
Elevated Mutation Burdens in Canadian Oat and Wheat Cultivars Released over the Past Century



Abstract
1. Introduction
2. Materials and Methods
2.1. Assayed Oat and Wheat Cultivars
2.2. Cultivar Yield Data Collection
2.3. RNA-Seq Analysis
2.4. SNP Calling
2.5. Identification of Deleterious SNPs
2.6. Gene Ontology (GO) and Expression Analysis
2.7. Mutation Burden Estimation and Its Association with Cultivar Features
2.8. Nucleotide Diversity and Genetic Association Analysis
3. Results
3.1. SNP Identification and Annotation
3.2. Deleterious Mutation
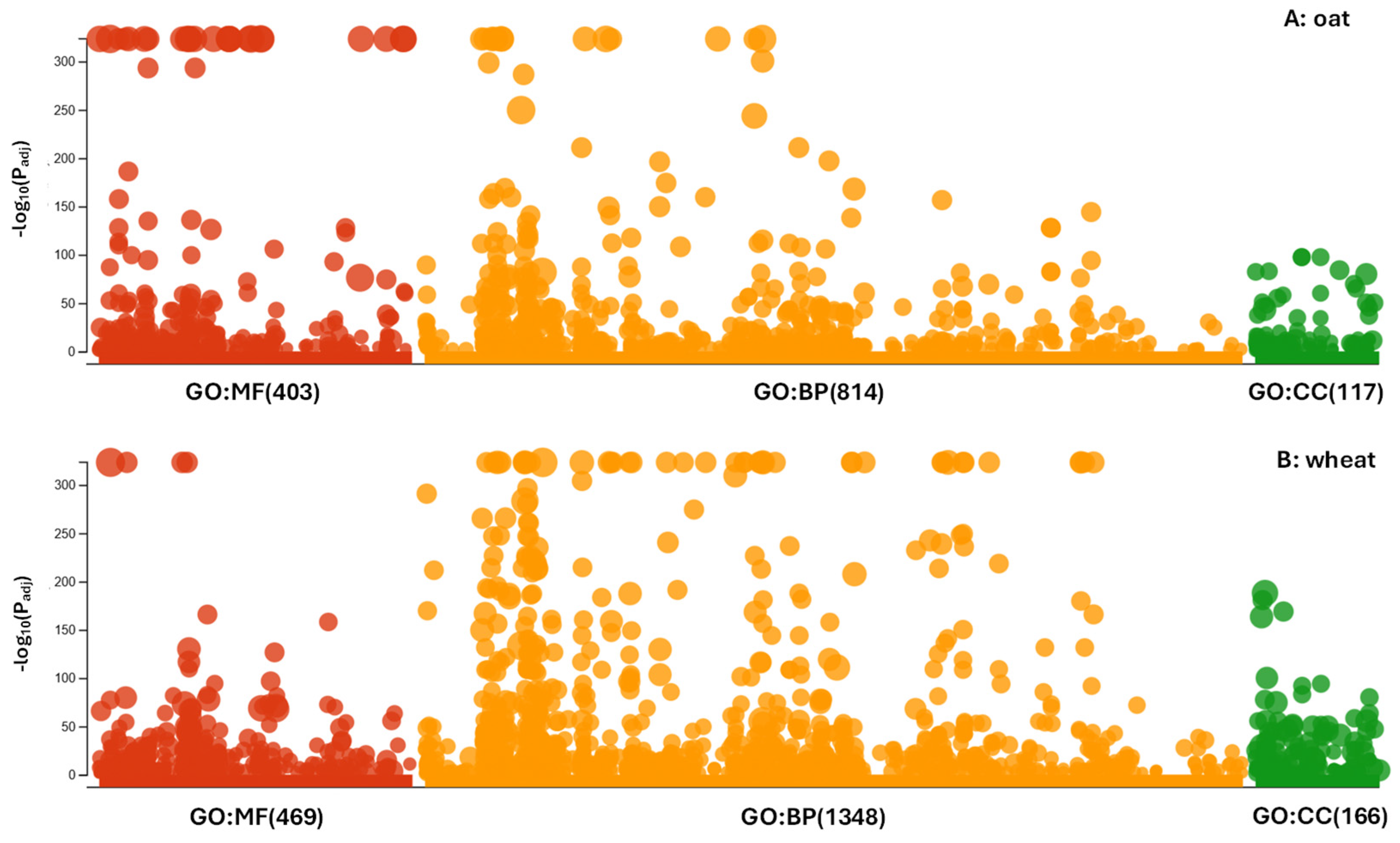
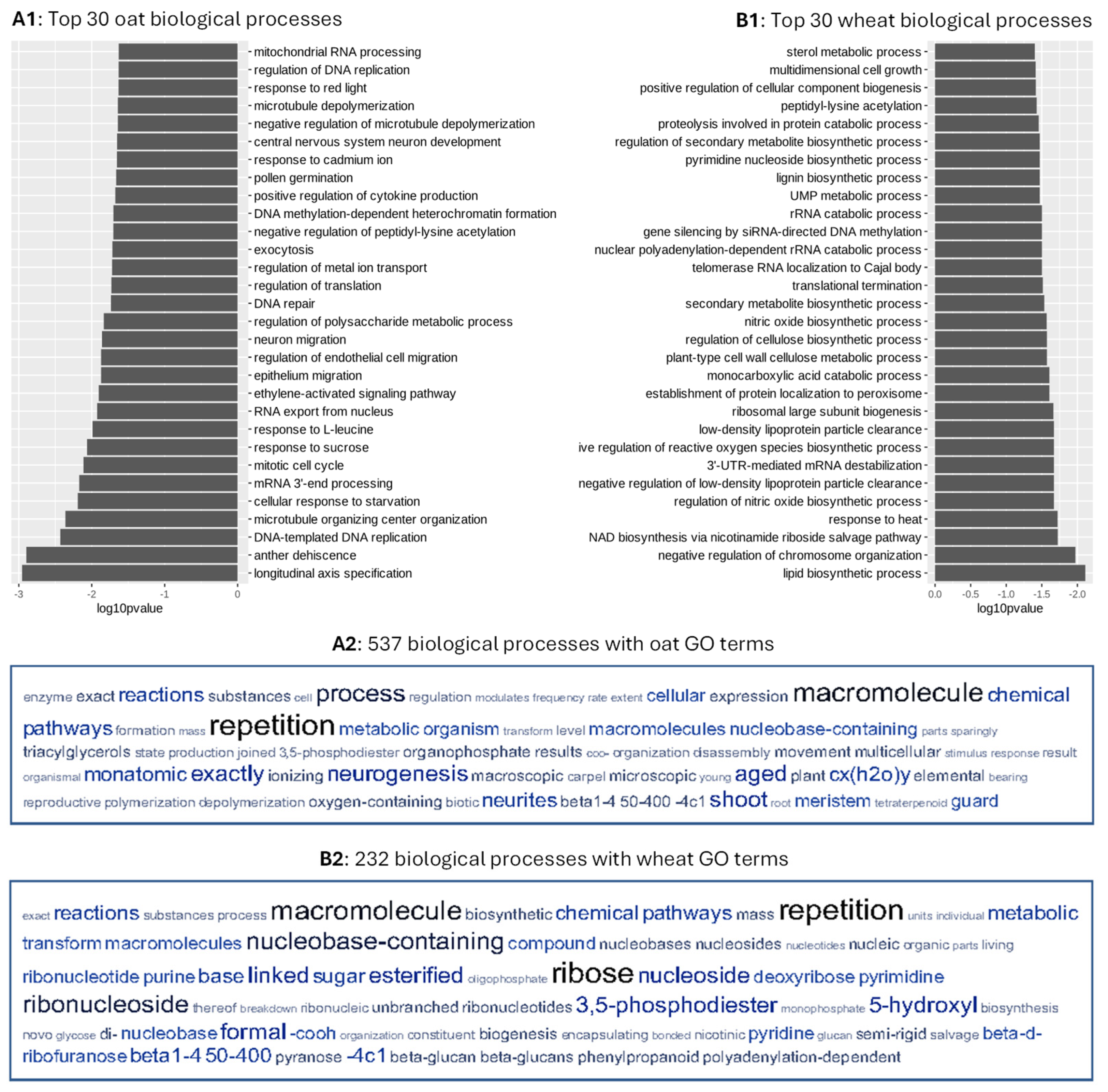
3.3. Ontology and Expression of the Associated Genes
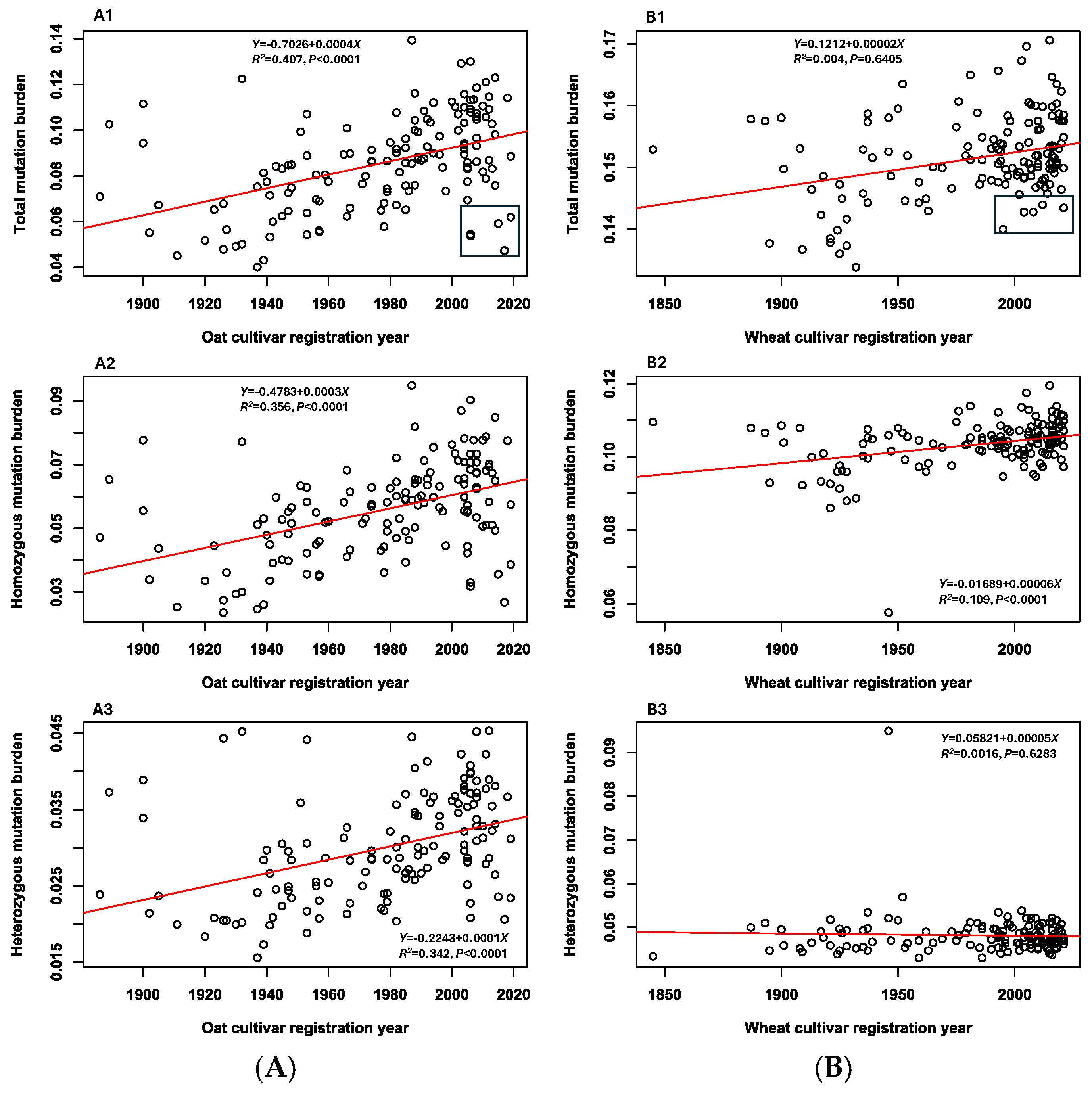
3.4. Mutation Burden
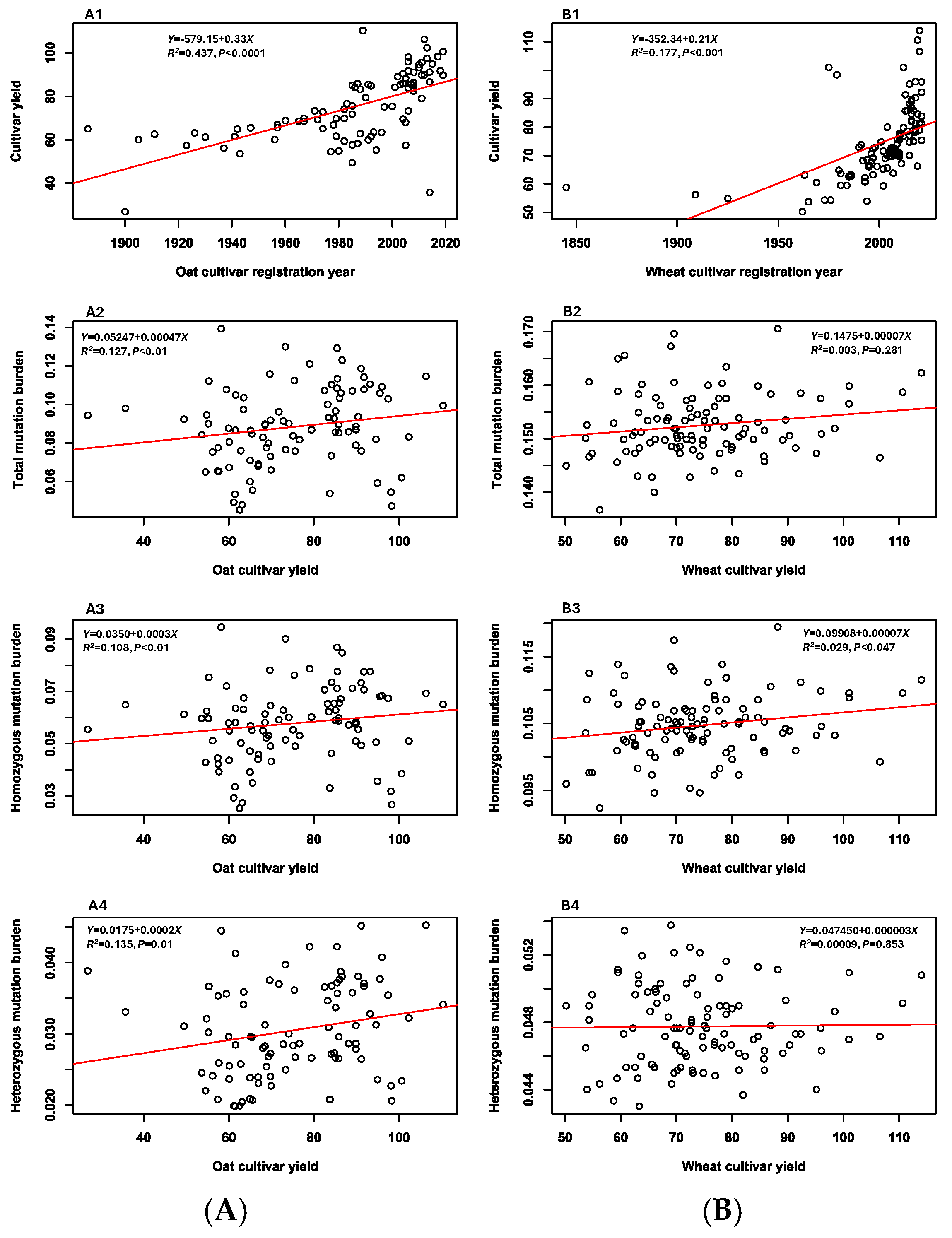
3.5. Associations Between Mutation Burdens and Cultivar Features
3.6. Nucleotide Diversity and Genetic Association
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAFC | Agriculture and Agri-Food Canada |
| BAM | Binary alignment map |
| CDC | Crop Development Centre |
| dSNP | Deleterious simple nucleotide polymorphism |
| FASTA | A text-based file format used to store nucleotide and protein sequences |
| FASTQ | A text-based file format used to store a nucleotide sequence and its quality scores |
| GO | Gene ontology |
| PCA | Principal component analysis |
| PCR | Polymerase chain reaction |
| PGRC | Plant Gene Resources of Canada |
| RNA-Seq | RNA sequencing |
| RS | Rejected substitution |
| SCIC | Saskatchewan Crop Insurance Corporation |
| SIFT | Sorting intolerant from tolerant |
| SNP | Single nucleotide polymorphism |
| VCF | Variant call format |
| VEP | Variant effect predictor |
References
- Muller, H.J. Our Load of Mutations. Am. J. Hum. Genet. 1950, 2, 111–176. [Google Scholar] [PubMed]
- Hedrick, P.W. Genetics of Populations, 2nd ed.; Jones and Bartlett Publishers: Sudbury, MA, USA, 2000. [Google Scholar]
- D’Antonio, M.; Benaglio, P.; Jakubosky, D.; Greenwald, W.W.; Matsui, H.; Donovan, M.K.R.; Li, H.; Smith, E.N.; D’Antonio-Chronowska, A.; Frazer, K.A. Insights into the Mutational Burden of Human Induced Pluripotent Stem Cells from an Integrative Multi-Omics Approach. Cell Rep. 2018, 24, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor Mutational Load Predicts Survival After Immunotherapy Across Multiple Cancer Types. Nat. Genet. 2019, 2, 202–206. [Google Scholar] [CrossRef]
- Sholl, L.M.; Hirsch, F.R.; Hwang, D.; Botling, J.; Lopez-Rios, F.; Bubendorf, L.; Mino-Kenudson, M.; Roden, A.C.; Beasley, M.B.; Borczuk, A.; et al. The Promises and Challenges of Tumor Mutation Burden as an Immunotherapy Biomarker: A Perspective from the International Association for the Study of Lung Cancer Pathology Committee. J. Thorac. Oncol. 2020, 15, 1409–1424. [Google Scholar] [CrossRef]
- Moyers, B.T.; Morrell, P.L.; McKay, J.K. Genetic Costs of Domestication and Improvement. J. Hered. 2018, 109, 103–116. [Google Scholar] [CrossRef]
- Kono, T.J.Y.; Liu, C.; Vonderharr, E.E.; Koenig, D.; Fay, J.C.; Smith, K.P.; Morrell, P.L. The Fate of Deleterious Variants in a Barley Genomic Prediction Population. Genetics 2019, 213, 1531–1544. [Google Scholar] [CrossRef]
- Dwivedi, S.L.; Heslop-Harrison, P.; Spillane, C.; McKeown, P.C.; Edwards, D.; Goldman, I.; Ortiz, R. Evolutionary Dynamics and Adaptive Benefits of Deleterious Mutations in Crop Gene Pools. Trends Plant Sci. 2023, 28, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, T.; Tang, H.; Huang, J.; Shi, S.; Wu, C.I. The Accumulation of Deleterious Mutations in Rice Genomes: A Hypothesis on the Cost of Domestication. Trends Genet. 2006, 22, 126–131. [Google Scholar] [CrossRef]
- Günther, T.; Schmid, K.J. Deleterious Amino Acid Polymorphisms in Arabidopsis thaliana and Rice. Theor. Appl. Genet. 2010, 121, 157–168. [Google Scholar] [CrossRef]
- Mezmouk, S.; Ross-Ibarra, J. The Pattern and Distribution of Deleterious Mutations in Maize. G3: Genes Genomes Genet. 2014, 4, 163–171. [Google Scholar] [CrossRef]
- Renaut, S.; Rieseberg, L.H. The Accumulation of Deleterious Mutations as a Consequence of Domestication and Improvement in Sunflowers and Other Compositae Crops. Mol. Biol. Evol. 2015, 32, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.J.Y.; Fu, F.; Mohammadi, M.; Hoffman, P.J.; Liu, C.; Stupar, R.M.; Smith, K.P.; Tiffin, P.; Fay, J.C.; Morrell, P.L. The Role of Deleterious Substitutions in Crop Genomes. Mol. Biol. Evol. 2016, 33, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.; Morrell, P.L.; Gaut, B.S. Deleterious Variants in Asian Rice and the Potential Cost of Domestication. Mol. Biol. Evol. 2017, 34, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Ramu, P.; Esuma, W.; Kawuki, R.; Rabbi, I.Y.; Egesi, C.; Bredeson, J.V.; Bart, R.S.; Verma, J.; Buckler, E.S.; Lu, F. Cassava Haplotype Map Highlights Fixation of Deleterious Mutations During Clonal Propagation. Nat. Genet. 2017, 49, 959–963. [Google Scholar] [CrossRef]
- Valluru, R.; Gazave, E.E.; Fernandes, S.B.; Ferguson, J.N.; Lozano, R.; Hirannaiah, P.; Zuo, T.; Brown, P.J.; Leakey, A.D.; Gore, M.A.; et al. Deleterious Mutation Burden and Its Association with Complex Traits in Sorghum (Sorghum bicolor). Genetics 2019, 211, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Gazave, E.; dos Santos, J.P.R.; Stetter, M.G.; Valluru, R.; Bandillo, N.; Fernandes, S.B.; Brown, P.J.; Shakoor, N.; Mockler, T.C.; et al. Comparative Evolutionary Genetics of Deleterious Load in Sorghum and Maize. Nat. Plants 2021, 7, 17–24. [Google Scholar] [CrossRef]
- Fu, Y.B.; Peterson, G.W.; Horbach, C. Deleterious and Adaptive Mutations in Plant Germplasm Conserved Ex Situ. Mol. Biol. Evol. 2023, 40, msad238. [Google Scholar] [CrossRef]
- Sun, S.; Wang, B.; Li, C.; Xu, G.; Yang, J.; Hufford, M.B.; Ross-Ibarra, J.; Wang, H.; Wang, L. Unraveling Prevalence and Effects of Deleterious Mutations in Maize Elite Lines Across Decades of Modern Breeding. Mol. Biol. Evol. 2023, 40, msad170. [Google Scholar] [CrossRef]
- Wu, Y.; Li, D.; Hu, Y.; Li, H.; Ramstein, G.P.; Zhou, S.; Zhang, X.; Bao, Z.; Zhang, Y.; Song, B.; et al. Phylogenomic Discovery of Deleterious Mutations Facilitates Hybrid Potato Breeding. Cell 2023, 186, 2313–2328. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and Intensity of Constraint in Mammalian Genomic Sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Henn, B.M.; Botigué, L.R.; Bustamante, C.D.; Clark, A.G.; Gravel, S. Estimating the Mutation Load in Human Genomes. Nat. Rev. Genet. 2015, 16, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Plekhanova, E.; Nuzhdin, S.V.; Utkin, L.V.; Samsonova, M.G. Prediction of Deleterious Mutations in Coding Regions of Mammals with Transfer Learning. Evol. Appl. 2019, 12, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, G.; Raffini, F.; Bosse, M.; Bortoluzzi, C.; Iannucci, A.; Trucchi, E.; Morales, H.; van Oosterhout, C. Genetic Load: Genomic Estimates and Applications in Non-Model Animals. Nat. Rev. Genet. 2022, 23, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Slinkard, A.E.; Knott, D.R. Harvest of Gold: The History of Field Crop Breeding in Canada; University Extension Press: Saskatoon, SK, Canada, 1995. [Google Scholar]
- DePauw, R.M.; Boughton, G.R.; Knott, D.R. Hard Red Spring Wheat. In Harvest of Gold: The History of Field Crop Breeding in Canada; Slinkard, A.E., Knott, D.R., Eds.; University Extension Press: Saskatoon, SK, Canada, 1995; pp. 5–35. [Google Scholar]
- Iqbal, M.; Moakhar, N.P.; Strenzke, K.; Haile, T.; Pozniak, C.; Hucl, P.; Spaner, D. Genetic Improvement in Grain Yield and Other Traits of Wheat Grown in Western Canada. Crop Sci. 2016, 56, 613–624. [Google Scholar] [CrossRef]
- Fu, Y.B.; Somers, D.J. Genome-Wide Reduction of Genetic Diversity in Wheat Breeding. Crop Sci. 2009, 49, 161–168. [Google Scholar] [CrossRef]
- Keneni, G.; Bekele, E.; Imtiaz, M.; Dagne, K. Genetic Vulnerability of Modern Crop Cultivars: Causes, Mechanism and Remedies. Int. J. Plant Res. 2012, 2, 69–79. [Google Scholar] [CrossRef]
- Fu, Y.B.; Dong, Y. Genetic Erosion Under Modern Plant Breeding: Case Studies in Canadian Crop Gene Pools. In Genetic Diversity and Erosion in Plants; Ahuja, M.R., Jain, S.M., Eds.; Springer: Cham, Switzerland, 2015; Chapter 4; pp. 89–104. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 June 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kamal, N.; Renhuldt, N.T.; Bentzer, J.; Gundlach, H.; Haberer, G.; Juhász, A.; Lux, T.; Bose, U.; Tye-Din, J.A.; Lang, D.; et al. The Mosaic Oat Genome Gives Insights into a Uniquely Healthy Cereal Crop. Nature 2022, 606, 113–119. [Google Scholar] [CrossRef]
- IWGSC; Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the Limits in Wheat Research and Breeding Using a Fully Annotated Reference Genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Naithani, S.; Geniza, M.; Jaiswal, P. Variant Effect Prediction Analysis Using Resources Available at Gramene Database. In Plant Genomics Databases: Methods and Protocols; van Dijk, A., Ed.; Humana Press: New York, NY, USA, 2017; pp. 270–297. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT Missense Predictions for Genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be Under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler—Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 22 September 2024).
- Fu, Y.B.; Peterson, G.W.; Scoles, G.; Rossnagel, B.; Schoen, D.J.; Richards, K.W. Allelic Diversity Changes in 96 Canadian Oat Cultivars Released from 1886 to 2001. Crop Sci. 2003, 43, 1989–1995. [Google Scholar] [CrossRef]
- Fu, Y.B.; Peterson, G.W.; Richards, K.W.; Somers, D.; DePauw, R.M.; Clarke, J.M. Allelic Reduction and Genetic Shift in the Canadian Hard Red Spring Wheat Germplasm Released from 1845 to 2004. Theor. Appl. Genet. 2005, 110, 1505–1516. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A High-Performance Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Charlesworth, D.; Morgan, M.T.; Charlesworth, B. Mutation Accumulation in Finite Outbreeding and Inbreeding Populations. Genet. Res. 1993, 61, 39–56. [Google Scholar] [CrossRef]
- Fu, Y.B.; Namkoong, G.; Carlson, J.E. Comparison of Breeding Strategies for Purging Inbreeding Depression via Simulation. Conserv. Biol. 1998, 12, 856–864. [Google Scholar] [CrossRef]
- Fu, Y.B. Patterns of Purging Deleterious Genes of Synergistic Interactions with Different Breeding Schemes. Theor. Appl. Genet. 1999, 98, 337–356. [Google Scholar] [CrossRef]
- National Research Council. Genetic Vulnerability of Major Crops; National Academy of Sciences: Washington, DC, USA, 1972.
- Duvick, D.N. Genetic Diversity in Major Farm Crops on the Farm and in Reserve. Econ. Bot. 1984, 38, 161–178. [Google Scholar] [CrossRef]
- Tripp, R. Biodiversity and Modern Crop Varieties: Sharpening the Debate. Agr. Hum. Values 1996, 13, 48–63. [Google Scholar] [CrossRef]
- Singh, J.; van der Knaap, E. Unintended Consequences of Plant Domestication. Plant Cell Physiol. 2022, 63, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, N.; Bao, Z.; Zhou, Q.; Guarracino, A.; Yang, Y.; Wang, P.; Zhang, Z.; Tang, D.; Zhang, P.; et al. Leveraging a Phased Pangenome for Haplotype Design of Hybrid Potato. Nature 2025, 640, 408–417. [Google Scholar] [CrossRef]
- Fu, Y.B. Variability in Predicted Deleterious Mutations Among Barley Accessions Conserved Ex Situ. Crop Sci. 2024, 64, 3372–3380. [Google Scholar] [CrossRef]
- Roles, A.J.; Conner, J.K. Fitness Effects of Mutation Accumulation in a Natural Outbred Population of Wild Radish (Raphanus raphanistrum): Comparison of Field and Greenhouse Environments. Evolution 2008, 62, 1066–1075. [Google Scholar] [CrossRef]
- Gao, C. Genome Engineering for Crop Improvement and Future Agriculture. Cell 2021, 184, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaye, C.; Kaur, R.; Singh, J. A Highly Efficient CRISPR-Cas9-based Gene-editing System in Oat (Avena sativa L.). Plant Biotechnol. J. 2025. [Google Scholar] [CrossRef]
- Li, S.; Lin, D.; Zhang, Y.; Deng, M.; Chen, Y.; Lv, B.; Li, B.; Lei, Y.; Wang, Y.; Zhao, L.; et al. Genome-Edited Powdery Mildew Resistance in Wheat Without Growth Penalties. Nature 2022, 602, 455–460. [Google Scholar] [CrossRef]
- Glaus, A.N.; Brechet, M.; Swinnen, G.; Lebeigle, L.; Iwaszkiewicz, J.; Ambrosini, G.; Julca, I.; Zhang, J.; Roberts, R.; Iseli, C.; et al. Repairing a Deleterious Domestication Variant in a Floral Regulator Gene of Tomato by Base Editing. Nat. Genet. 2025, 57, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Yang, M.-H.; Zeng, F.; Biligetu, B. Searching for an Accurate Marker-Based Prediction of an Individual Quantitative Trait in Molecular Plant Breeding. Front. Plant Sci. 2017, 8, 1182. [Google Scholar] [CrossRef]
- Johnsson, M.; Gaynor, R.C.; Jenko, J.; Gorjanc, G.; de Koning, D.-J.; Hickey, J.M. Removal of Alleles by Genome Editing (RAGE) Against Deleterious Load. Genet. Sel. Evol. 2019, 51, 14. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | 141 Oat Cultivars | 142 Wheat Cultivars |
|---|---|---|
| SNP calling and filtering | ||
| Total SNPs without missing values | 253,264 | 270,622 |
| SNP annotation with VEP (most severe consequences) | ||
| Missense_variant (MV) | 74,655 | 94,003 |
| Proportion of MV in total SNPs | 0.2948 | 0.3474 |
| Synonymous_variant (SV) | 125,064 | 144,071 |
| Proportion of SV in total SNPs | 0.4938 | 0.5324 |
| Splice_acceptor_variant | 155 | 279 |
| Splice_donor_variant | 137 | 341 |
| Stop_gained | 582 | 520 |
| Stop_lost | 147 | 84 |
| Start_lost | 57 | 51 |
| Splice_region_variant | 659 | 1214 |
| Stop_retained_variant | 184 | 127 |
| Coding_sequence_variant | 0 | 3 |
| 5_prime_UTR_variant | 14,382 | 29,115 |
| 3_prime_UTR_variant | 52,173 | 57,649 |
| Non_coding_transcript_exon_variant | 0 | 190 |
| Intron_variant | 5598 | 8104 |
| Upstream_gene_variant | 83,198 | 71,843 |
| Downstream_gene_variant | 131,064 | 114,488 |
| Intergenic_variant | 2937 | 20,233 |
| Loss-of-function variant * | ||
| Total count | 1921 | 2616 |
| Proportion | 0.0076 | 0.0097 |
| SIFT analysis with CT ** | ||
| SIFT-deleterious SNPs (SDS) | 12,182 | 12,855 |
| Proportion of SDS in total SNPs | 0.0481 | 0.0475 |
| Deleterious_low_confidence SNPs | 4026 | 4664 |
| Tolerated SNPs | 152,099 | 57,736 |
| Tolerated_low_confidence SNPs | NA *** | 17,559 |
| Deleterious SNPs by SIFT+RS | ||
| SDS+RS-filtered SNPs (RSD) | 5726 | 3022 |
| Proportion of RSD in total SNPs | 0.02261 | 0.01117 |
| Fixed RSD | 3 | 16 |
| Proportion of fixed RSD in total SNPs | 0.000012 | 0.000059 |
| Weakly deleterious with RS < 1 | 2348 | 2834 |
| Mildly deleterious with RS of 1–3 | 2295 | 161 |
| Highly deleterious with RS > 3 | 1083 | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Crown Copyright: @ His Majesty the King in Right of Canada, 2025. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.-B.; Horbach, C. Elevated Mutation Burdens in Canadian Oat and Wheat Cultivars Released over the Past Century. Cells 2025, 14, 844. https://doi.org/10.3390/cells14110844
Fu Y-B, Horbach C. Elevated Mutation Burdens in Canadian Oat and Wheat Cultivars Released over the Past Century. Cells. 2025; 14(11):844. https://doi.org/10.3390/cells14110844
Chicago/Turabian StyleFu, Yong-Bi, and Carolee Horbach. 2025. "Elevated Mutation Burdens in Canadian Oat and Wheat Cultivars Released over the Past Century" Cells 14, no. 11: 844. https://doi.org/10.3390/cells14110844
APA StyleFu, Y.-B., & Horbach, C. (2025). Elevated Mutation Burdens in Canadian Oat and Wheat Cultivars Released over the Past Century. Cells, 14(11), 844. https://doi.org/10.3390/cells14110844