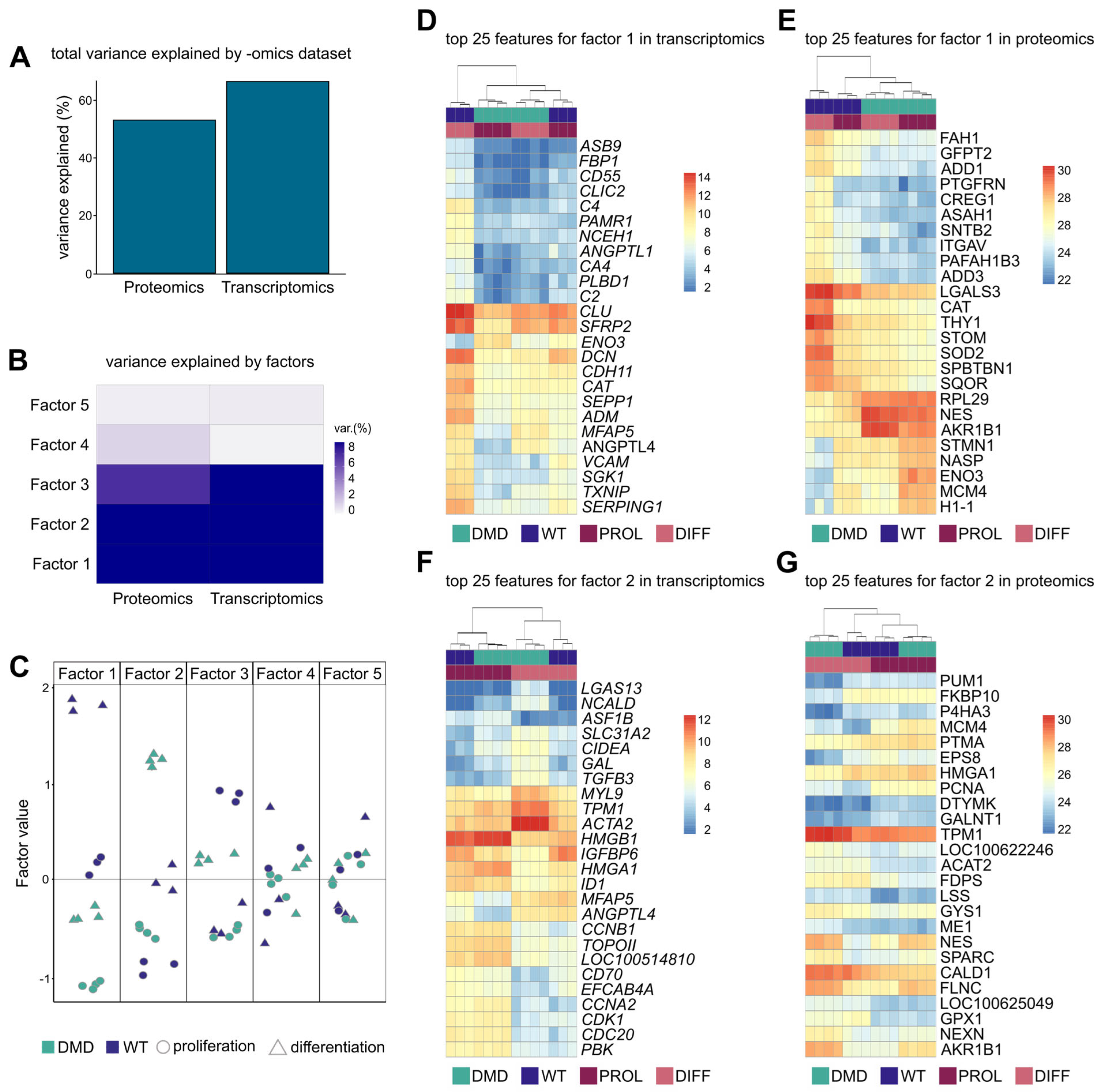
3.9. Integration of Transcriptomic and Proteomic Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Actin |
| AKR1C1 | Aldo-Keto Reductase Family 1 Member C1 |
| ALDH | Aldehyde dehydrogenase |
| BP | Biological process |
| C3 | Complement Component 3 |
| CCK | Cholecystokinin |
| CFH | Complement Factor H |
| CNN1 | Calponin 1 |
| CORT | Cortistatin |
| CYLC1 | Cytoplasmic Linker Protein 1 |
| DAB | Diaminobenzidine tetrahydrochloride dihydrate |
| DES | Desmin |
| DGC | Dystrophin-associated glycoprotein complex |
| DIFF | Differentiation |
| DMD | Duchenne muscular dystrophy |
| ECM | Extracellular matrix |
| FMO1 | Flavin-Containing Monooxygenase 1 |
| GO | Gene Ontology |
| GTP | Guanosine Triphosphate |
| HECW2 | HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 2 |
| HIF1α | Hypoxia-Inducible Factor 1 Alpha |
| IgG | Immunoglobulin G |
| INT | Intercostalis muscle |
| ITG | Integrin |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| LF | Latent factor |
| LGALS13 | Lectin, Galactoside-Binding, Soluble, 13 |
| MACF1 | Microtubule Actin Crosslinking Factor 1 |
| MACS | Magnetically activated cell sorting |
| MARK2 | Microtubule Affinity-Regulating Kinase 2 |
| MOFA | Multi-omics factor analysis |
| MYF5 | Myogenic regulatory factor 5 |
| MYH | Myosin Heavy Chain |
| MYL | Myosin Light Chain |
| MYLPF | Myosin Light Chain Phosphatase, F subunit |
| MYOD1 | Myoblast determination protein 1 |
| MYOG | Muscle-specific transcription factor myogenin |
| NCALD | Neurocalcin Delta |
| PAQR6 | Progestin and AdipoQ Receptor Family Member 6 |
| PAX7 | Paired box protein 7 |
| PECT | Pectoralis muscle |
| PPL | Plakophilin 1 |
| PROL | Proliferation |
| SCs | Satellite cells |
| SLC46A3 | Solute Carrier Family 46 Member 3 |
| TAGLN | Transgelin |
| TFM | Traction force microscopy |
| TGF-β | Transforming Growth Factor Beta |
| TNN | Troponin |
| TPM | Tropomyosin 1 |
| WT | Wild type |
References
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Hildyard, J.C.W.; Roskrow, L.E.; Wells, D.J.; Piercy, R.J. Spatiotemporal analysis of dystrophin expression during muscle repair. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kodippili, K.; Rudnicki, M.A. Satellite cell contribution to disease pathology in Duchenne muscular dystrophy. Front. Physiol. 2023, 14, 1180980. [Google Scholar] [CrossRef]
- Gosselin, M.R.F.; Mournetas, V.; Borczyk, M.; Verma, S.; Occhipinti, A.; Rog, J.; Bozycki, L.; Korostynski, M.; Robson, S.C.; Angione, C.; et al. Loss of full-length dystrophin expression results in major cell-autonomous abnormalities in proliferating myoblasts. eLife 2022, 11, e75521. [Google Scholar] [CrossRef]
- Kottlors, M.; Kirschner, J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bankole, L.C.; Feasson, L.; Ponsot, E.; Kadi, F. Fibre type-specific satellite cell content in two models of muscle disease. Histopathology 2013, 63, 826–832. [Google Scholar] [CrossRef]
- Reimann, J.; Irintchev, A.; Wernig, A. Regenerative capacity and the number of satellite cells in soleus muscles of normal and mdx mice. Neuromuscul. Disord. 2000, 10, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.F., Jr.; Souza, L.S.; Almeida, C.F.; Ishiba, R.; Fernandes, S.A.; Guerrieri, D.A.; Santos, A.L.F.; Onofre-Oliveira, P.C.G.; Vainzof, M. Muscle satellite cells and impaired late stage regeneration in different murine models for muscular dystrophies. Sci. Rep. 2019, 9, 11842. [Google Scholar] [CrossRef]
- Heslop, L.; Morgan, J.E.; Partridge, T.A. Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J. Cell Sci. 2000, 113 Pt 12, 2299–2308. [Google Scholar] [CrossRef]
- Luz, M.A.; Marques, M.J.; Santo Neto, H. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz. J. Med. Biol. Res. 2002, 35, 691–695. [Google Scholar] [CrossRef]
- Dumont, N.A.; Rudnicki, M.A. Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. npj Regen. Med. 2016, 1, 16006. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Bencze, M.; Borok, M.J.; Der Vartanian, A.; Gattazzo, F.; Mademtzoglou, D.; Perez-Diaz, S.; Prola, A.; Reyes-Fernandez, P.C.; Rotini, A.; et al. Perspectives on skeletal muscle stem cells. Nat. Commun. 2021, 12, 692. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Muntoni, F.; Zammit, P.S. Defining and identifying satellite cell-opathies within muscular dystrophies and myopathies. Exp. Cell Res. 2022, 411, 112906. [Google Scholar] [CrossRef]
- Ganassi, M.; Zammit, P.S. Involvement of muscle satellite cell dysfunction in neuromuscular disorders: Expanding the portfolio of satellite cell-opathies. Eur. J. Transl. Myol. 2022, 32, 10064. [Google Scholar] [CrossRef]
- Emery, A.E.H. Population Frequencies of Inherited Neuromuscular Diseases—A World Survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Happi Mbakam, C.; Tremblay, J.P. Gene therapy for Duchenne muscular dystrophy: An update on the latest clinical developments. Expert Rev. Neurother. 2023, 23, 905–920. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Fontelonga, T.M.; Jordan, B.; Nunes, A.M.; Barraza-Flores, P.; Bolden, N.; Wuebbles, R.D.; Griner, L.M.; Hu, X.; Ferrer, M.; Marugan, J.; et al. Sunitinib promotes myogenic regeneration and mitigates disease progression in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2019, 28, 2120–2132. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; De Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar]
- Wahlgren, L.; Kroksmark, A.K.; Tulinius, M.; Sofou, K. One in five patients with Duchenne muscular dystrophy dies from other causes than cardiac or respiratory failure. Eur. J. Epidemiol. 2022, 37, 147–156. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Models Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Zaynitdinova, M.I.; Lavrov, A.V.; Smirnikhina, S.A. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 2021, 30, 709–725. [Google Scholar] [CrossRef]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Lindner, M.; Chirivi, M.; Lange, A.; Kaufhold, C.; Mayer, C.; Medugorac, I.; Kessler, B.; et al. A scalable, clinically severe pig model for Duchenne muscular dystrophy. Dis. Models Mech. 2021, 14, dmm049285. [Google Scholar] [CrossRef] [PubMed]
- Stirm, M.; Klymiuk, N.; Nagashima, H.; Kupatt, C.; Wolf, E. Pig models for translational Duchenne muscular dystrophy research. Trends Mol. Med. 2024, 30, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Klymiuk, N.; Blutke, A.; Graf, A.; Krause, S.; Burkhardt, K.; Wuensch, A.; Krebs, S.; Kessler, B.; Zakhartchenko, V.; Kurome, M.; et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum. Mol. Genet. 2013, 22, 4368–4382. [Google Scholar] [CrossRef]
- Ding, S.; Wang, F.; Liu, Y.; Li, S.; Zhou, G.; Hu, P. Characterization and isolation of highly purified porcine satellite cells. Cell Death Discov. 2017, 3, 17003. [Google Scholar] [CrossRef]
- Holenstein, C.N.; Silvan, U.; Snedeker, J.G. High-resolution traction force microscopy on small focal adhesions—Improved accuracy through optimal marker distribution and optical flow tracking. Sci. Rep. 2017, 7, 41633. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Schell, C.; Huber, T.B.; Şimşek, A.N.; Hersch, N.; Merkel, R.; Gompper, G.; Sabass, B. Traction force microscopy with optimized regularization and automated Bayesian parameter selection for comparing cells. Sci. Rep. 2019, 9, 539. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Bagnoli, J.W.; Ziegenhain, C.; Janjic, A.; Wange, L.E.; Vieth, B.; Parekh, S.; Geuder, J.; Hellmann, I.; Enard, W. Sensitive and powerful single-cell RNA sequencing using mcSCRB-seq. Nat. Commun. 2018, 9, 2937. [Google Scholar] [CrossRef]
- Andrews, S. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 October 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Signorelli, M.; Tsonaka, R.; Aartsma-Rus, A.; Spitali, P. Multiomic characterization of disease progression in mice lacking dystrophin. PLoS ONE 2023, 18, e0283869. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Ropka-Molik, K.; Eckert, R.; Piorkowska, K. The expression pattern of myogenic regulatory factors MyoD, Myf6 and Pax7 in postnatal porcine skeletal muscles. Gene Expr. Patterns 2011, 11, 79–83. [Google Scholar] [CrossRef]
- Zammit, P.S.; Relaix, F.; Nagata, Y.; Ruiz, A.P.; Collins, C.A.; Partridge, T.A.; Beauchamp, J.R. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006, 119, 1824–1832. [Google Scholar] [CrossRef]
- Jiang, C.; Wen, Y.; Kuroda, K.; Hannon, K.; Rudnicki, M.A.; Kuang, S. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Models Mech. 2014, 7, 997–1004. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Wang, K.; Li, L.; Yuan, H.; Liu, X.; Liu, Y.; Guan, W. Isolation, culture and biological characteristics of multipotent porcine skeletal muscle satellite cells. Cell Tissue Bank. 2017, 18, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Asfour, H.A.; Allouh, M.Z.; Said, R.S. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp. Biol. Med. 2018, 243, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, T.; Toyono, T.; Inoue, A.; Matsubara, T.; Kawamoto, T.; Kokabu, S. Factors Regulating or Regulated by Myogenic Regulatory Factors in Skeletal Muscle Stem Cells. Cells 2022, 11, 1493. [Google Scholar] [CrossRef]
- Bruyere, C.; Versaevel, M.; Mohammed, D.; Alaimo, L.; Luciano, M.; Vercruysse, E.; Gabriele, S. Actomyosin contractility scales with myoblast elongation and enhances differentiation through YAP nuclear export. Sci. Rep. 2019, 9, 15565. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Zettler, S.; Renner, S.; Kemter, E.; Hinrichs, A.; Klymiuk, N.; Backman, M.; Riedel, E.O.; Mueller, C.; Streckel, E.; Braun-Reichhart, C.; et al. A decade of experience with genetically tailored pig models for diabetes and metabolic research. Anim. Reprod. 2020, 17, e20200064. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Stockl, J.B.; Kurome, M.; Kessler, B.; Zakhartchenko, V.; Kemter, E.; Blum, H.; Arnold, G.J.; et al. Pig models for Duchenne muscular dystrophy—From disease mechanisms to validation of new diagnostic and therapeutic concepts. Neuromuscul. Disord. 2022, 32, 543–556. [Google Scholar] [CrossRef]
- Lo Mauro, A.; Aliverti, A. Physiology of respiratory disturbances in muscular dystrophies. Breathe 2016, 12, 318–327. [Google Scholar] [CrossRef]
- Baig, M.A.; Bordoni, B. Anatomy, Shoulder and Upper Limb, Pectoral Muscles. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2024. [Google Scholar]
- Burmeister, D.M.; Supp, D.M.; Clark, R.A.; Tredget, E.E.; Powell, H.M.; Enkhbaatar, P.; Bohannon, J.K.; Cancio, L.C.; Hill, D.M.; Nygaard, R.M. Advantages and Disadvantages of Using Small and Large Animals in Burn Research: Proceedings of the 2021 Research Special Interest Group. J. Burn Care Res. 2022, 43, 1032–1041. [Google Scholar] [CrossRef]
- Hou, N.; Du, X.; Wu, S. Advances in pig models of human diseases. Anim. Models Exp. Med. 2022, 5, 141–152. [Google Scholar] [CrossRef]
- Agley, C.C.; Rowlerson, A.M.; Velloso, C.P.; Lazarus, N.L.; Harridge, S.D. Isolation and quantitative immunocytochemical characterization of primary myogenic cells and fibroblasts from human skeletal muscle. J. Vis. Exp. 2015, 95, e52049. [Google Scholar] [CrossRef]
- Syverud, B.C.; Lee, J.D.; VanDusen, K.W.; Larkin, L.M. Isolation and Purification of Satellite Cells for Skeletal Muscle Tissue Engineering. J. Regen. Med. 2014, 3, 117. [Google Scholar] [CrossRef] [PubMed]
- Dan-Jumbo, S.O.; Riley, S.E.; Cortes-Araya, Y.; Ho, W.; Lee, S.; Thrower, T.; Esteves, C.L.; Donadeu, F.X. Derivation and long-term maintenance of porcine skeletal muscle progenitor cells. Sci. Rep. 2024, 14, 9370. [Google Scholar] [CrossRef]
- Vacanti, V.; Kong, E.; Suzuki, G.; Sato, K.; Canty, J.M.; Lee, T. Phenotypic changes of adult porcine mesenchymal stem cells induced by prolonged passaging in culture. J. Cell. Physiol. 2005, 205, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, J.; Gou, L.; Zhu, Y.; Zhong, W.; Guo, H.; Du, Y. Integrating transcriptomic and proteomic data for a comprehensive molecular perspective on the association between sarcopenia and osteoporosis. Arch. Gerontol. Geriatr. 2024, 125, 105486. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Budnik, B. A review of the current state of single-cell proteomics and future perspective. Anal. Bioanal. Chem. 2023, 415, 6889–6899. [Google Scholar] [CrossRef]
- Cha, J.; Lee, I. Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp. Mol. Med. 2020, 52, 1798–1808. [Google Scholar] [CrossRef]
- Blau, H.M.; Webster, C.; Pavlath, G.K. Defective myoblasts identified in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 1983, 80, 4856–4860. [Google Scholar] [CrossRef]
- Hurko, O.; McKee, L.; Zuurveld, J.; Swick, H.M. Comparison of Duchenne and normal myoblasts from a heterozygote. Neurology 1987, 37, 675. [Google Scholar] [CrossRef]
- Mournetas, V.; Massouridès, E.; Dupont, J.B.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis modelled by human pluripotent stem cells: A multi-omic study of Duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Górecki, D.C. Dystrophin: The dead calm of a dogma. Rare Dis. 2016, 4, e1153777. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Filippelli, R.L.; Chang, N.C. Empowering Muscle Stem Cells for the Treatment of Duchenne Muscular Dystrophy. Cells Tissues Organs 2022, 211, 641–654. [Google Scholar] [CrossRef]
- Wang, Y.X.; Feige, P.; Brun, C.E.; Hekmatnejad, B.; Dumont, N.A.; Renaud, J.M.; Faulkes, S.; Guindon, D.E.; Rudnicki, M.A. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell 2019, 24, 419–432.e416. [Google Scholar] [CrossRef]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Hofemeier, A.D.; Muenker, T.M.; Herkenrath, F.; Ristau, M.; Brandt, M.; Shahriyari, M.; Tiburcy, M.; Zimmermann, W.H.; Lenz, C.; Mamchaoui, K.; et al. Dystrophin is a mechanical tension modulator. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pang, K.T.; Loo, L.S.W.; Chia, S.; Ong, F.Y.T.; Yu, H.; Walsh, I. Insight into muscle stem cell regeneration and mechanobiology. Stem Cell Res. Ther. 2023, 14, 129. [Google Scholar] [CrossRef]
- Loreti, M.; Sacco, A. The jam session between muscle stem cells and the extracellular matrix in the tissue microenvironment. npj Regen. Med. 2022, 7, 16. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Daley, W.P.; Peters, S.B.; Larsen, M. Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 2008, 121, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Biressi, S.; Miyabara, E.H.; Gopinath, S.D.; Carlig, P.M.; Rando, T.A. A Wnt-TGFbeta2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 2014, 6, 267ra176. [Google Scholar] [CrossRef]
- Giovarelli, M.; Arnaboldi, F.; Zecchini, S.; Cornaghi, L.B.; Nava, A.; Sommariva, M.; Clementi, E.G.I.; Gagliano, N. Characterisation of Progressive Skeletal Muscle Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8735. [Google Scholar] [CrossRef]
- Ramirez, M.P.; Anderson, M.J.M.; Kelly, M.D.; Sundby, L.J.; Hagerty, A.R.; Wenthe, S.J.; Odde, D.J.; Ervasti, J.M.; Gordon, W.R. Dystrophin missense mutations alter focal adhesion tension and mechanotransduction. Proc. Natl. Acad. Sci. USA 2022, 119, e2205536119. [Google Scholar] [CrossRef]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Bönnemann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef]
- Yamamoto, M.; Legendre, N.P.; Biswas, A.A.; Lawton, A.; Yamamoto, S.; Tajbakhsh, S.; Kardon, G.; Goldhamer, D.J. Loss of MyoD and Myf5 in Skeletal Muscle Stem Cells Results in Altered Myogenic Programming and Failed Regeneration. Stem Cell Rep. 2018, 10, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S.; Carvajal, J.J.; Golding, J.P.; Morgan, J.E.; Summerbell, D.; Zolnerciks, J.; Partridge, T.A.; Rigby, P.W.; Beauchamp, J.R. Myf5 expression in satellite cells and spindles in adult muscle is controlled by separate genetic elements. Dev. Biol. 2004, 273, 454–465. [Google Scholar] [CrossRef]
- Abdul-Hussein, S.; van der Ven, P.F.M.; Tajsharghi, H. Expression profiles of muscle disease-associated genes and their isoforms during differentiation of cultured human skeletal muscle cells. BMC Musculoskelet. Disord. 2012, 13, 262. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-H.; Paprzycki, L.; Legrand, A.; Declèves, A.-E.; Heher, P.; Limpens, M.; Belayew, A.; Banerji, C.R.S.; Zammit, P.S.; Tassin, A. Hypoxia enhances human myoblast differentiation: Involvement of HIF1α and impact of DUX4, the FSHD causal gene. Skelet. Muscle 2023, 13, 21. [Google Scholar] [CrossRef]
- Lundby, C.; Calbet, J.A.; Robach, P. The response of human skeletal muscle tissue to hypoxia. Cell. Mol. Life Sci. 2009, 66, 3615–3623. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fassler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, G.; Amato, A.; Tarone, G.; Vita, G.; Monici, M.C.; Magaudda, L.; Brancaccio, M.; Sidoti, A.; Trimarchi, F.; Favaloro, A.; et al. Distribution and localization of vinculin-talin-integrin system and dystrophin-glycoprotein complex in human skeletal muscle. Immunohistochemical study using confocal laser scanning microscopy. Cells Tissues Organs 2003, 175, 151–164. [Google Scholar] [CrossRef]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef]
- Steingruber, L.; Krabichler, F.; Franzmeier, S.; Wu, W.; Schlegel, J.; Koch, M. ALDH1A1 and ALDH1A3 paralogues of aldehyde dehydrogenase 1 control myogenic differentiation of skeletal muscle satellite cells by retinoic acid-dependent and -independent mechanisms. Cell Tissue Res. 2023, 394, 515–528. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Ladakis, D.C.; Pedrini, E.; Reyes-Mantilla, M.I.; Sanjayan, M.; Smith, M.D.; Fitzgerald, K.C.; Pardo, C.A.; Reich, D.S.; Absinta, M.; Bhargava, P. Metabolomics of Multiple Sclerosis Lesions Demonstrates Lipid Changes Linked to Alterations in Transcriptomics-Based Cellular Profiles. Neurol. Neuroimmunol. Neuroinflamm. 2024, 11, e200219. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Wu, T.; Dhindsa, J.; El Fadel, O.; Le, A.; Perez, A.; Amoh, B.; Tarkunde, A.; Zhu, K.F.; Avalos, M.; et al. Longitudinal multi-omics in alpha-synuclein Drosophila model discriminates disease- from age-associated pathologies in Parkinson’s disease. npj Park. Dis. 2025, 11, 46. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91–92, 152–166. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bandla, C.; Kundu, D.J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N.S.; Prakash, A.; Walzer, M.; Wang, S.; et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 2025, 53, D543–D553. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzmeier, S.; Chakraborty, S.; Mortazavi, A.; Stöckl, J.B.; Jiang, J.; Pfarr, N.; Sabass, B.; Fröhlich, T.; Kaufhold, C.; Stirm, M.; et al. Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy. Cells 2025, 14, 892. https://doi.org/10.3390/cells14120892
Franzmeier S, Chakraborty S, Mortazavi A, Stöckl JB, Jiang J, Pfarr N, Sabass B, Fröhlich T, Kaufhold C, Stirm M, et al. Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy. Cells. 2025; 14(12):892. https://doi.org/10.3390/cells14120892
Chicago/Turabian StyleFranzmeier, Sophie, Shounak Chakraborty, Armina Mortazavi, Jan B. Stöckl, Jianfei Jiang, Nicole Pfarr, Benedikt Sabass, Thomas Fröhlich, Clara Kaufhold, Michael Stirm, and et al. 2025. "Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy" Cells 14, no. 12: 892. https://doi.org/10.3390/cells14120892
APA StyleFranzmeier, S., Chakraborty, S., Mortazavi, A., Stöckl, J. B., Jiang, J., Pfarr, N., Sabass, B., Fröhlich, T., Kaufhold, C., Stirm, M., Wolf, E., Schlegel, J., & Matiasek, K. (2025). Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy. Cells, 14(12), 892. https://doi.org/10.3390/cells14120892