
Broken Balance: Emerging Cross-Talk Between Proteostasis and Lipostasis in Neurodegenerative Diseases


Abstract
1. Introduction
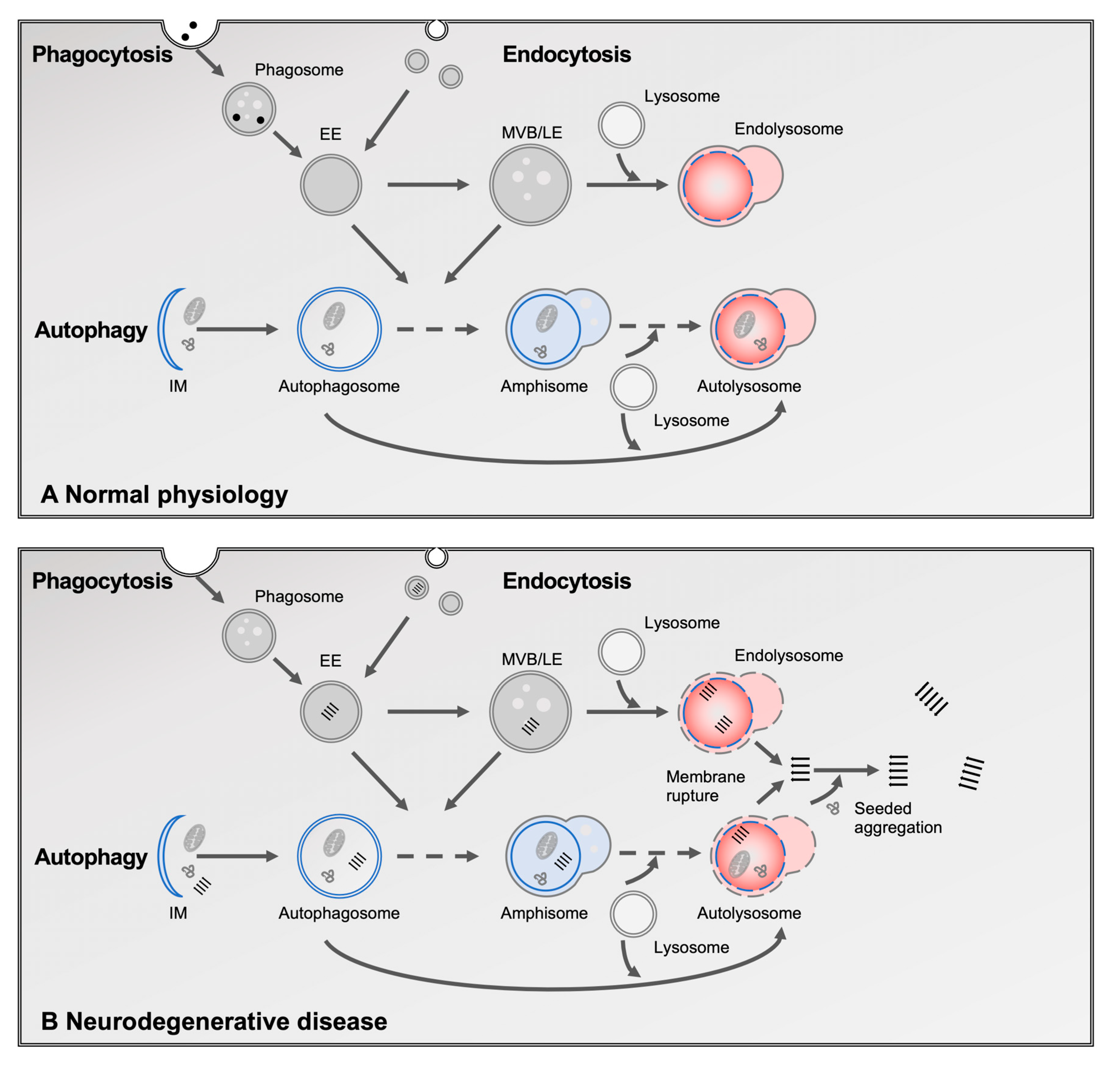
2. The Role of Lysosomes in Protein Homeostasis
3. The Role of the Lysosomal Degradation System in Neurodegenerative Diseases
4. The Role of Lysosomes in Lipid Homeostasis
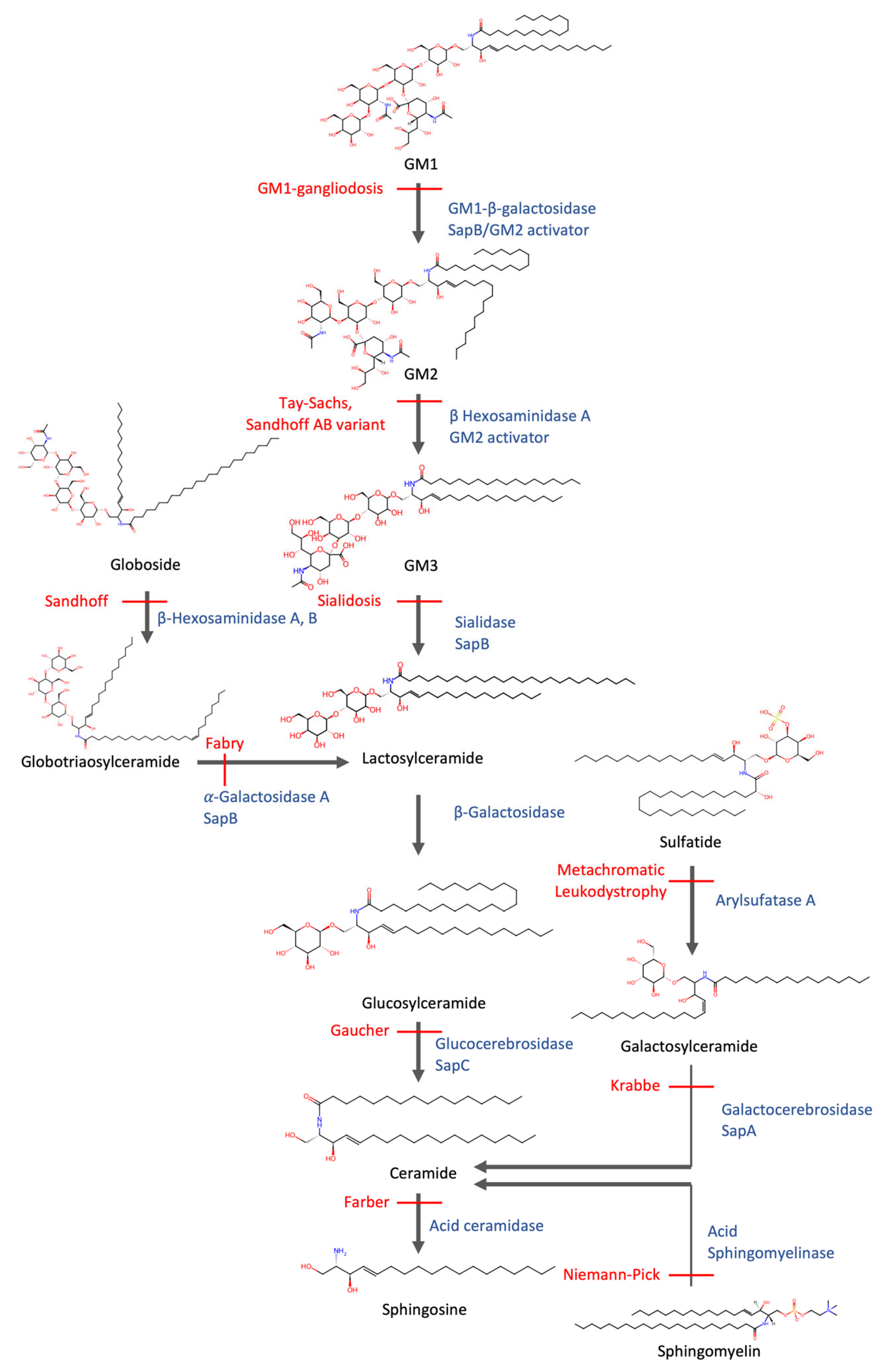
5. Failure of Lipid Degradation Leads to Lipidosis
6. Commonalities Between Sphingolipidosis and Neurodegenerative Diseases
6.1. GBA1
6.2. NPC1
6.3. SMPD1
6.4. GALC
6.5. GRN
7. Genetic Screens Reveal Regulators of Lipid and Protein Homeostasis
8. Dose-Dependent Effects and the Aging Brain
9. Conclusions
10. Outlook
- Leverage lipidomics for biomarker discovery: High-resolution lipid profiling holds promise for identifying early diagnostic and prognostic markers of neurodegenerative diseases. Achieving this goal will require the standardization of lipidomics methodologies across laboratories and platforms.
- Investigate the role of lipid droplets in neural cells: Accumulation of lipid droplets in neurons and glia has been linked to neurodegeneration [109]. Understanding the dynamics and function of lipid droplets could uncover new therapeutic targets.
- Evaluate the therapeutic potential of fatty acid substitution, including polyunsaturated fatty acids: Dietary supplementation with, e.g., polyunsaturated fatty acids may help rebalance lipid metabolism and reduce neuroinflammation, particularly in aging and early disease stages.
- Understanding disease specificity: Several lysosomal genes have been linked to distinct neurodegenerative diseases (e.g., GBA1 to PD, GRN to FTD), but the basis for this specificity remains unclear. Future work should explore how factors such as particular lipid metabolites, cell type, or potential regional differences in brain lipid composition might influence which disease phenotype emerges in response to a given lysosomal defect.
- Clarify the interplay between genetic and environmental factors in lipid–protein homeostasis: Determining how risk genes and environmental stressors influence lipostasis and proteostasis in neurons will be essential for identifying susceptible populations and designing preventive interventions.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Jucker, M. The prion principle and Alzheimer’s disease. Science 2024, 385, 1278–1279. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, M.; Bezard, E.; Dehay, B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 2014, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Winkens, C.; Sankaran, C.; Schätzl, H.M. From Seeds to Fibrils and Back: Fragmentation as an Overlooked Step in the Propagation of Prions and Prion-Like Proteins. Biomolecules 2020, 10, 1305. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Wang, C.; Tang, Y.; Mok, S.A.; Tsai, R.M.; Rojas, J.C.; Karydas, A.; Miller, B.L.; Boxer, A.L.; et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 2020, 15, 2. [Google Scholar] [CrossRef]
- Yan, M.; Zheng, T. Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem. Int. 2021, 145, 104988. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef]
- Schulze, H.; Sandhoff, K. Lysosomal Lipid Storage Diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004804. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Naslavsky, N.; Caplan, S. The enigmatic endosome–sorting the ins and outs of endocytic trafficking. J. Cell Sci. 2018, 131, jcs216499. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2018, 218, 757–770. [Google Scholar] [CrossRef]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 101–118. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Hernández, F.; Lucas, J.; Gómez-Ramos, P.; Morán, M.; Ávila, J. FTDP-17 Mutations in tau Transgenic Mice Provoke Lysosomal Abnormalities and Tau Filaments in Forebrain. Mol. Cell. Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, E.; Magrin, C.; Ciccaldo, M.; Sola, M.; Bellotto, M.; Molinari, M.; Papin, S.; Paganetti, P. Tau accumulation in degradative organelles is associated to lysosomal stress. Sci. Rep. 2023, 13, 18024. [Google Scholar] [CrossRef]
- Lee, J.-H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The Role of VPS35 in the Pathobiology of Parkinson’s Disease. Cell. Mol. Neurobiol. 2020, 41, 199–227. [Google Scholar] [CrossRef]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK 2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020, 6, eabb2454. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Lee, H.-J.; Cho, E.-D.; Lee, K.W.; Kim, J.-H.; Cho, S.-G.; Lee, S.-J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Nathaniel, D.L.; Raghavan, P.; Nelson, M.; Tian, R.; Tse, E.; Hong, J.Y.; See, S.K.; Mok, S.-A.; Hein, M.Y.; et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J. Biol. Chem. 2019, 294, 18952–18966. [Google Scholar] [CrossRef]
- Dimou, E.; Katsinelos, T.; Meisl, G.; Tuck, B.J.; Keeling, S.; Smith, A.E.; Hidari, E.; Lam, J.Y.; Burke, M.; Lövestam, S.; et al. Super-resolution imaging unveils the self-replication of tau aggregates upon seeding. Cell Rep. 2023, 42, 112725. [Google Scholar] [CrossRef] [PubMed]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gan, M.; Yen, S.-H.; McLean, P.J.; Dickson, D.W. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of α-synuclein aggregates. Sci. Rep. 2017, 7, 7690. [Google Scholar] [CrossRef]
- Polanco, J.C.; Hand, G.R.; Briner, A.; Li, C.; Götz, J. Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta. Neuropathol. 2021, 141, 235–256. [Google Scholar] [CrossRef]
- Rose, K.; Jepson, T.; Shukla, S.; Maya-Romero, A.; Kampmann, M.; Xu, K.; Hurley, J.H. Tau fibrils induce nanoscale membrane damage and nucleate cytosolic tau at lysosomes. Proc. Natl. Acad. Sci. USA 2024, 121, e2315690121. [Google Scholar] [CrossRef]
- Sandhof, C.A.; Hoppe, S.O.; Druffel-Augustin, S.; Gallrein, C.; Kirstein, J.; Voisine, C.; Nussbaum-Krammer, C. Reducing INS-IGF1 signaling protects against non-cell autonomous vesicle rupture caused by SNCA spreading. Autophagy 2019, 16, 878–899. [Google Scholar] [CrossRef]
- Sanyal, A.; Scanavachi, G.; Somerville, E.; Saminathan, A.; Nair, A.; Correia, R.F.B.D.C.; Aylan, B.; Sitarska, E.; Oikonomou, A.; Hatzakis, N.S.; et al. Neuronal constitutive endolysosomal perforations enable α-synuclein aggregation by internalized PFFs. J. Cell Biol. 2024, 224, e202401136. [Google Scholar] [CrossRef]
- Tuck, B.J.; Miller, L.V.; Katsinelos, T.; Smith, A.E.; Wilson, E.L.; Keeling, S.; Cheng, S.; Vaysburd, M.J.; Knox, C.; Tredgett, L.; et al. Cholesterol determines the cytosolic entry and seeded aggregation of tau. Cell Rep. 2022, 39, 110776. [Google Scholar] [CrossRef]
- Ebner, M.; Fröhlich, F.; Haucke, V. Mechanisms and functions of lysosomal lipid homeostasis. Cell Chem. Biol. 2025, 32, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Tanaka, Y.; Saftig, P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003, 13, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Li, M.; Yang, C.; Wang, X. M05B5.4 (lysosomal phospholipase A2) promotes disintegration of autophagic vesicles to maintain C. elegans development. Autophagy 2021, 18, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Hinkovska-Galcheva, V.; Kelly, R.; Abe, A.; Shayman, J.A.; Tesmer, J.J.G. Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 2015, 6, 6250. [Google Scholar] [CrossRef]
- He, M.; Kuk, A.C.Y.; Ding, M.; Chin, C.F.; Galam, D.L.; Nah, J.M.; Tan, B.C.; Yeo, H.L.; Chua, G.L.; Benke, P.I.; et al. Spns1 is a lysophospholipid transporter mediating lysosomal phospholipid salvage. Proc. Natl. Acad. Sci. USA 2022, 119, e2210353119. [Google Scholar] [CrossRef]
- Laqtom, N.N.; Dong, W.; Medoh, U.N.; Cangelosi, A.L.; Dharamdasani, V.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Heinze, I.; Tang, R.; et al. CLN3 is required for the clearance of glycerophosphodiesters from lysosomes. Nature 2022, 609, 1005–1011. [Google Scholar] [CrossRef]
- Zhang, H. Lysosomal acid lipase and lipid metabolism: New mechanisms, new questions, and new therapies. Curr. Opin. Infect. Dis. 2018, 29, 218–223. [Google Scholar] [CrossRef]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef]
- Trinh, M.N.; Brown, M.S.; Seemann, J.; Goldstein, J.L.; Lu, F. Lysosomal cholesterol export reconstituted from fragments of Niemann-Pick C1. eLife 2018, 7, e38564. [Google Scholar] [CrossRef]
- Höglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. PRINCIPLES OF LYSOSOMAL MEMBRANE DIGESTION: Stimulation of Sphingolipid Degradation by Sphingolipid Activator Proteins and Anionic Lysosomal Lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. In Sphingolipids as Signaling and Regulatory Molecules; Springer Nature: New York, NY, USA, 2010; pp. 1–23. [Google Scholar] [CrossRef]
- Sandhoff, K.; Kolter, T.; Harzer, K.; Schepers, U.; Remmel, N. Sphingolipid Activator Proteins; The Online Metabolic and Molecular Bases of Inherited Disease; McGraw Hill Medical: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Abdul-Hammed, M.; Breiden, B.; Adebayo, M.A.; Babalola, J.O.; Schwarzmann, G.; Sandhoff, K. Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J. Lipid Res. 2010, 51, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef] [PubMed]
- Sastry, P. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Martens, Y.A.; Bu, G. Lipoproteins in the Central Nervous System: From Biology to Pathobiology. Annu. Rev. Biochem. 2022, 91, 731–759. [Google Scholar] [CrossRef]
- Gulbins, E.; Kolesnick, R. Raft ceramide in molecular medicine. Oncogene 2003, 22, 7070–7077. [Google Scholar] [CrossRef]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin Fat Facts: An Overview of Lipids and Fatty Acid Metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 175–191. [Google Scholar] [CrossRef]
- Bi, X.; Liao, G. Cholesterol in Niemann-Pick Type C disease. Subcell. Biochem. 2010, 51, 319–335. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef]
- Harvald, E.B.; Olsen, A.S.B.; Færgeman, N.J. Autophagy in the light of sphingolipid metabolism. Apoptosis 2015, 20, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.; Aasly, J.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Fernandez-Gonzalez, I.; Riera, J.; Montpeyo, M.; Albert-Bayo, M.; Lopez-Royo, T.; Castillo-Sanchez, P.; Carnicer-Caceres, C.; Arranz-Amo, J.A.; Castillo-Ribelles, L.; et al. Lysosomal lipid alterations caused by glucocerebrosidase deficiency promote lysosomal dysfunction, chaperone-mediated-autophagy deficiency, and alpha-synuclein pathology. NPJ Park. Dis. 2022, 8, 126. [Google Scholar] [CrossRef]
- Bae, E.-J.; Yang, N.-Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.-J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Davis, O.B.; Shin, H.R.; Lim, C.-Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev. Cell 2020, 56, 260–276.e7. [Google Scholar] [CrossRef]
- Zoncu, R.; Perera, R.M. Built to last: Lysosome remodeling and repair in health and disease. Trends Cell Biol. 2022, 32, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, L.; Sharma, J.; di Ronza, A.; Zhang, P.; Eblimit, A.; Pal, R.; Roman, D.; Collette, J.R.; Booth, C.; Chang, K.T.; et al. A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer. J. Clin. Investig. 2020, 130, 4118–4132. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Suzuki, K.; Hulette, C.M.; Murayama, S. Aberrant Phosphorylation of α-Synuclein in Human Niemann-Pick Type C1 Disease. J. Neuropathol. Exp. Neurol. 2004, 63, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann—Pick disease type C. Brain 1995, 118, 119–129. [Google Scholar] [CrossRef]
- Chiba, Y.; Komori, H.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Adachi, K.; Nanba, E.; Hosokawa, M.; Enokido, Y.; Kouchi, Z.; et al. Niemann-Pick disease type C1 predominantly involving the frontotemporal region, with cortical and brainstem Lewy bodies: An autopsy case. Neuropathology 2014, 34, 49–57. [Google Scholar] [CrossRef]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef]
- Kluenemann, H.H.; Nutt, J.G.; Davis, M.Y.; Bird, T.D. Parkinsonism syndrome in heterozygotes for Niemann–Pick C1. J. Neurol. Sci. 2013, 335, 219–220. [Google Scholar] [CrossRef]
- Bencheikh, B.O.A.; Senkevich, K.; Rudakou, U.; Yu, E.; Mufti, K.; Ruskey, J.A.; Asayesh, F.; Laurent, S.B.; Spiegelman, D.; Fahn, S.; et al. Variants in the Niemann–Pick type C gene NPC1 are not associated with Parkinson’s disease. Neurobiol. Aging 2020, 93, 143.e1–143.e4. [Google Scholar] [CrossRef]
- Somerville, E.N.; Krohn, L.; Yu, E.; Rudakou, U.; Senkevich, K.; Ruskey, J.A.; Asayesh, F.; Ahmad, J.; Spiegelman, D.; Dauvilliers, Y.; et al. NPC1 variants are not associated with Parkinson’s disease, REM-sleep behavior disorder or dementia with Lewy bodies in European cohorts. Neurobiol. Aging 2023, 127, 94–98. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mallet, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.-N.; Liany, H.; Bei, J.-X.; Yu, X.-Q.; Liu, J.; Au, W.-L.; Prakash, K.M.; Tan, L.C.; Tan, E.-K. A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef] [PubMed]
- Feltri, M.L.; Weinstock, N.I.; Favret, J.; Dhimal, N.; Wrabetz, L.; Shin, D. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia 2021, 69, 2309–2331. [Google Scholar] [CrossRef] [PubMed]
- White, A.B.; Givogri, M.I.; Lopez-Rosas, A.; Cao, H.; van Breemen, R.; Thinakaran, G.; Bongarzone, E.R. Psychosine Accumulates in Membrane Microdomains in the Brain of Krabbe Patients, Disrupting the Raft Architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef]
- Hatton, C.; Ghanem, S.S.; Koss, D.J.; Abdi, I.Y.; Gibbons, E.; Guerreiro, R.; Bras, J.; Walker, L.; Gelpi, E.; Heywood, W.; et al. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain 2022, 145, 1257–1263. [Google Scholar] [CrossRef]
- Abdelkarim, H.; Marshall, M.S.; Scesa, G.; Smith, R.A.; Rue, E.; Marshall, J.; Elackattu, V.; Stoskute, M.; Issa, Y.; Santos, M.; et al. α-Synuclein interacts directly but reversibly with psychosine: Implications for α-synucleinopathies. Sci. Rep. 2018, 8, 12462. [Google Scholar] [CrossRef]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Smith, K.R.; Damiano, J.; Franceschetti, S.; Carpenter, S.; Canafoglia, L.; Morbin, M.; Rossi, G.; Pareyson, D.; Mole, S.E.; Staropoli, J.F.; et al. Strikingly Different Clinicopathological Phenotypes Determined by Progranulin-Mutation Dosage. Am. J. Hum. Genet. 2012, 90, 1102–1107. [Google Scholar] [CrossRef]
- Logan, T.; Simon, M.J.; Rana, A.; Cherf, G.M.; Srivastava, A.; Davis, S.S.; Low, R.L.Y.; Chiu, C.-L.; Fang, M.; Huang, F.; et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 2021, 184, 4651–4668.e25. [Google Scholar] [CrossRef]
- Katsumata, Y.; Shade, L.M.; Hohman, T.J.; Schneider, J.A.; Bennett, D.A.; Farfel, J.M.; Kukull, W.A.; Fardo, D.W.; Nelson, P.T. Multiple gene variants linked to Alzheimer’s-type clinical dementia via GWAS are also associated with non-Alzheimer’s neuropathologic entities. Neurobiol. Dis. 2022, 174, 105880. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sullivan, P.M.; Sun, L.; Hu, F. The interaction between progranulin and prosaposin is mediated by granulins and the linker region between saposin B and C. J. Neurochem. 2017, 143, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Bhagwagar, S.; Nies, S.H.; Ye, H.; Han, X.; Chiasseu, M.T.; Wang, G.; Mackenzie, I.R.; Strittmatter, S.M. Reduced progranulin increases tau and α-synuclein inclusions and alters mouse tauopathy phenotypes via glucocerebrosidase. Nat. Commun. 2024, 15, 1434. [Google Scholar] [CrossRef] [PubMed]
- Marian, O.C.; Teo, J.D.; Lee, J.Y.; Song, H.; Kwok, J.B.; Landin-Romero, R.; Halliday, G.; Don, A.S. Disrupted myelin lipid metabolism differentiates frontotemporal dementia caused by GRN and C9orf72 gene mutations. Acta Neuropathol. Commun. 2023, 11, 52. [Google Scholar] [CrossRef]
- Yong, J.E.; Villalta, J.; Vu, N.; A Kukurugya, M.; Olsson, N.; López, M.P.; Lazzari-Dean, J.R.; Hake, K.E.; McAllister, F.; Bennett, B.D.; et al. Impairment of lipid homeostasis causes lysosomal accumulation of endogenous protein aggregates through ESCRT disruption. eLife 2024, 12, RP86194. [Google Scholar] [CrossRef]
- Sandhof, C.A.; Martin, N.; Tittelmeier, J.; Schlueter, A.; Pezzali, M.; Schoendorf, D.C.; Lange, T.; Reinhardt, P.; Ried, J.S.; Liang, S.; et al. A Novel C. elegans Model for Tau spreading Reveals Genes Critical for Endolysosomal Integrity and Seeded Tau Aggregation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Sandhof, C.A.; Martin, N.; El-Kabarity, D.; Ngonza-Nito, S.-B.; Melki, R.; Nussbaum-Krammer, C. Sphingolipid imbalance aggravates tau pathology by endomembrane rigidification and rupture. eLife 2025, 14, RP106865. [Google Scholar] [CrossRef]
- Fabri, J.H.T.M.; de Sá, N.P.; Malavazi, I.; Del Poeta, M. The dynamics and role of sphingolipids in eukaryotic organisms upon thermal adaptation. Prog. Lipid Res. 2020, 80, 101063. [Google Scholar] [CrossRef]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal dysfunction in neurodegeneration: Emerging concepts and methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef]
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain 2022, 145, 3472–3487. [Google Scholar] [CrossRef]
- Tang, H.; Huang, X.; Pang, S. Regulation of the lysosome by sphingolipids: Potential role in aging. J. Biol. Chem. 2022, 298, 102118. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta Biomembr. 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed]
- Uranbileg, B.; Isago, H.; Sakai, E.; Kubota, M.; Saito, Y.; Kurano, M. Alzheimer’s disease manifests abnormal sphingolipid metabolism. Front. Aging Neurosci. 2024, 16, 1368839. [Google Scholar] [CrossRef]
- Roca-Agujetas, V.; Barbero-Camps, E.; de Dios, C.; Podlesniy, P.; Abadin, X.; Morales, A.; Marí, M.; Trullàs, R.; Colell, A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 15. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Lysosomes in senescence and aging. EMBO Rep. 2023, 24, e57265. [Google Scholar] [CrossRef]
- Lan, Z.-Q.; Ge, Z.-Y.; Lv, S.-K.; Zhao, B.; Li, C.-X. The regulatory role of lipophagy in central nervous system diseases. Cell Death Discov. 2023, 9, 229. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene | Lipid Storage Disorder | Lipid Accumulation or Imbalance | Neurodegeneration-Linked Pathology | Shared Mechanisms |
|---|---|---|---|---|
| GBA1 | Gaucher disease | glucosylceramide, glucosylsphingosine | ↑ PD risk; α-synuclein aggregation and transmission | autophagy impairment, lysosomal rupture, proteostasis failure |
| NPC1 | Niemann–Pick type C | unesterified cholesterol, glycosphingolipids | lysosomal rupture; debated PD association | autophagy impairment, lysosomal enzyme mistrafficking, membrane rupture |
| SMPD1 | Niemann–Pick type A/B | sphingomyelin | ↑ PD risk (variants) | autophagy impairment, lysosomal stress |
| GALC | Krabbe disease | psychosine (galactosyl-sphingosine) | ↑ PD risk; aggregation and prion-like propagation of α-synuclein | lysosomal stress and membrane destabilization |
| GRN | NCL and GRN-FTD | glucosylceramide; loss of myelin lipids (e.g., sulfatide) | TDP-43 aggregation, cortical neuronal loss; ↑ AD and PD risk | disrupted GCase activity (see GBA1), myelin loss |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tittelmeier, J.; Nussbaum-Krammer, C. Broken Balance: Emerging Cross-Talk Between Proteostasis and Lipostasis in Neurodegenerative Diseases. Cells 2025, 14, 845. https://doi.org/10.3390/cells14110845
Tittelmeier J, Nussbaum-Krammer C. Broken Balance: Emerging Cross-Talk Between Proteostasis and Lipostasis in Neurodegenerative Diseases. Cells. 2025; 14(11):845. https://doi.org/10.3390/cells14110845
Chicago/Turabian StyleTittelmeier, Jessica, and Carmen Nussbaum-Krammer. 2025. "Broken Balance: Emerging Cross-Talk Between Proteostasis and Lipostasis in Neurodegenerative Diseases" Cells 14, no. 11: 845. https://doi.org/10.3390/cells14110845
APA StyleTittelmeier, J., & Nussbaum-Krammer, C. (2025). Broken Balance: Emerging Cross-Talk Between Proteostasis and Lipostasis in Neurodegenerative Diseases. Cells, 14(11), 845. https://doi.org/10.3390/cells14110845