
GCN5 Is a Master Regulator of Gene Expression in the Malaria Parasite Plasmodium falciparum

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PfGCN5 KD Results in Growth Defects
2.2. PfGCN5 KD Led to Globally Reduced H3K9ac and H3K4me3 Levels
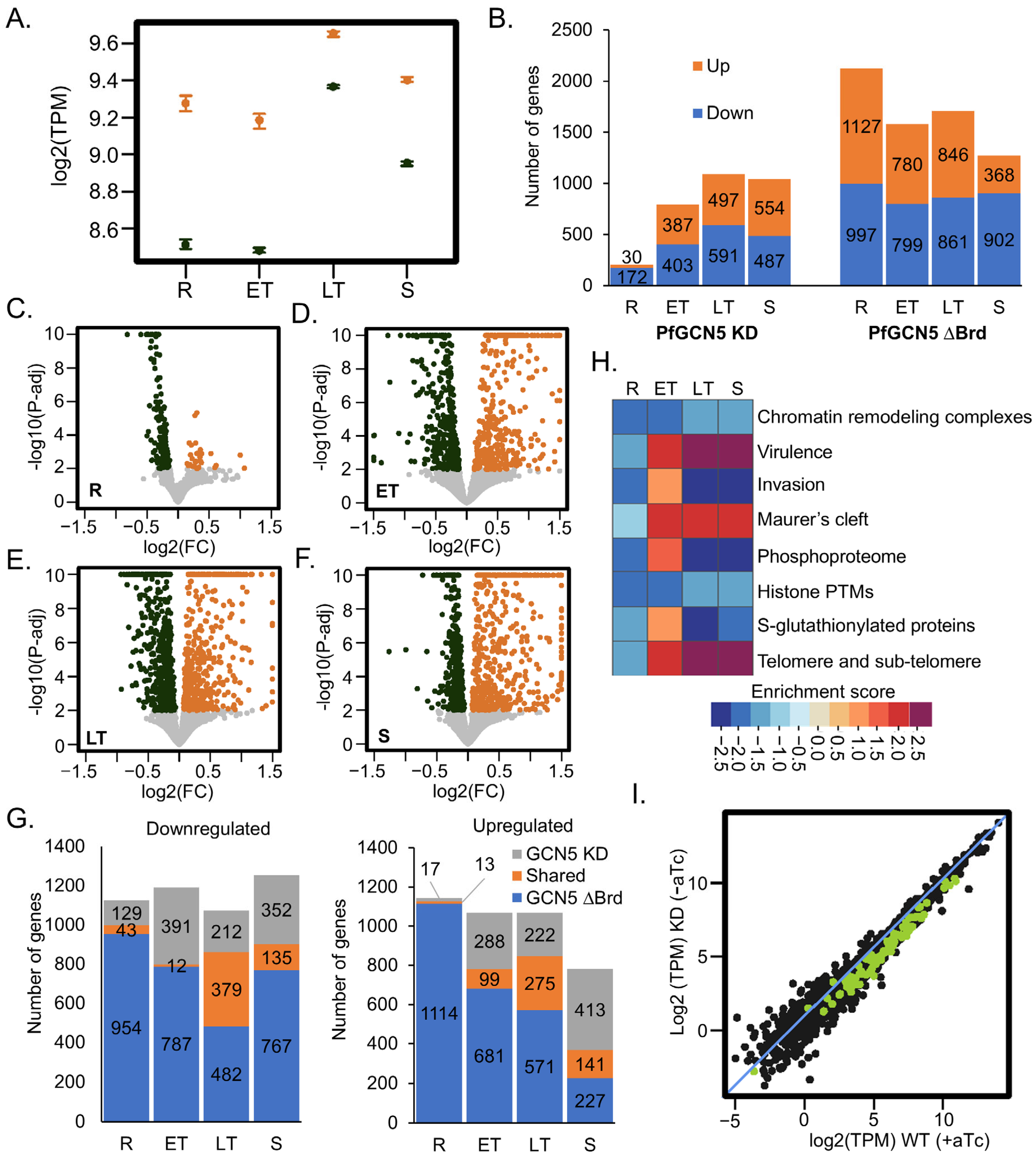
2.3. PfGCN5 KD Profoundly Affects Global Transcription and Specific Cellular Pathways
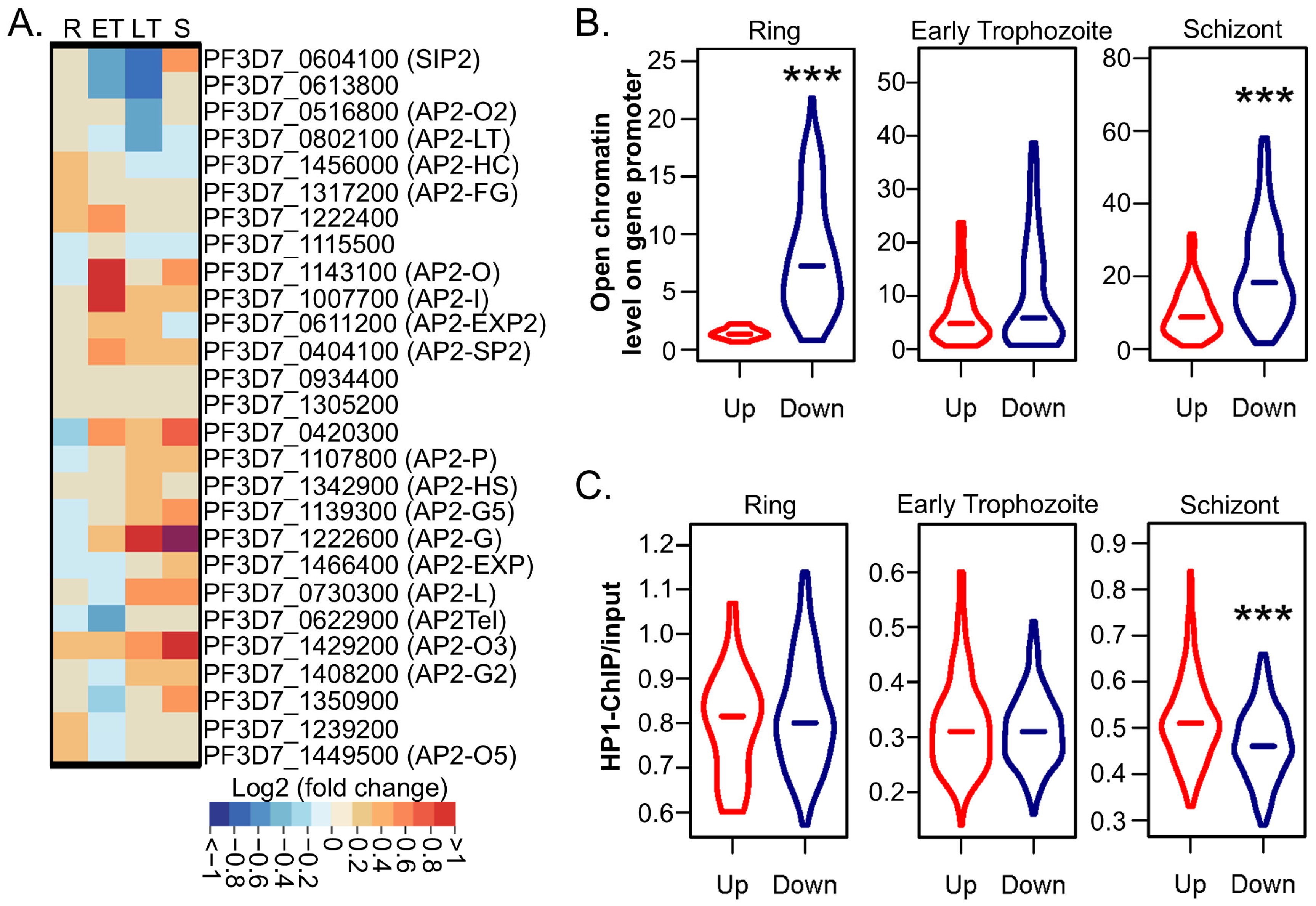
2.4. PfGCN5 KD Disturbs the Transcription of AP2 TFs
2.5. PfGCN5 KD Broadly Alters Chromatin Structures
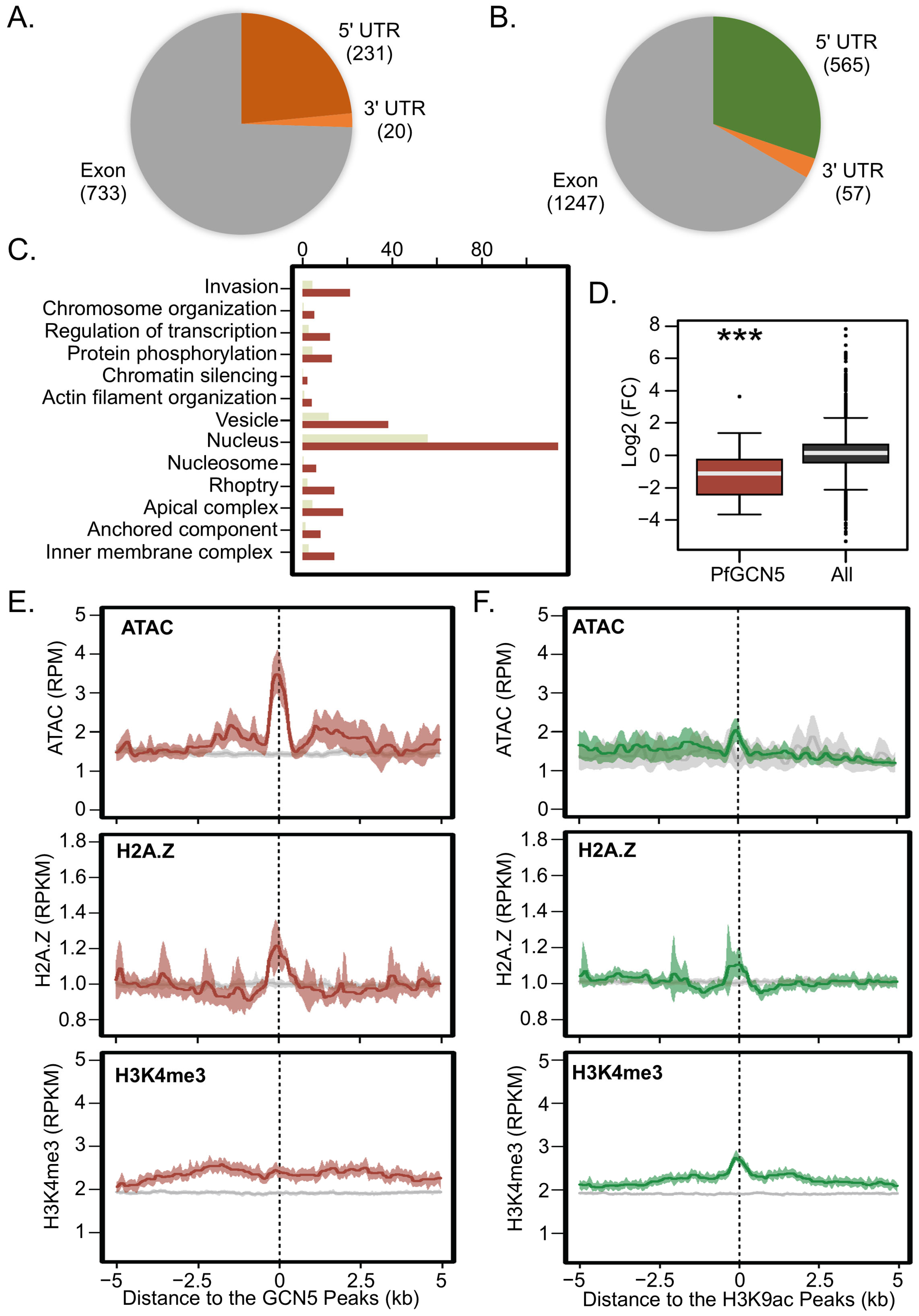
2.6. CUT&Tag-seq Is More Sensitive for Profiling Histone PTMs and “Writers”
2.7. Extensive Regulation by Enrichment of H3K9ac/PfGCN5 at Promoters
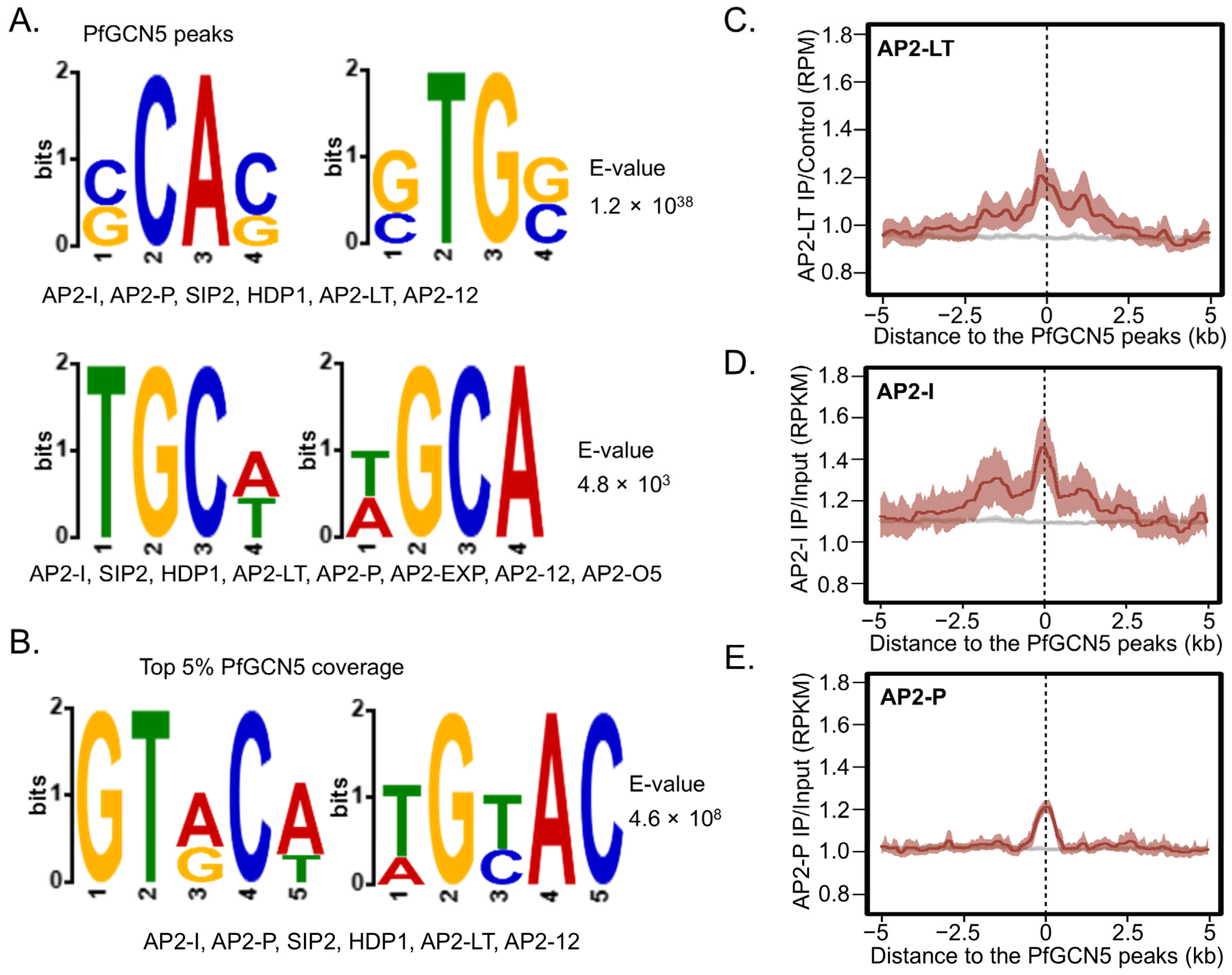
2.8. The PfGCN5 Complex Is Potentially Recruited by Specific AP2 TFs
3. Discussion
4. Limitations of the Study
5. Materials and Methods
5.1. Parasite Culture
5.2. Growth Phenotype Analysis
5.3. Histone PTMs
5.4. Transcriptome Analysis
5.5. Association Between Transcription Alterations and Chromatin Structures upon PfGCN5 KD
5.6. ChIP-seq and CUT&Tag-seq
5.7. Analyses of ChIP-seq and CUT&Tag-seq Data
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.A.; Duggan, L.; Cote, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F.; et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997, 11, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.C.; Dent, S.Y.R. Conservation and diversity of the eukaryotic SAGA coactivator complex across kingdoms. Epigenetics Chromatin 2021, 14, 26. [Google Scholar] [CrossRef]
- Huisinga, K.L.; Pugh, B.F. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 2004, 13, 573–585. [Google Scholar] [CrossRef]
- Vlachonasios, K.E.; Thomashow, M.F.; Triezenberg, S.J. Disruption mutations of ADA2b and GCN5 transcriptional adaptor genes dramatically affect Arabidopsis growth, development, and gene expression. Plant Cell 2003, 15, 626–638. [Google Scholar] [CrossRef]
- Bonnet, J.; Wang, C.Y.; Baptista, T.; Vincent, S.D.; Hsiao, W.C.; Stierle, M.; Kao, C.F.; Tora, L.; Devys, D. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014, 28, 1999–2012. [Google Scholar] [CrossRef]
- Baptista, T.; Grunberg, S.; Minoungou, N.; Koster, M.J.E.; Timmers, H.T.M.; Hahn, S.; Devys, D.; Tora, L. SAGA Is a General Cofactor for RNA Polymerase II Transcription. Mol. Cell 2017, 68, 130–143.e135. [Google Scholar] [CrossRef]
- Koutelou, E.; Hirsch, C.L.; Dent, S.Y. Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 2010, 22, 374–382. [Google Scholar] [CrossRef]
- Mao, Y.; Pavangadkar, K.A.; Thomashow, M.F.; Triezenberg, S.J. Physical and functional interactions of Arabidopsis ADA2 transcriptional coactivator proteins with the acetyltransferase GCN5 and with the cold-induced transcription factor CBF1. Biochim. Biophys. Acta 2006, 1759, 69–79. [Google Scholar] [CrossRef]
- Moraga, F.; Aquea, F. Composition of the SAGA complex in plants and its role in controlling gene expression in response to abiotic stresses. Front. Plant Sci. 2015, 6, 865. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, W.C.; Sutter, B.M.; Ruess, H.; Barnes, S.D.; Malladi, V.S.; Tu, B.P. Glucose starvation induces a switch in the histone acetylome for activation of gluconeogenic and fat metabolism genes. Mol. Cell 2022, 82, 60–74.e65. [Google Scholar] [CrossRef] [PubMed]
- WHO. Would Malaria Report 2024; WHO: Geneva, Switzerland, 2024. [Google Scholar]
- Hall, N.; Karras, M.; Raine, J.D.; Carlton, J.M.; Kooij, T.W.; Berriman, M.; Florens, L.; Janssen, C.S.; Pain, A.; Christophides, G.K.; et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 2005, 307, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Bozdech, Z.; Llinas, M.; Pulliam, B.L.; Wong, E.D.; Zhu, J.; DeRisi, J.L. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1, E5. [Google Scholar] [CrossRef]
- Le Roch, K.G.; Zhou, Y.; Blair, P.L.; Grainger, M.; Moch, J.K.; Haynes, J.D.; De La Vega, P.; Holder, A.A.; Batalov, S.; Carucci, D.J.; et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003, 301, 1503–1508. [Google Scholar] [CrossRef]
- Coulson, R.M.; Hall, N.; Ouzounis, C.A. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 2004, 14, 1548–1554. [Google Scholar] [CrossRef]
- Balaji, S.; Babu, M.M.; Iyer, L.M.; Aravind, L. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005, 33, 3994–4006. [Google Scholar] [CrossRef]
- Campbell, T.L.; De Silva, E.K.; Olszewski, K.L.; Elemento, O.; Llinas, M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 2010, 6, e1001165. [Google Scholar] [CrossRef]
- Morillo, R.A.C.; Tong, X.R.; Xie, W.; Abel, S.; Orchard, L.M.; Daher, W.; Patel, D.J.; Llinás, M.; Le Roch, K.G.; Kafsack, B.F.C. The transcriptional regulator HDP1 controls expansion of the inner membrane complex during early sexual differentiation of malaria parasites. Nat. Microbiol. 2022, 7, 289–299. [Google Scholar] [CrossRef]
- Hollin, T.; Chahine, Z.; Le Roch, K.G. Epigenetic Regulation and Chromatin Remodeling in Malaria Parasites. Annu. Rev. Microbiol. 2023, 77, 255–276. [Google Scholar] [CrossRef]
- de Boer, C.G.; Hughes, T.R. YeTFaSCo: A database of evaluated yeast transcription factor sequence specificities. Nucleic Acids Res. 2012, 40, D169–D179. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Miao, J. Chromatin-mediated epigenetic regulation in the malaria parasite Plasmodium falciparum. Eukaryot. Cell 2010, 9, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.S.; Bozdech, Z.; Bartfai, R. Epigenetic memory takes center stage in the survival strategy of malaria parasites. Curr. Opin. Microbiol. 2014, 20, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rubio, J.J.; Mancio-Silva, L.; Scherf, A. Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe 2009, 5, 179–190. [Google Scholar] [CrossRef]
- Salcedo-Amaya, A.M.; van Driel, M.A.; Alako, B.T.; Trelle, M.B.; van den Elzen, A.M.; Cohen, A.M.; Janssen-Megens, E.M.; van de Vegte-Bolmer, M.; Selzer, R.R.; Iniguez, A.L.; et al. Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2009, 106, 9655–9660. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Bol, S.M.; Prudhomme, J.; Vert, J.P.; Noble, W.S.; Le Roch, K.G. Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 2014, 24, 974–988. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Bottius, E.; Pirrit, L.A.; Deitsch, K.W.; Scheidig, C.; Guinet, F.; Nehrbass, U.; Wellems, T.E.; Scherf, A. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 2000, 407, 1018–1022. [Google Scholar] [CrossRef]
- Bunnik, E.M.; Venkat, A.; Shao, J.; McGovern, K.E.; Batugedara, G.; Worth, D.; Prudhomme, J.; Lapp, S.A.; Andolina, C.; Ross, L.S.; et al. Comparative 3D genome organization in apicomplexan parasites. Proc. Natl. Acad. Sci. USA 2019, 116, 3183–3192. [Google Scholar] [CrossRef]
- Bunnik, E.M.; Cook, K.B.; Varoquaux, N.; Batugedara, G.; Prudhomme, J.; Cort, A.; Shi, L.; Andolina, C.; Ross, L.S.; Brady, D.; et al. Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages. Nat. Commun. 2018, 9, 1910. [Google Scholar] [CrossRef]
- Crowley, V.M.; Rovira-Graells, N.; Ribas de Pouplana, L.; Cortes, A. Heterochromatin formation in bistable chromatin domains controls the epigenetic repression of clonally variant Plasmodium falciparum genes linked to erythrocyte invasion. Mol. Microbiol. 2011, 80, 391–406. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Hernandez-Rivas, R.; Ralph, S.A.; Montiel-Condado, D.; Ruvalcaba-Salazar, O.K.; Rojas-Meza, A.P.; Mancio-Silva, L.; Leal-Silvestre, R.J.; Gontijo, A.M.; Shorte, S.; et al. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Chookajorn, T.; Dzikowski, R.; Frank, M.; Li, F.; Jiwani, A.Z.; Hartl, D.L.; Deitsch, K.W. Epigenetic memory at malaria virulence genes. Proc. Natl. Acad. Sci. USA 2007, 104, 899–902. [Google Scholar] [CrossRef]
- Perez-Toledo, K.; Rojas-Meza, A.P.; Mancio-Silva, L.; Hernandez-Cuevas, N.A.; Delgadillo, D.M.; Vargas, M.; Martinez-Calvillo, S.; Scherf, A.; Hernandez-Rivas, R. Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res. 2009, 37, 2596–2606. [Google Scholar] [CrossRef]
- Flueck, C.; Bartfai, R.; Volz, J.; Niederwieser, I.; Salcedo-Amaya, A.M.; Alako, B.T.; Ehlgen, F.; Ralph, S.A.; Cowman, A.F.; Bozdech, Z.; et al. Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog. 2009, 5, e1000569. [Google Scholar] [CrossRef]
- Brancucci, N.M.B.; Bertschi, N.L.; Zhu, L.; Niederwieser, I.; Chin, W.H.; Wampfler, R.; Freymond, C.; Rottmann, M.; Felger, I.; Bozdech, Z.; et al. Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe 2014, 16, 165–176. [Google Scholar] [CrossRef]
- Gupta, A.P.; Chin, W.H.; Zhu, L.; Mok, S.; Luah, Y.H.; Lim, E.H.; Bozdech, Z. Dynamic epigenetic regulation of gene expression during the life cycle of malaria parasite Plasmodium falciparum. PLoS Pathog. 2013, 9, e1003170. [Google Scholar] [CrossRef]
- Bartfai, R.; Hoeijmakers, W.A.; Salcedo-Amaya, A.M.; Smits, A.H.; Janssen-Megens, E.; Kaan, A.; Treeck, M.; Gilberger, T.W.; Francoijs, K.J.; Stunnenberg, H.G. H2A.Z demarcates intergenic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010, 6, e1001223. [Google Scholar] [CrossRef]
- Tang, J.; Chisholm, S.A.; Yeoh, L.M.; Gilson, P.R.; Papenfuss, A.T.; Day, K.P.; Petter, M.; Duffy, M.F. Histone modifications associated with gene expression and genome accessibility are dynamically enriched at Plasmodium falciparum regulatory sequences. Epigenetics Chromatin 2020, 13, 50. [Google Scholar] [CrossRef]
- Fan, Q.; An, L.; Cui, L. Plasmodium falciparum histone acetyltransferase, a yeast GCN5 homologue involved in chromatin remodeling. Eukaryot. Cell 2004, 3, 264–276. [Google Scholar] [CrossRef]
- Cui, L.; Miao, J.; Furuya, T.; Li, X.; Su, X.Z.; Cui, L. PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot. Cell 2007, 6, 1219–1227. [Google Scholar] [CrossRef]
- Miao, J.; Fan, Q.; Cui, L.; Li, X.; Wang, H.; Ning, G.; Reese, J.C.; Cui, L. The MYST family histone acetyltransferase regulates gene expression and cell cycle in malaria parasite Plasmodium falciparum. Mol. Microbiol. 2010, 78, 883–902. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Fan, Q.; Miao, J. Histone lysine methyltransferases and demethylases in Plasmodium falciparum. Int. J. Parasitol. 2008, 38, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Merrick, C.J.; Duraisingh, M.T. Epigenetics in Plasmodium: What do we really know? Eukaryot. Cell 2010, 9, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.F.; Selvarajah, S.A.; Josling, G.A.; Petter, M. The role of chromatin in Plasmodium gene expression. Cell Microbiol. 2012, 14, 819–828. [Google Scholar] [CrossRef]
- Gupta, A.P.; Bozdech, Z. Epigenetic landscapes underlining global patterns of gene expression in the human malaria parasite, Plasmodium falciparum. Int. J. Parasitol. 2017, 47, 399–407. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Vert, J.P.; Noble, W.S.; Le Roch, K.G. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. falciparum. Bioessays 2015, 37, 182–194. [Google Scholar] [CrossRef]
- Batugedara, G.; Lu, X.M.; Bunnik, E.M.; Le Roch, K.G. The Role of Chromatin Structure in Gene Regulation of the Human Malaria Parasite. Trends Parasitol. 2017, 33, 364–377. [Google Scholar] [CrossRef]
- Hollin, T.; Le Roch, K.G. From Genes to Transcripts, a Tightly Regulated Journey in Plasmodium. Front. Cell. Infect. Microbiol. 2020, 10, 618454. [Google Scholar] [CrossRef]
- Hoeijmakers, W.A.M.; Miao, J.; Schmidt, S.; Toenhake, C.G.; Shrestha, S.; Venhuizen, J.; Henderson, R.; Birnbaum, J.; Ghidelli-Disse, S.; Drewes, G.; et al. Epigenetic reader complexes of the human malaria parasite, Plasmodium falciparum. Nucleic Acids Res. 2019, 47, 11574–11588. [Google Scholar] [CrossRef]
- Miao, J.; Wang, C.Q.; Lucky, A.B.; Liang, X.Y.; Min, H.; Adapa, S.R.; Jiang, R.; Kim, K.; Cui, L.W. A unique GCN5 histone acetyltransferase complex controls erythrocyte invasion and virulence in the malaria parasite Plasmodium falciparum. PLoS Pathog. 2021, 17, e1009351. [Google Scholar] [CrossRef]
- Li, S.; Shogren-Knaak, M.A. The Gcn5 bromodomain of the SAGA complex facilitates cooperative and cross-tail acetylation of nucleosomes. J. Biol. Chem. 2009, 284, 9411–9417. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Miao, J.; Furuya, T.; Fan, Q.; Li, X.; Rathod, P.K.; Su, X.Z.; Cui, L. Histone acetyltransferase inhibitor anacardic acid causes changes in global gene expression during in vitro Plasmodium falciparum development. Eukaryot. Cell 2008, 7, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Lucky, A.B.; Wang, C.; Shakri, A.R.; Kalamuddin, M.; Chim-Ong, A.; Li, X.; Miao, J. Plasmodium falciparum GCN5 plays a key role in regulating artemisinin resistance-related stress responses. Antimicrob. Agents Chemother. 2023, 67, e00577-23. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, K.; Tehlan, A.; Sunita; Sudhakar, R.; Kaur, I.; Sijwali, P.S.; Krishnamachari, A.; Dhar, S.K. Plasmodium falciparum GCN5 acetyltransferase follows a novel proteolytic processing pathway that is essential for its function. J. Cell Sci. 2020, 133, jcs236489. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Janssens, D.H.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 2020, 15, 3264–3283. [Google Scholar] [CrossRef]
- Lucky, A.B.; Wang, C.Q.; Liu, M.; Liang, X.Y.; Min, H.; Fan, Q.; Siddiqui, F.A.; Adapa, S.R.; Li, X.L.; Jiang, R.H.Y.; et al. A type II protein arginine methyltransferase regulates merozoite invasion in Plasmodium falciparum. Commun. Biol. 2023, 6, 659. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Cowman, A.F.; Berry, D.; Baum, J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 2012, 198, 961–971. [Google Scholar] [CrossRef]
- Flueck, C.; Bartfai, R.; Niederwieser, I.; Witmer, K.; Alako, B.T.; Moes, S.; Bozdech, Z.; Jenoe, P.; Stunnenberg, H.G.; Voss, T.S. A major role for the Plasmodium falciparum ApiAP2 protein PfSIP2 in chromosome end biology. PLoS Pathog. 2010, 6, e1000784. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Miranda, M.; Vembar, S.S.; Delgadillo, D.M.; Avila-Lopez, P.A.; Herrera-Solorio, A.M.; Lozano Amado, D.; Vargas, M.; Hernandez-Rivas, R. PfAP2Tel, harbouring a non-canonical DNA-binding AP2 domain, binds to Plasmodium falciparum telomeres. Cell. Microbiol. 2017, 19, e12742. [Google Scholar] [CrossRef] [PubMed]
- Carrington, E.; Cooijmans, R.H.M.; Keller, D.; Toenhake, C.G.; Bartfai, R.; Voss, T.S. The ApiAP2 factor PfAP2-HC is an integral component of heterochromatin in the malaria parasite Plasmodium falciparum. iScience 2021, 24, 102444. [Google Scholar] [CrossRef]
- Shang, X.M.; Wang, C.H.; Fan, Y.T.; Guo, G.Q.; Wang, F.; Zhao, Y.M.; Sheng, F.; Tang, J.X.; He, X.Q.; Yu, X.Y.; et al. Genome-wide landscape of ApiAP2 transcription factors reveals a heterochromatin-associated regulatory network during Plasmodium falciparum blood-stage development. Nucleic Acids Res. 2022, 50, 3413–3431. [Google Scholar] [CrossRef]
- Subudhi, A.K.; Green, J.L.; Satyam, R.; Salunke, R.P.; Lenz, T.; Shuaib, M.; Isaioglou, I.; Abel, S.; Gupta, M.; Esau, L.; et al. DNA-binding protein PfAP2-P regulates parasite pathogenesis during malaria parasite blood stages. Nat. Microbiol. 2023, 8, 2154–2169. [Google Scholar] [CrossRef]
- Santos, J.M.; Josling, G.; Ross, P.; Joshi, P.; Orchard, L.; Campbell, T.; Schieler, A.; Cristea, I.M.; Llinas, M. Red Blood Cell Invasion by the Malaria Parasite Is Coordinated by the PfAP2-I Transcription Factor. Cell Host Microbe 2017, 21, 731–741.e710. [Google Scholar] [CrossRef]
- Josling, G.A.; Russell, T.J.; Venezia, J.; Orchard, L.; van Biljon, R.; Painter, H.J.; Llinas, M. Dissecting the role of PfAP2-G in malaria gametocytogenesis. Nat. Commun. 2020, 11, 1503. [Google Scholar] [CrossRef]
- Kafsack, B.F.; Rovira-Graells, N.; Clark, T.G.; Bancells, C.; Crowley, V.M.; Campino, S.G.; Williams, A.E.; Drought, L.G.; Kwiatkowski, D.P.; Baker, D.A.; et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 2014, 507, 248–252. [Google Scholar] [CrossRef]
- Toenhake, C.G.; Fraschka, S.A.; Vijayabaskar, M.S.; Westhead, D.R.; van Heeringen, S.J.; Bartfai, R. Chromatin Accessibility-Based Characterization of the Gene Regulatory Network Underlying Plasmodium falciparum Blood-Stage Development. Cell Host Microbe 2018, 23, 557–569.e559. [Google Scholar] [CrossRef]
- Karmodiya, K.; Pradhan, S.J.; Joshi, B.; Jangid, R.; Reddy, P.C.; Galande, S. A comprehensive epigenome map of Plasmodium falciparum reveals unique mechanisms of transcriptional regulation and identifies H3K36me2 as a global mark of gene suppression. Epigenetics Chromatin 2015, 8, 32. [Google Scholar] [CrossRef]
- Bailey, T.L. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Bonnell, V.A.; Zhang, Y.; Brown, A.S.; Horton, J.; Josling, G.A.; Chiu, T.P.; Rohs, R.; Mahony, S.; Gordan, R.; Llinas, M. DNA sequence and chromatin differentiate sequence-specific transcription factor binding in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2024, 52, 10161–10179. [Google Scholar] [CrossRef]
- Rajaram, K.; Liu, H.B.; Prigge, S.T. Redesigned TetR-Aptamer System to Control Gene Expression in Plasmodium falciparum. mSphere 2020, 5, e00457-20. [Google Scholar] [CrossRef]
- Ganesan, S.M.; Falla, A.; Goldfless, S.J.; Nasamu, A.S.; Niles, J.C. Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat. Commun. 2016, 7, 10727. [Google Scholar] [CrossRef]
- Falekun, S.; Sepulveda, J.; Jami-Alahmadi, Y.; Park, H.; Wohlschlegel, J.A.; Sigala, P.A. Divergent acyl carrier protein decouples mitochondrial Fe-S cluster biogenesis from fatty acid synthesis in malaria parasites. eLife 2021, 10, e71636. [Google Scholar] [CrossRef]
- Nasamu, A.S.; Falla, A.; Pasaje, C.F.A.; Wall, B.A.; Wagner, J.C.; Ganesan, S.M.; Goldfless, S.J.; Niles, J.C. An integrated platform for genome engineering and gene expression perturbation in. Sci. Rep. 2021, 11, 342. [Google Scholar] [CrossRef]
- Clements, R.L.; Morano, A.A.; Navarro, F.M.; McGee, J.P.; Du, E.W.; Streva, V.A.; Lindner, S.E.; Dvorin, J.D. Identification of basal complex protein that is essential for maturation of transmission-stage malaria parasites. Proc. Natl. Acad. Sci. USA 2022, 119, e2204167119. [Google Scholar] [CrossRef]
- Ke, H.J.; Dass, S.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. The mitochondrial ribosomal protein L13 is critical for the structural and functional integrity of the mitochondrion in. J. Biol. Chem. 2018, 293, 8128–8137. [Google Scholar] [CrossRef]
- Kudyba, H.M.; Cobb, D.W.; Vega-Rodríguez, J.; Muralidharan, V. Some conditions apply: Systems for studying protein function. PLoS Pathog. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, L.; Wang, Q.; He, C.; Dender, L.F.; Gong, F. A role for Gcn5 in heterochromatin structure, gene silencing and NER at the HML locus in budding yeast. bioRxiv 2021. bioRxiv:2021.02.28.433071. [Google Scholar] [CrossRef]
- Duraisingh, M.T.; Voss, T.S.; Marty, A.J.; Duffy, M.F.; Good, R.T.; Thompson, J.K.; Freitas-Junior, L.H.; Scherf, A.; Crabb, B.S.; Cowman, A.F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dixon, S.E.; Ting, L.M.; Liu, T.K.; Jeffers, V.; Croken, M.M.; Calloway, M.; Cannella, D.; Hakimi, M.A.; Kim, K.; et al. Lysine acetyltransferase GCN5b interacts with AP2 factors and is required for Toxoplasma gondii proliferation. PLoS Pathog. 2014, 10, e1003830. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.K.; Chen, K.J.; Wang, H.; Li, X.M.; Chen, Z.C. Structure of the NuA4 acetyltransferase complex bound to the nucleosome. Nature 2022, 610, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.; Lambert, J.P.; Gerdes, M.; Al-Madhoun, A.S.; Skerjanc, I.S.; Figeys, D.; Baetz, K. Functional dissection of the NuA4 histone acetyltransferase reveals its role as a genetic hub and that Eaf1 is essential for complex integrity. Mol. Cell. Biol. 2008, 28, 2244–2256. [Google Scholar] [CrossRef]
- Kalamuddin, M.; Shakri, A.R.; Wang, C.; Min, H.; Li, X.; Cui, L.; Miao, J. MYST regulates DNA repair and forms a NuA4-like complex in the malaria parasite Plasmodium falciparum. mSphere 2024, 9, e00140-24. [Google Scholar] [CrossRef]
- LaCount, D.J.; Vignali, M.; Chettier, R.; Phansalkar, A.; Bell, R.; Hesselberth, J.R.; Schoenfeld, L.W.; Ota, I.; Sahasrabudhe, S.; Kurschner, C.; et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 2005, 438, 103–107. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Miao, J.; Cui, L. Rapid isolation of single malaria parasite-infected red blood cells by cell sorting. Nat. Protoc. 2011, 6, 140–146. [Google Scholar] [CrossRef]
- Boyle, M.J.; Wilson, D.W.; Richards, J.S.; Riglar, D.T.; Tetteh, K.K.; Conway, D.J.; Ralph, S.A.; Baum, J.; Beeson, J.G. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc. Natl. Acad. Sci. USA 2010, 107, 14378–14383. [Google Scholar] [CrossRef]
- Miao, J.; Li, J.; Fan, Q.; Li, X.; Li, X.; Cui, L. The Puf-family RNA-binding protein PfPuf2 regulates sexual development and sex differentiation in the malaria parasite Plasmodium falciparum. J. Cell Sci. 2010, 123, 1039–1049. [Google Scholar] [CrossRef]
- Shrestha, S.; Li, X.; Ning, G.; Miao, J.; Cui, L. The RNA-binding protein Puf1 functions in the maintenance of gametocytes in Plasmodium falciparum. J. Cell Sci. 2016, 129, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Fan, Q.; Cui, L.; Li, J.; Li, J.; Cui, L. The malaria parasite Plasmodium falciparum histones: Organization, expression, and acetylation. Gene 2006, 369, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Fraschka, S.A.; Filarsky, M.; Hoo, R.; Niederwieser, I.; Yam, X.Y.; Brancucci, N.M.B.; Mohring, F.; Mushunje, A.T.; Huang, X.; Christensen, P.R.; et al. Comparative Heterochromatin Profiling Reveals Conserved and Unique Epigenome Signatures Linked to Adaptation and Development of Malaria Parasites. Cell Host Microbe 2018, 23, 407–420.e408. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucky, A.B.; Shakri, A.R.; Liang, X.; Min, H.; Li, X.-L.; Adapa, S.R.; Jiang, R.H.Y.; Cui, L.; Wang, C.; Miao, J. GCN5 Is a Master Regulator of Gene Expression in the Malaria Parasite Plasmodium falciparum. Cells 2025, 14, 876. https://doi.org/10.3390/cells14120876
Lucky AB, Shakri AR, Liang X, Min H, Li X-L, Adapa SR, Jiang RHY, Cui L, Wang C, Miao J. GCN5 Is a Master Regulator of Gene Expression in the Malaria Parasite Plasmodium falciparum. Cells. 2025; 14(12):876. https://doi.org/10.3390/cells14120876
Chicago/Turabian StyleLucky, Amuza Byaruhanga, Ahmad Rushdi Shakri, Xiaoying Liang, Hui Min, Xiao-Lian Li, Swamy Rakesh Adapa, Rays H. Y. Jiang, Liwang Cui, Chengqi Wang, and Jun Miao. 2025. "GCN5 Is a Master Regulator of Gene Expression in the Malaria Parasite Plasmodium falciparum" Cells 14, no. 12: 876. https://doi.org/10.3390/cells14120876
APA StyleLucky, A. B., Shakri, A. R., Liang, X., Min, H., Li, X.-L., Adapa, S. R., Jiang, R. H. Y., Cui, L., Wang, C., & Miao, J. (2025). GCN5 Is a Master Regulator of Gene Expression in the Malaria Parasite Plasmodium falciparum. Cells, 14(12), 876. https://doi.org/10.3390/cells14120876