
Soluble β-Amyloid Oligomers Selectively Upregulate TRPC3 in Excitatory Neurons via Calcineurin-Coupled NFAT

 , , , ,
, , , ,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Human Brain Specimens for Western Blots, RT-qPCR, and Immunohistochemistry
2.3. Primary Hippocampal Cell Cultures
2.4. Naturally Secreted AβO-Containing Conditioned Medium (7PA2 CM), Immunodepletion, and Synthetic Aβ Oligomerization
2.5. RNA Isolation and RT-qPCR
2.6. siRNA Transfection in Primary Neurons
2.7. Fluorescent Immunocytochemistry
2.8. The Quantification of Images Captured by the KEYENCE Microscope
2.9. Western Bslotting
2.10. Calcineurin Enzymatic Activity Assay
2.11. ChIP-PCR
2.12. Single-Nucleus RNAseq Data Analysis
2.13. Electrophysiology
2.14. Calcium Imaging
2.15. Confocal Ca2+ Imaging (For Data Presented in Figure 7E)
2.16. Statistics
3. Results
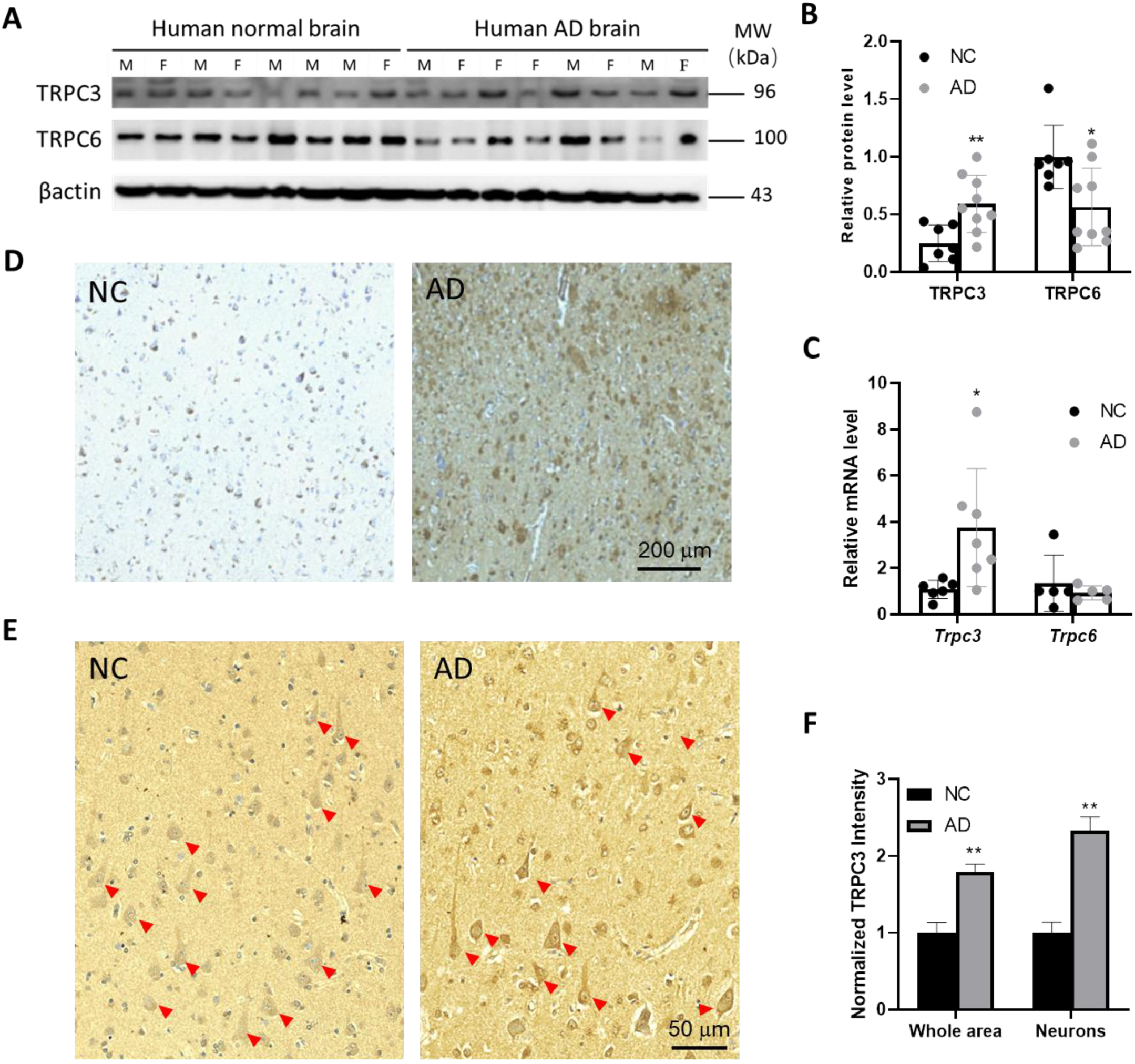
3.1. TRPC3 Expression Is Elevated in Post-Mortem Human AD Brains
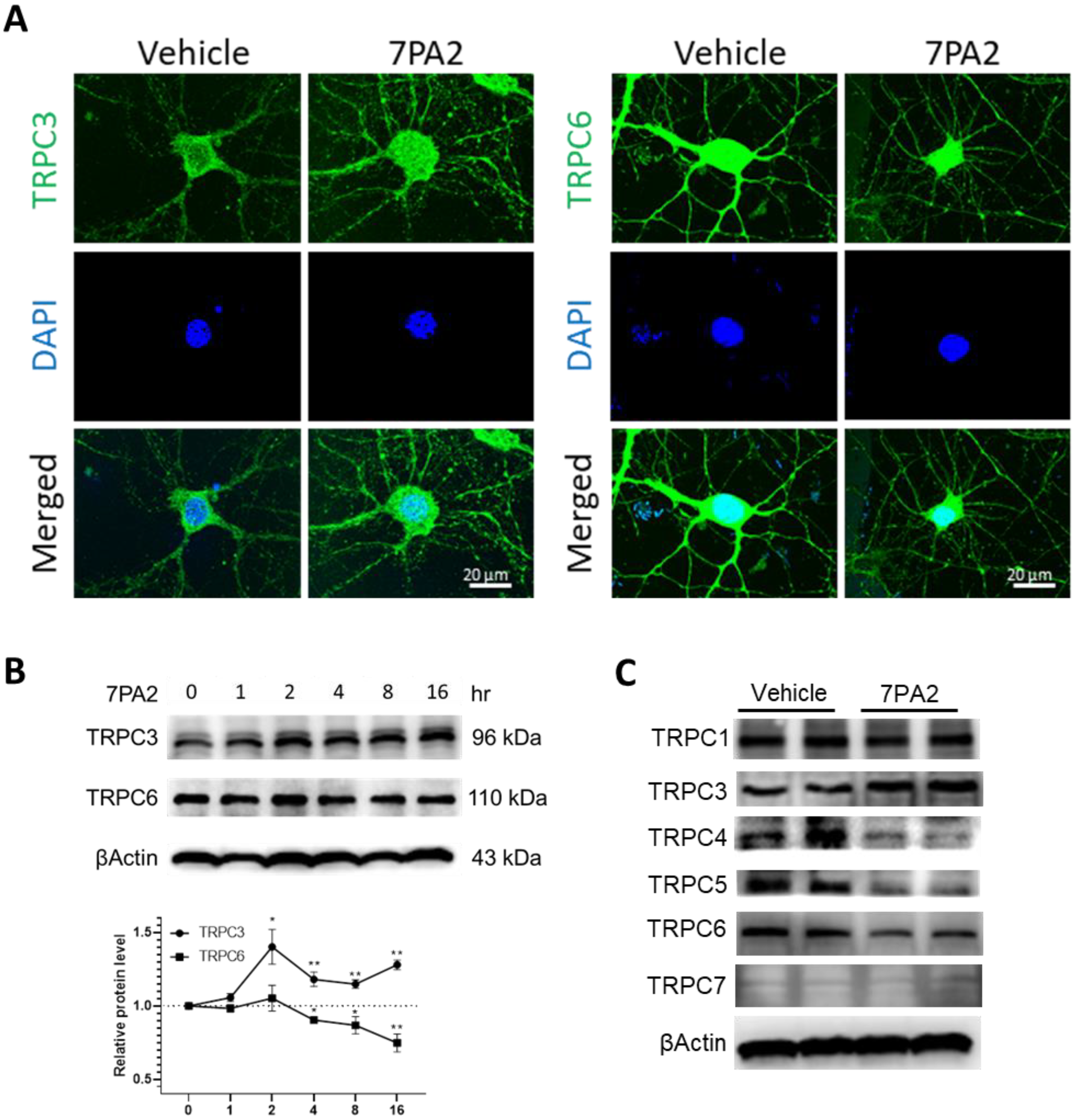
3.2. AβOs Aberrantly Upregulate TRPC3 and Downregulate TRPC6 Expression in Mature Hippocampal Neurons
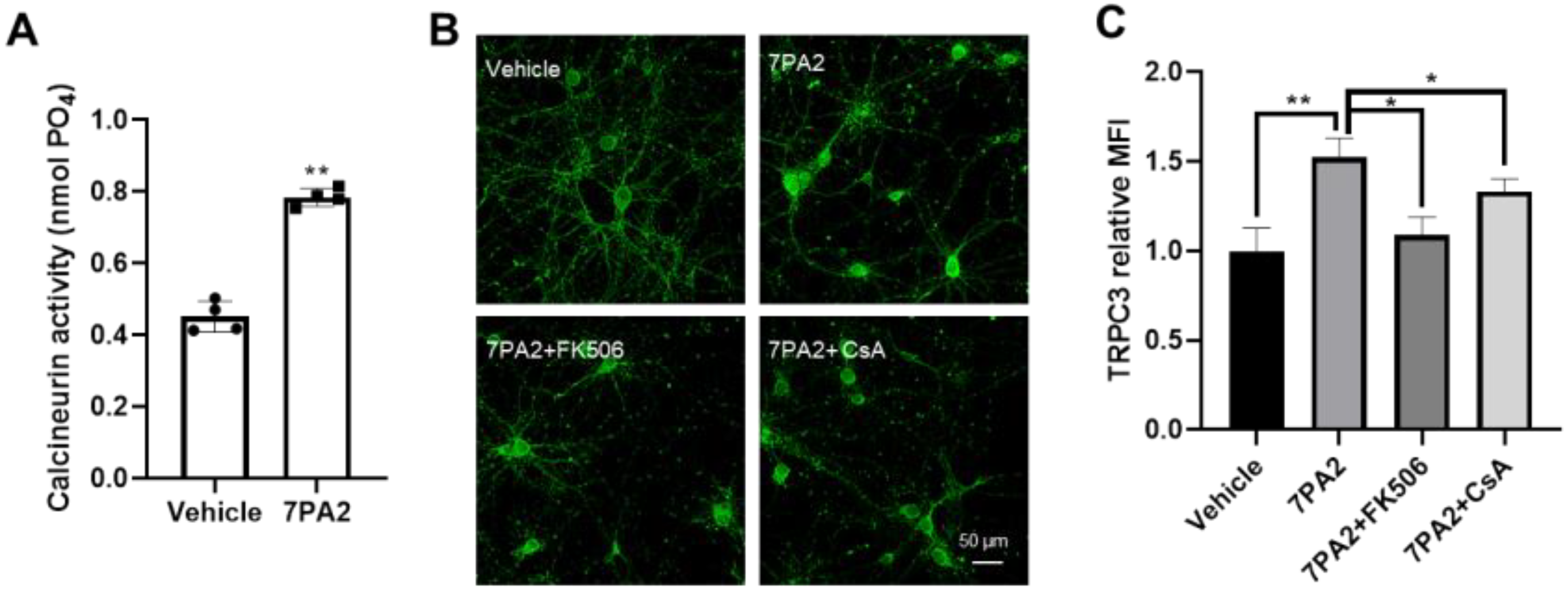
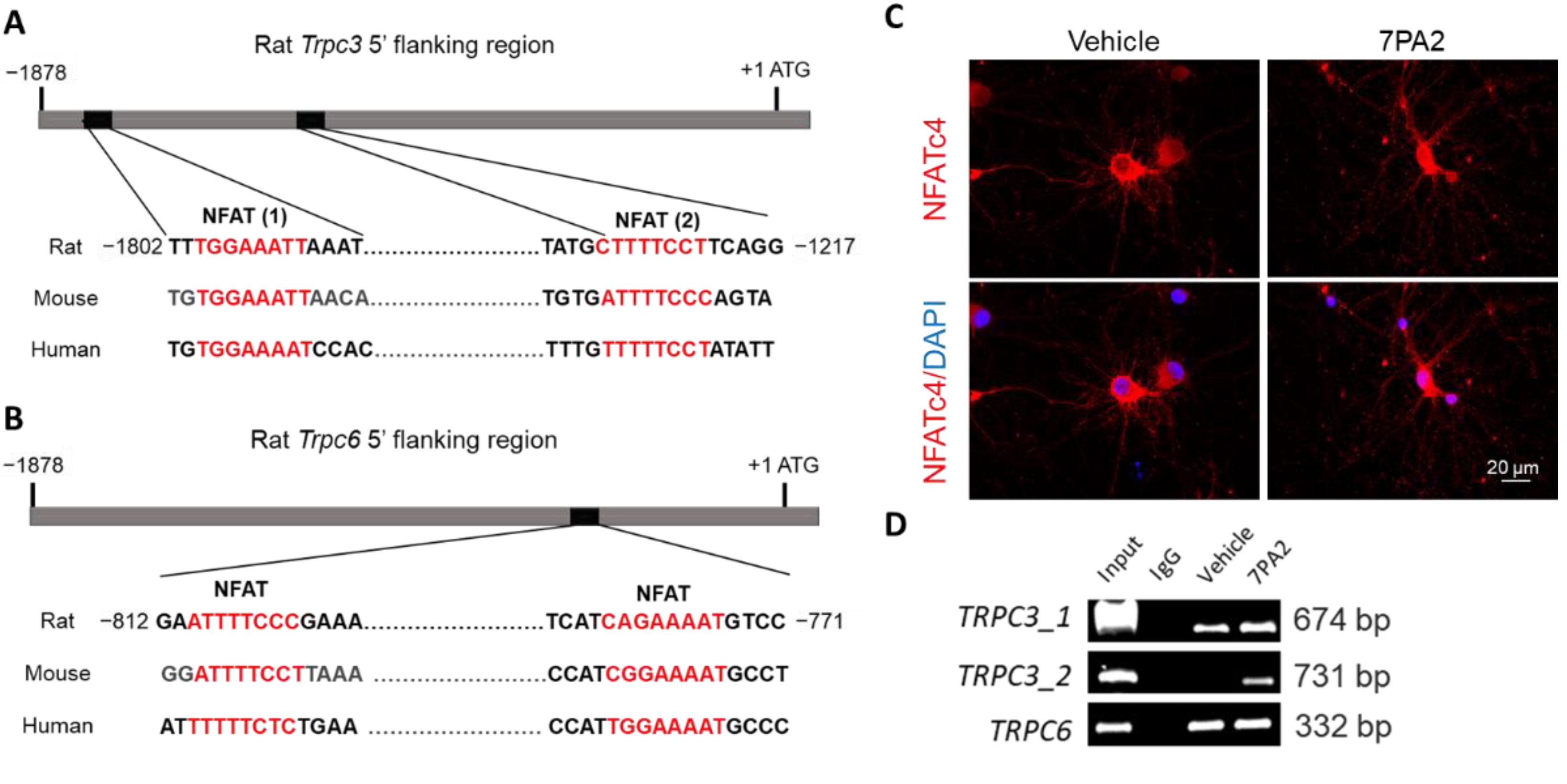
3.3. AβOs Transcriptionally Upregulate Trpc3 Gene Expression via Ca2+-Dependent Calcineurin–NFAT Mechanism
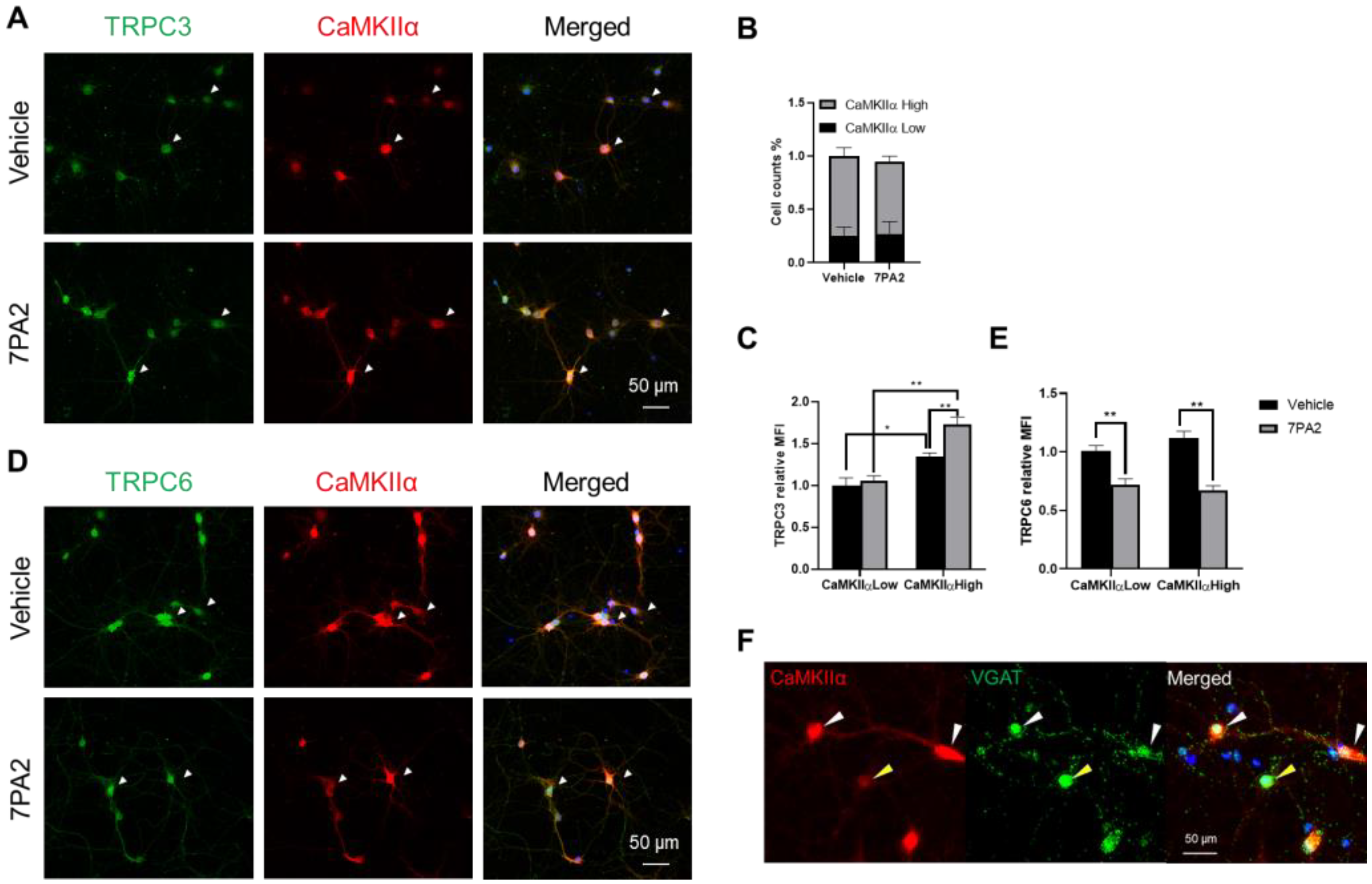
3.4. AβOs Upregulate TRPC3 Exclusively in Excitatory Neurons
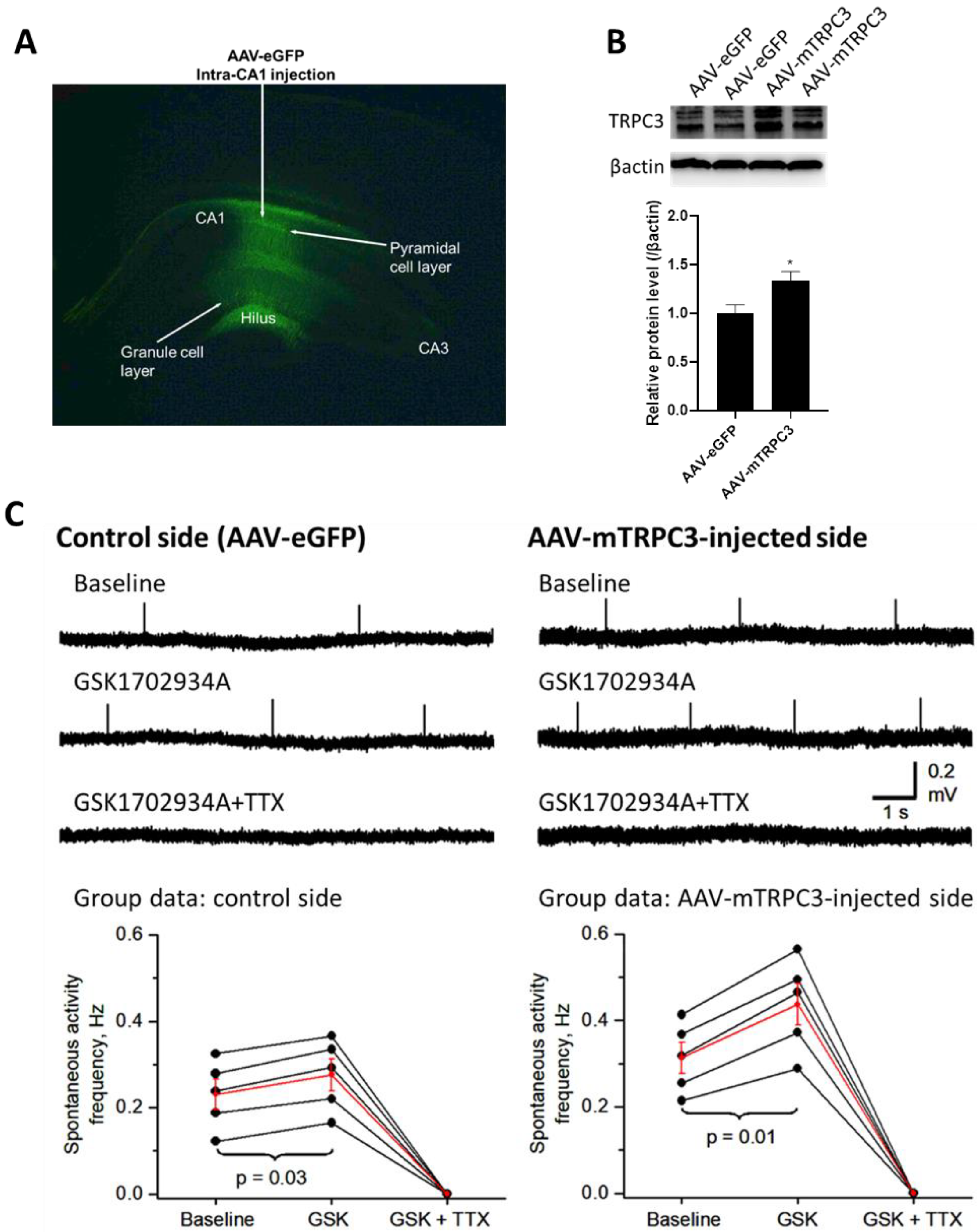
3.5. Hippocampal CA1-Overexpressed TRPC3 Induces Neuronal Hyperexcitability
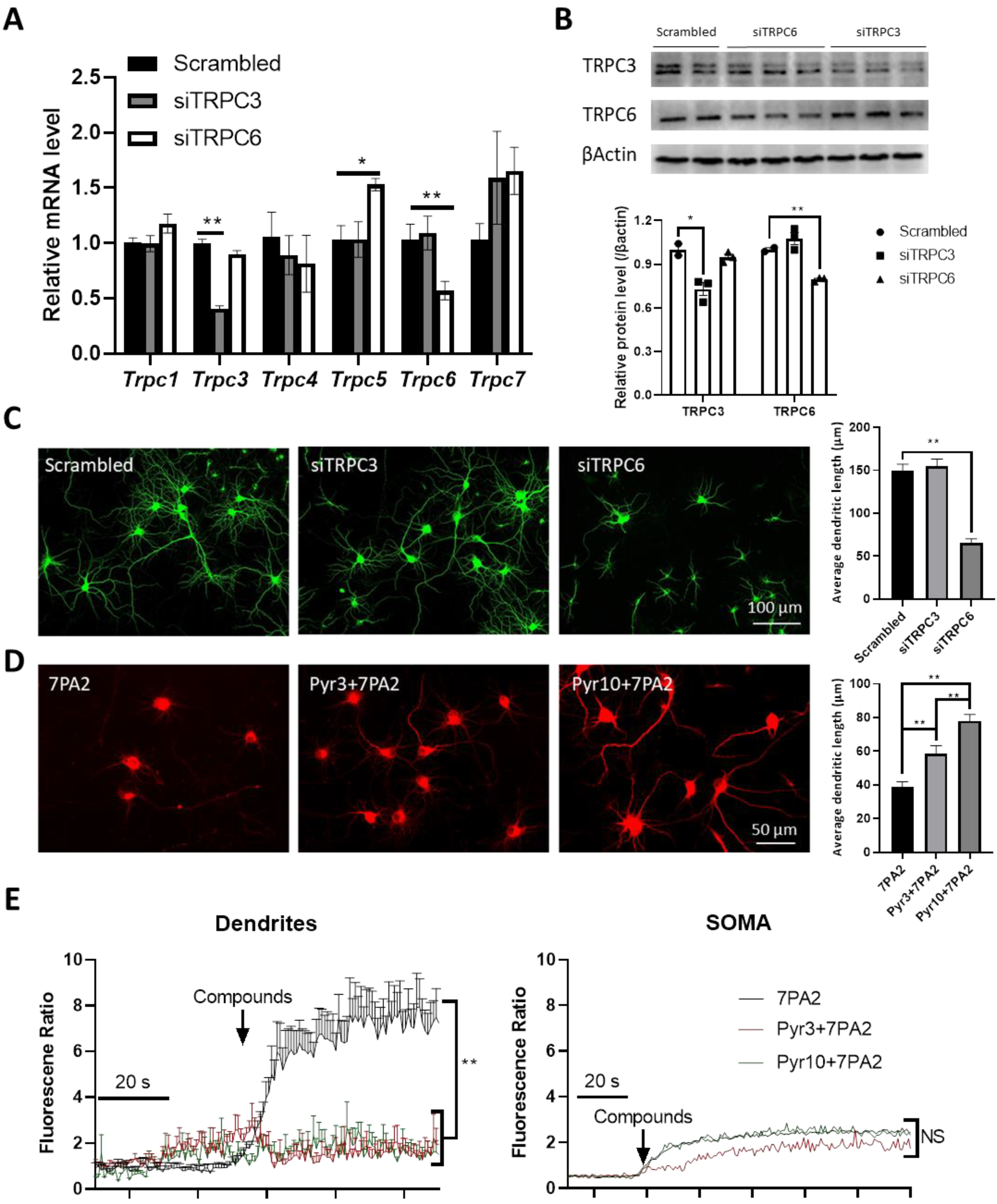
3.6. Selective TRPC3 Inhibition Renders Synaptic Protection Against AβOs
4. Discussion
4.1. Differential Regulation of TRPC3 and TRPC6 in AD
4.2. Potential Implications of the Upregulated TRPC3 and Downregulated TRPC6 in AD: Gain-of-Function vs. Loss-of-Function in Ca2+ Dysregulation and Hyperexcitability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, W.M.; de Vugt, M.E.; Smets, E.M.; Blom, M.; Teunissen, C.E. Towards a future where Alzheimer’s disease pathology is stopped before the onset of dementia. Nat. Aging 2023, 3, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hyman, B.T.; Spires-Jones, T.L. Alzheimer’s disease: Synapses gone cold. Mol. Neurodegener. 2011, 6, 63. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef]
- Tu, S.; Okamoto, S.-I.; Lipton, S.A.; Xu, H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Vbeyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [PubMed]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Shrestha, B.R.; Vitolo, O.V.; Joshi, P.; Lordkipanidze, T.; Shelanski, M.; Dunaevsky, A. Amyloid β peptide adversely affects spine number and motility in hippocampal neurons. Mol. Cell. Neurosci. 2006, 33, 274–282. [Google Scholar]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar]
- Alzheimer’s Association Calcium Hypothesis Workgroup; Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182.e17. [Google Scholar]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Stutzmann, G.E. Early calcium dysregulation in Alzheimer’s disease: Setting the stage for synaptic dysfunction. Sci. China Life Sci. 2011, 54, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Du, W.; Wang, Y. Functional roles of TRPC channels in the developing brain. Pflügers Arch.-Eur. J. Physiol. 2009, 458, 283–289. [Google Scholar] [CrossRef]
- Gualdani, R.; Gailly, P. How TRPC channels modulate hippocampal function. Int. J. Mol. Sci. 2020, 21, 3915. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient receptor potential canonical (TRPC) channels: Then and now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wajima, T.; Hara, Y.; Nishida, M.; Mori, Y. Transient receptor potential channels in Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2007, 1772, 958–967. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Singh, B.B. TRPC channels and their implications for neurological diseases. CNS Neurol. Disord.-Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2010, 9, 94–104. [Google Scholar] [CrossRef]
- Birnbaumer, L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 395–426. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sukumaran, P.; Bandyopadhyay, B.C.; Singh, B.B. Physiological function and characterization of TRPCs in neurons. Cells 2014, 3, 455–475. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Dolios, G.; Wang, R.; Liao, F.-F. Soluble beta-amyloid peptides, but not insoluble fibrils, have specific effect on neuronal microRNA expression. PLoS ONE 2014, 9, e90770. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.-Q.; Heldt, S.A.; Xu, H.; Liao, F.-F. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Huang, L.; Chen, J.; Li, J.J.; Wang, R.; Kim, E.; Chen, Y.; Justicia, C.; Sakata, K. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 2017, 22, 990–1001. [Google Scholar] [CrossRef]
- Podlisny, M.B.; Ostaszewski, B.L.; Squazzo, S.L.; Koo, E.H.; Rydell, R.E.; Teplow, D.B.; Selkoe, D.J. Aggregation of Secreted Amyloid β-Protein into Sodium Dodecyl Sulfate-stable Oligomers in Cell Culture. J. Biol. Chem. 1995, 270, 9564–9570. [Google Scholar] [CrossRef]
- Ishii, A.; Pathoulas, J.A.; MoustafaFathy Omar, O.; Ge, Y.; Yao, A.Y.; Pantalena, T.; Singh, N.; Zhou, J.; He, W.; Murphy, P.; et al. Contribution of amyloid deposition from oligodendrocytes in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 83. [Google Scholar] [CrossRef]
- Ding, S.; Wei, W.; Zhou, F.-M. Molecular and functional differences in voltage-activated sodium currents between GABA projection neurons and dopamine neurons in the substantia nigra. J. Neurophysiol. 2011, 106, 3019–3034. [Google Scholar] [CrossRef]
- Zhang, S.; Romero, L.O.; Deng, S.; Wang, J.; Li, Y.; Yang, L.; Hamilton, D.J.; Miller, D.D.; Liao, F.F.; Cordero-Morales, J.F.; et al. Discovery of a highly selective and potent TRPC3 inhibitor with high metabolic stability and low toxicity. ACS Med. Chem. Lett. 2021, 12, 572–578. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Caceres, A.; Banker, G.; Binder, L. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J. Neurosci. 1986, 6, 714–722. [Google Scholar] [CrossRef]
- Mitchell, P.; Hanson, J.; Quets-Nguyen, A.; Bergeron, M.; Smith, R. A quantitative method for analysis of in vitro neurite outgrowth. J. Neurosci. Methods 2007, 164, 350–362. [Google Scholar] [CrossRef]
- DeGiosio, R.A.; Grubisha, M.J.; MacDonald, M.L.; McKinney, B.C.; Camacho, C.J.; Sweet, R.A. More than a marker: Potential pathogenic functions of MAP2. Front. Mol. Neurosci. 2022, 15, 974890. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H.; et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, J.; Lisek, M.; Boczek, T. Targeting CaN/NFAT in Alzheimer’s brain degeneration. Front. Immunol. 2023, 14, 1281882. [Google Scholar] [CrossRef] [PubMed]
- Mody, I.; MacDonald, J.F. NMDA receptor-dependent excitotoxicity: The role of intracellular Ca2+ release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e1948. [Google Scholar] [CrossRef]
- Dawson, T.M.; Steiner, J.P.; Dawson, V.L.; Dinerman, J.L.; Uhl, G.R.; Snyder, S.H. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 9808–9812. [Google Scholar] [CrossRef]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Repnik, U.; Česen, M.H.; Turk, B. The endolysosomal system in cell death and survival. Cold Spring Harb. Perspect. Biol. 2013, 5, a008755. [Google Scholar] [CrossRef]
- Freund, T.F.; Buzsáki, G. Interneurons of the hippocampus. Hippocampus 1996, 6, 347–470. [Google Scholar] [CrossRef]
- Li, Y.; Jia, Y.-C.; Cui, K.; Li, N.; Zheng, Z.-Y.; Wang, Y.-Z.; Yuan, X.-B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 2005, 434, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Feng, S.; Ge, R.; Du, W.; Zhang, X.; He, Z.; Wang, Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J. Cell science 2008, 121, 2301–2307. [Google Scholar] [PubMed]
- Zhou, J.; Du, W.; Zhou, K.; Tai, Y.; Yao, H.; Jia, Y.; Ding, Y.; Wang, Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 2008, 11, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Tacer, K.F.; Bezprozvanny, I. Store-operated calcium channel complex in postsynaptic spines: A new therapeutic target for Alzheimer’s disease treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef]
- Zerov, N.; Pupugaeva, E. Role of neuronal TRPC6 channels in synapse development, memory formation and animal behavior. Int. J. Mol. Sci. 2023, 24, 15415. [Google Scholar]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.; Kappe, C.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Wang, Z.; Zhang, S.; Ding, D.; Lin, G.; Zhang, H.; Boda, V.K.; Kong, D.; Ortyl, T.C.; et al. TRPC3 suppression ameliorates synaptic dysfunctions and memory deficits in Alzheimer’s disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Oveisgharan, S.; Arvanitakis, Z.; Yu, L.; Farfel, J.; Schneider, J.A.; Bennett, D.A. Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol. 2018, 136, 887–900. [Google Scholar] [CrossRef]
- Filon, J.R.; Intorcia, A.J.; Sue, L.I.; Vazquez Arreola, E.; Wilson, J.; Davis, K.J.; Sabbagh, M.N.; Belden, C.M.; Caselli, R.J.; Adler, C.H.; et al. Gender Differences in Alzheimer Disease: Brain Atrophy, Histopathology Burden, and Cognition. J. Neuropathol. Exp. Neurol. 2016, 75, 748–754. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- Schwartz, N.; Schohl, A.; Ruthazer, E.S. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 2009, 62, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Sompol, P.; Furman, J.L.; Pleiss, M.M.; Kraner, S.D.; Artiushin, I.A.; Batten, S.R.; Quintero, J.E.; Simmerman, L.A.; Beckett, T.L.; Lovell, M.A.; et al. Calcineurin/NFAT Signaling in Activated Astrocytes Drives Network Hyperexcitability in Aβ-Bearing Mice. J. Neurosci. 2017, 37, 6132–6148. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Dai, Y.-S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiac hypertrophy is activated by TRPC in the adult mouse heart. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1660. [Google Scholar]
- Talukdar, P.D.; Chatterji, U. Transcriptional co-activators: Emerging roles in signaling pathways and potential therapeutic targets for diseases. Signal Transduct. Target. Ther. 2023, 8, 427. [Google Scholar] [CrossRef]
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J. Clin. Investig. 2010, 120, 3480–3492. [Google Scholar]
- Chauvet, S.; Boonen, M.; Chevallet, M.; Jarvis, L.; Abebe, A.; Benharouga, M.; Faller, P.; Jadot, M.; Bouron, A. The Na+/K+-ATPase and the amyloid-beta peptide aβ1–40 control the cellular distribution, abundance and activity of TRPC6 channels. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 2957–2965. [Google Scholar] [CrossRef]
- Saqib, U.; Munjuluri, S.; Sarkar, S.; Biswas, S.; Mukherjee, O.; Satsangi, H.; Baig, M.S.; Obukhov, A.G.; Hajela, K. Transient receptor potential canonical 6 (TRPC6) channel in the pathogenesis of diseases: A jack of many trades. Inflammation 2023, 46, 1144–1160. [Google Scholar]
- Li, H.; Huang, J.; Du, W.; Jia, C.; Yao, H.; Wang, Y. TRPC 6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. J. Neurochem. 2012, 123, 1010–1018. [Google Scholar]
- Guo, C.; Ma, Y.; Ma, S.; Mu, F.; Deng, J.; Duan, J.; Xiong, L.; Yin, Y.; Wang, Y.; Xi, M.; et al. The role of TRPC6 in the neuroprotection of calycosin against cerebral ischemic injury. Sci. Rep. 2017, 7, 3039. [Google Scholar]
- Shekhar, S.; Liu, Y.; Wang, S.; Zhang, H.; Fang, X.; Zhang, J.; Fan, L.; Zheng, B.; Roman, R.J.; Wang, Z.; et al. Novel mechanistic insights and potential therapeutic impact of TRPC6 in neurovascular coupling and ischemic stroke. Int. J. Mol. Sci. 2021, 22, 2074. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Z.; Hatcher, J.T.; Chen, Q.-H.; Chen, L.; Wurster, R.D.; Chan, S.L.; Cheng, Z. Deletion of TRPC6 attenuates NMDA receptor-mediated Ca2+ entry and Ca2+-induced neurotoxicity following cerebral ischemia and oxygen-glucose deprivation. Front. Neurosci. 2017, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 2015, 20, 1350–1365. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.C.; Ali, G.; Ali Moussa, H.Y.; Gupta, V.; de la Fuente, A.; Kim, H.-G.; Stanton, L.W.; Park, Y. Deletion of TRPC6, an autism risk gene, induces hyperexcitability in cortical neurons derived from human pluripotent stem cells. Mol. Neurobiol. 2023, 60, 7297–7308. [Google Scholar] [CrossRef]
- Álvarez-Miguel, I.; Cidad, P.; Pérez-García, M.T.; López-López, J.R. Differences in TRPC3 and TRPC6 channels assembly in mesenteric vascular smooth muscle cells in essential hypertension. J. Physiol. 2017, 595, 1497–1513. [Google Scholar] [CrossRef]
- Kollewe, A.; Schwarz, Y.; Oleinikov, K.; Raza, A.; Haupt, A.; Wartenberg, P.; Wyatt, A.; Boehm, U.; Ectors, F.; Bildl, W.; et al. Subunit composition, molecular environment, and activation of native TRPC channels encoded by their interactomes. Neuron 2022, 110, 4162–4175.E7. [Google Scholar] [CrossRef]
- Chung, Y.H.; Ahn, H.S.; Kim, D.; Shin, D.H.; Kim, S.S.; Kim, K.Y.; Lee, W.B.; Cha, C.I. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res. 2006, 1085, 132–137. [Google Scholar] [CrossRef]
- Nagy, G.A.; Botond, G.; Borhegyi, Z.; Plummer, N.W.; Freund, T.F.; Hájos, N. DAG-sensitive and Ca2+ permeable TRPC6 channels are expressed in dentate granule cells and interneurons in the hippocampal formation. Hippocampus 2013, 23, 221–232. [Google Scholar] [CrossRef]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef]
- Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: What are the drivers behind this aberrant phenotype? Transl. Psychiatry 2022, 12, 257. [Google Scholar] [CrossRef]
- Nagib, M.M.; Zhang, S.; Yasmen, N.; Li, L.; Hou, R.; Yu, Y.; Boda, V.K.; Wu, Z.; Li, W.; Jiang, J. Inhibition of TRPC3 channels by a novel pyrazole compound confers antiseizure effects. Epilepsia 2022, 63, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.B.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Bu, F.; Sun, G.; Tian, J.-B.; Ting, S.-M.; Li, J.; Aronowski, J.; Birnbaumer, L.; Freichel, M.; Zhu, M.X. Contribution of TRPC channels in neuronal excitotoxicity associated with neurodegenerative disease and ischemic stroke. Front. Cell Dev. Biol. 2021, 8, 618663. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Wu, H.; Zheng, F. Investigating Contributions of Canonical Transient Receptor Potential Channel 3 to Hippocampal Hyperexcitability and Seizure-Induced Neuronal Cell Death. Int. J. Mol. Sci. 2024, 25, 6260. [Google Scholar] [CrossRef]
- Kim, D.-S.; Ryu, H.J.; Kim, J.-E.; Kang, T.-C. The reverse roles of transient receptor potential canonical channel-3 and-6 in neuronal death following pilocarpine-induced status epilepticus. Cell. Mol. Neurobiol. 2013, 33, 99–109. [Google Scholar] [CrossRef]
- Zeng, C.; Zhou, P.; Jiang, T.; Yuan, C.; Ma, Y.; Feng, L.; Liu, R.; Tang, W.; Long, X.; Xiao, B.; et al. Upregulation and diverse roles of TRPC3 and TRPC6 in synaptic reorganization of the mossy fiber pathway in temporal lobe epilepsy. Mol. Neurobiol. 2015, 52, 562–572. [Google Scholar] [CrossRef]
- Li, H.-S.; Xu, X.-Z.S.; Montell, C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 1999, 24, 261–273. [Google Scholar] [CrossRef]
- Amaral, M.D.; Pozzo-Miller, L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 2007, 27, 5179–5189. [Google Scholar] [CrossRef]
- Li, Y.; Calfa, G.; Inoue, T.; Amaral, M.D.; Pozzo-Miller, L. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J. Neurophysiol. 2010, 103, 2846–2856. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| hTrpc3 | TGCAGAAGGAGAAGGCTTC | ACGTGTTGGCTGATTGAGA |
| hTrpc6 | GTGGCCTATGTCAAGTATAA | GGCAGTAGATAAAGAGGAAT |
| hβActin | CCCGCGAGTACAACCTTCT | CGTCATCCATGGCGAACT |
| rTrpc1 | CACGATTTCCCTCTCAGC | AGTTCCTGAACACCGTTTGG |
| rTrpc3 | CCACATGCAGTGAGACTTTGACTC | AGGCCAACCTTGGGATCATTT |
| rTrpc4 | AAGGATTAGCTTCACGGGGTG | CCTCCTCCTGGGCGTGTTTC |
| rTrpc5 | TGAGTCGTCAGGCAAACGGTC | AGAAATTTGGAATTTTGGGAAGTC |
| rTrpc6 | AGAAATTTGGAATTTTGGGAAGTC | TCCTTATCAATCTGGGCCTGC |
| rTrpc7 | TCCCTTTAACCTGGTGCCGAGTC | TTCAGCATGCCCATTTCCAGG |
| rβActin | CCCGCGAGTACAACCTTCT | CGTCATCCATGGCGAACT |
| Chip_Trpc3_1 | CGTTGGTTACAGCCAACCTC | GCCCTTACTGGTGGGGTATT |
| Chip_Trpc3_2 | GGCTGTCAGGGAACTGTCTC | GAAATCACCCCCTGCTGGAA |
| Chip_Trpc6 | TTAGGACAAGCAGAGCCACG | GGGCTAACTGCTCCCAAAGT |
| siTRPC3 | UCAUCUUCCUGGGUCUGCUUGUGUU | AACACAAGCAGACCCAGGAAGAUGA |
| siTRPC6 | GGAGCUCAGAAGAUUUCCAUUUAAA | UUUAAAUGGAAAUCUUCUGAGCUCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Ding, D.; Wang, J.; Chen, L.; Dong, Q.; Khamrai, M.; Zhou, Y.; Ishii, A.; Sakata, K.; Li, W.; et al. Soluble β-Amyloid Oligomers Selectively Upregulate TRPC3 in Excitatory Neurons via Calcineurin-Coupled NFAT. Cells 2025, 14, 843. https://doi.org/10.3390/cells14110843
Wang Z, Ding D, Wang J, Chen L, Dong Q, Khamrai M, Zhou Y, Ishii A, Sakata K, Li W, et al. Soluble β-Amyloid Oligomers Selectively Upregulate TRPC3 in Excitatory Neurons via Calcineurin-Coupled NFAT. Cells. 2025; 14(11):843. https://doi.org/10.3390/cells14110843
Chicago/Turabian StyleWang, Zhengjun, Dongyi Ding, Jiaxing Wang, Ling Chen, Qingming Dong, Moumita Khamrai, Yuyang Zhou, Akihiro Ishii, Kazuko Sakata, Wei Li, and et al. 2025. "Soluble β-Amyloid Oligomers Selectively Upregulate TRPC3 in Excitatory Neurons via Calcineurin-Coupled NFAT" Cells 14, no. 11: 843. https://doi.org/10.3390/cells14110843
APA StyleWang, Z., Ding, D., Wang, J., Chen, L., Dong, Q., Khamrai, M., Zhou, Y., Ishii, A., Sakata, K., Li, W., Du, J., Vaithianathan, T., Zhou, F.-M., & Liao, F.-F. (2025). Soluble β-Amyloid Oligomers Selectively Upregulate TRPC3 in Excitatory Neurons via Calcineurin-Coupled NFAT. Cells, 14(11), 843. https://doi.org/10.3390/cells14110843