


Brain Organoids and Assembloids—From Disease Modeling to Drug Discovery


Abstract

1. Introduction
2. Stem Cells
2.1. Embryonic Stem Cells
2.2. Human Induced Pluripotent Stem Cells
2.3. Neural Progenitor or Stem Cells
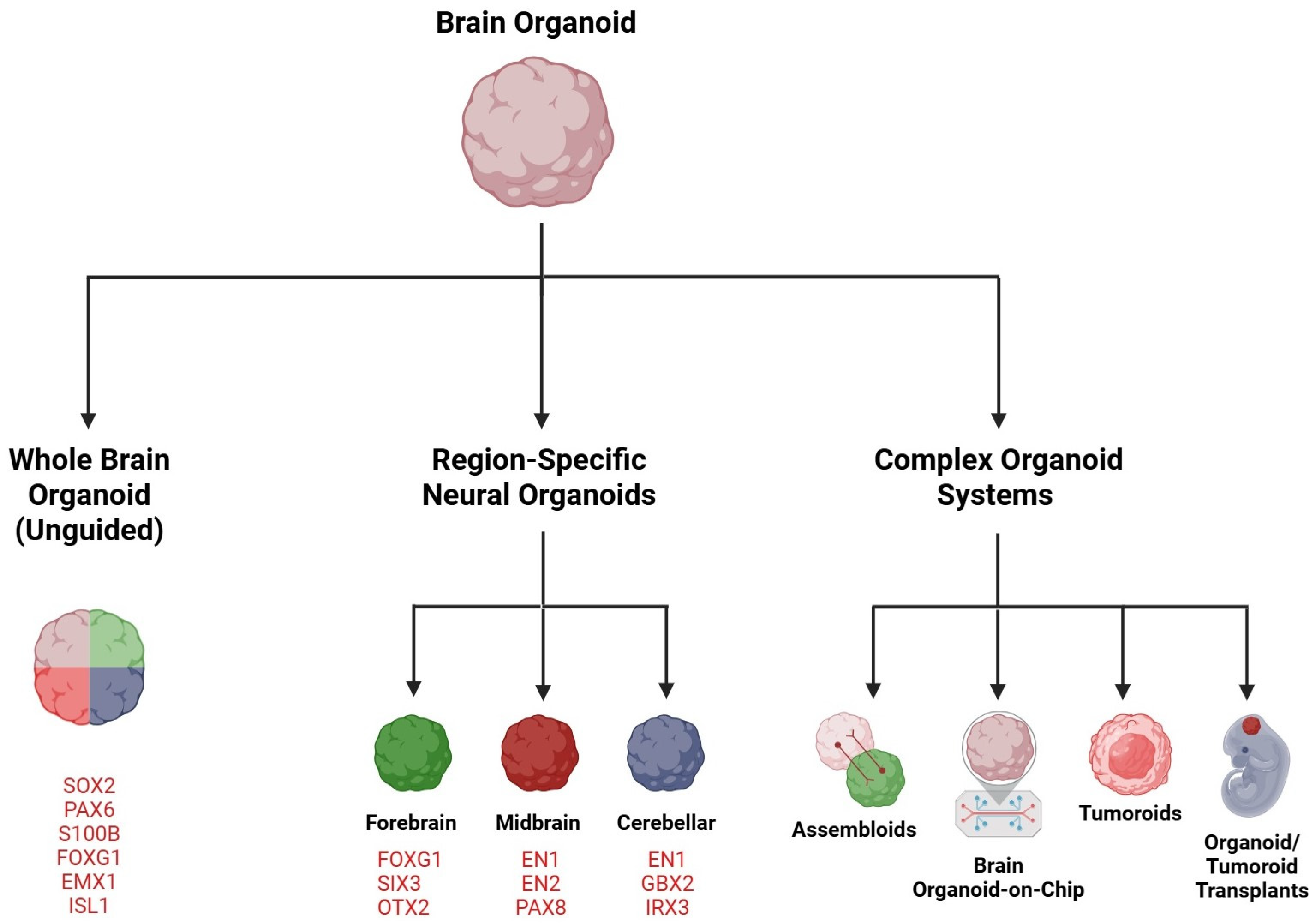
3. Brain Organoids
3.1. Whole Brain or Cerebral Organoid
3.2. Forebrain Organoid
3.3. Midbrain Organoid
3.4. Cerebellar Organoid
4. Complex Organoid Systems
4.1. Assembloids
4.2. Brain Organoid-on-a-Chip
5. Brain Tumoroids
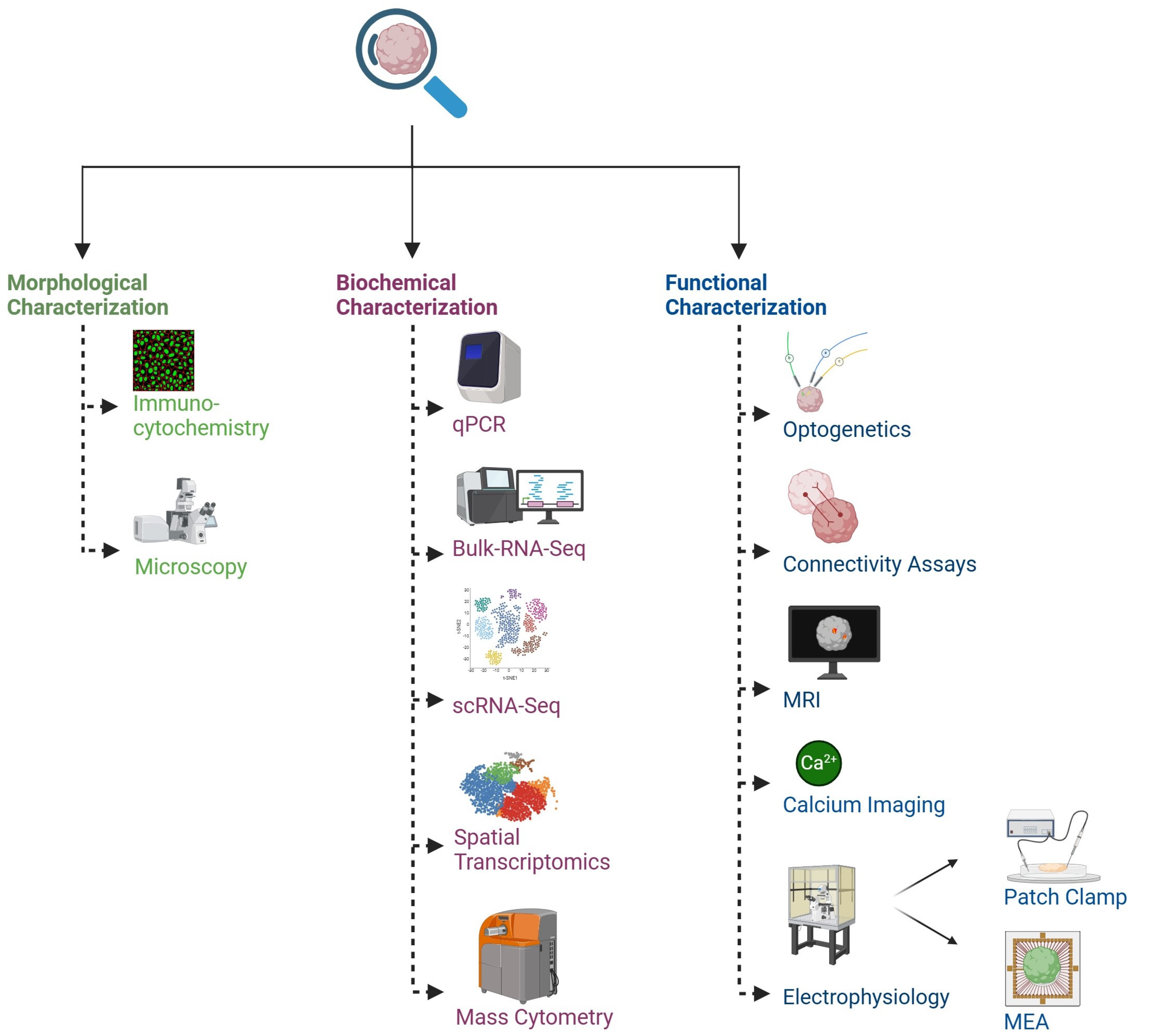
6. Functional Assays and Omics Used in Brain Organoid Characterization
7. Brain Organoids as Disease Models—Cancer
7.1. Gliomas
7.2. Medulloblastoma
8. Brain Organoids as Disease Models—Neurodevelopmental Disorders
8.1. Autism Spectrum Disorder
8.2. Epilepsy
8.3. Cerebral Palsy
8.4. Attention Deficit Hyperactivity Disorders
9. Assembloids and Rare Diseases of the Nervous System
10. Brain Organoids in Drug Discovery
11. Discussion
12. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Qian, X.; Song, H.; Ming, G.L. Brain organoids: Advances, applications and challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [PubMed]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Bagley, J.A.; Reumann, D.; Bian, S.; Levi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Lebkowski, J.; McNiece, I. Stem Cells. Biol. Blood Marrow Transplant. 2010, 16, S115–S118. [Google Scholar] [CrossRef] [PubMed]
- Poliwoda, S.; Noor, N.; Downs, E.; Schaaf, A.; Cantwell, A.; Ganti, L.; Kaye, A.D.; Mosel, L.I.; Carroll, C.B.; Viswanath, O.; et al. Stem cells: A comprehensive review of origins and emerging clinical roles in medical practice. Orthop. Rev. 2022, 14, 37498. [Google Scholar] [CrossRef]
- Daley, G.Q. Stem cells and the evolving notion of cellular identity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140376. [Google Scholar] [CrossRef]
- Kolios, G.; Moodley, Y. Introduction to stem cells and regenerative medicine. Respiration 2013, 85, 3–10. [Google Scholar] [CrossRef]
- Ilic, D.; Polak, J.M. Stem cells in regenerative medicine: Introduction. Br. Med. Bull. 2011, 98, 117–126. [Google Scholar] [CrossRef]
- De Miguel, M.P.; Fuentes-Julian, S.; Alcaina, Y. Pluripotent stem cells: Origin, maintenance and induction. Stem Cell Rev. Rep. 2010, 6, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Poniewierska, A.; Suszynska, M.; Ratajczak, J. Pluripotent and multipotent stem cells in adult tissues. Adv. Med. Sci. 2012, 57, 1–17. [Google Scholar] [CrossRef]
- Majo, F.; Rochat, A.; Nicolas, M.; Jaoude, G.A.; Barrandon, Y. Oligopotent stem cells are distributed throughout the mammalian ocular surface. Nature 2008, 456, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Rudnicki, M.A. The emerging biology of muscle stem cells: Implications for cell-based therapies. Bioessays 2013, 35, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Golchin, A.; Chatziparasidou, A.; Ranjbarvan, P.; Niknam, Z.; Ardeshirylajimi, A. Embryonic Stem Cells in Clinical Trials: Current Overview of Developments and Challenges. Adv. Exp. Med. Biol. 2021, 1312, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Bongso, A. Blastocyst culture for deriving human embryonic stem cells. Methods Mol. Biol. 2006, 331, 13–22. [Google Scholar] [CrossRef]
- Hambiliki, F.; Strom, S.; Zhang, P.; Stavreus-Evers, A. Co-localization of NANOG and OCT4 in human pre-implantation embryos and in human embryonic stem cells. J. Assist. Reprod. Genet. 2012, 29, 1021–1028. [Google Scholar] [CrossRef]
- Wang, Z.; Oron, E.; Nelson, B.; Razis, S.; Ivanova, N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 2012, 10, 440–454. [Google Scholar] [CrossRef]
- Baharvand, H.; Ashtiani, S.K.; Valojerdi, M.R.; Shahverdi, A.; Taee, A.; Sabour, D. Establishment and in vitro differentiation of a new embryonic stem cell line from human blastocyst. Differentiation 2004, 72, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, E.; Jung, J.; Benvenisty, N. High prevalence of acquired cancer-related mutations in 146 human pluripotent stem cell lines and their differentiated derivatives. Nat. Biotechnol. 2024, 42, 1667–1671. [Google Scholar] [CrossRef]
- Doetschman, T.C.; Eistetter, H.; Katz, M.; Schmidt, W.; Kemler, R. The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium. J. Embryol. Exp. Morphol. 1985, 87, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, T.; Iiboshi, Y.; Oka, M.; Papst, P.J.; Meacham, A.M.; Zon, L.I.; Terada, N. Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett. 2001, 497, 15–19. [Google Scholar] [CrossRef]
- Thoma, E.C.; Maurus, K.; Wagner, T.U.; Schartl, M. Parallel differentiation of embryonic stem cells into different cell types by a single gene-based differentiation system. Cell Reprogram. 2012, 14, 106–111. [Google Scholar] [CrossRef]
- Shiroi, A.; Ueda, S.; Ouji, Y.; Saito, K.; Moriya, K.; Sugie, Y.; Fukui, H.; Ishizaka, S.; Yoshikawa, M. Differentiation of embryonic stem cells into insulin-producing cells promoted by Nkx2.2 gene transfer. World J. Gastroenterol. 2005, 11, 4161–4166. [Google Scholar] [CrossRef]
- Heydarkhan-Hagvall, S.; Gluck, J.M.; Delman, C.; Jung, M.; Ehsani, N.; Full, S.; Shemin, R.J. The effect of vitronectin on the differentiation of embryonic stem cells in a 3D culture system. Biomaterials 2012, 33, 2032–2040. [Google Scholar] [CrossRef]
- Ludwig, T.E.; Andrews, P.W.; Barbaric, I.; Benvenisty, N.; Bhattacharyya, A.; Crook, J.M.; Daheron, L.M.; Draper, J.S.; Healy, L.E.; Huch, M.; et al. ISSCR standards for the use of human stem cells in basic research. Stem Cell Rep. 2023, 18, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Medicine National Academies of Sciences, Engineering; Policy and Global Affairs; Law Committee on Science, Technology; Regulatory Issues Associated with Neural Chimeras and Organoids Committee on Ethical. The National Academies Collection: Reports funded by National Institutes of Health. In The Emerging Field of Human Neural Organoids, Transplants, and Chimeras: Science, Ethics, and Governance; National Academies Press (US): Washington, DC, USA, 2021. [Google Scholar]
- Sohn, Y.D.; Han, J.W.; Yoon, Y.S. Generation of induced pluripotent stem cells from somatic cells. Prog. Mol. Biol. Transl. Sci. 2012, 111, 1–26. [Google Scholar] [CrossRef]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Rais, Y.; Zviran, A.; Geula, S.; Gafni, O.; Chomsky, E.; Viukov, S.; Mansour, A.A.; Caspi, I.; Krupalnik, V.; Zerbib, M.; et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature 2013, 502, 65–70. [Google Scholar] [CrossRef]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Mancarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Edel, M.J.; Menchon, C.; Menendez, S.; Consiglio, A.; Raya, A.; Izpisua Belmonte, J.C. Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes. Dev. 2010, 24, 561–573. [Google Scholar] [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef]
- Temple, S. The development of neural stem cells. Nature 2001, 414, 112–117. [Google Scholar] [CrossRef]
- Merkle, F.T.; Tramontin, A.D.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc. Natl. Acad. Sci. USA 2004, 101, 17528–17532. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cerdeno, V.; Noctor, S.C. Neural Progenitor Cell Terminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef]
- Galiakberova, A.A.; Dashinimaev, E.B. Neural Stem Cells and Methods for Their Generation From Induced Pluripotent Stem Cells in vitro. Front. Cell Dev. Biol. 2020, 8, 815. [Google Scholar] [CrossRef]
- Pașca, S.P.; Arlotta, P.; Bateup, H.S.; Camp, J.G.; Cappello, S.; Gage, F.H.; Knoblich, J.A.; Kriegstein, A.R.; Lancaster, M.A.; Ming, G.L.; et al. A nomenclature consensus for nervous system organoids and assembloids. Nature 2022, 609, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Birtele, M.; Lancaster, M.; Quadrato, G. Modelling human brain development and disease with organoids. Nat. Rev. Mol. Cell Biol. 2025, 26, 389–412. [Google Scholar] [CrossRef] [PubMed]
- Pașca, S.P. The rise of three-dimensional human brain cultures. Nature 2018, 553, 437–445. [Google Scholar] [CrossRef]
- Watanabe, K.; Kamiya, D.; Nishiyama, A.; Katayama, T.; Nozaki, S.; Kawasaki, H.; Watanabe, Y.; Mizuseki, K.; Sasai, Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat. Neurosci. 2005, 8, 288–296. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Brauninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Nowakowski, T.J. Human brain development through the lens of cerebral organoid models. Brain Res. 2019, 1725, 146470. [Google Scholar] [CrossRef] [PubMed]
- Atamian, A.; Birtele, M.; Hosseini, N.; Nguyen, T.; Seth, A.; Del Dosso, A.; Paul, S.; Tedeschi, N.; Taylor, R.; Coba, M.P.; et al. Human cerebellar organoids with functional Purkinje cells. Cell Stem Cell 2024, 31, 39–51.e36. [Google Scholar] [CrossRef] [PubMed]
- Bystron, I.; Blakemore, C.; Rakic, P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008, 9, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Sloan, S.A.; Andersen, J.; Pasca, A.M.; Birey, F.; Pasca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef]
- Revah, O.; Gore, F.; Kelley, K.W.; Andersen, J.; Sakai, N.; Chen, X.; Li, M.Y.; Birey, F.; Yang, X.; Saw, N.L.; et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 2022, 610, 319–326. [Google Scholar] [CrossRef]
- Ruchalski, K.; Hathout, G.M. A medley of midbrain maladies: A brief review of midbrain anatomy and syndromology for radiologists. Radiol. Res. Pract. 2012, 2012, 258524. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G.L. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 2018, 13, 565–580. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. npj Park. Disease 2019, 5, 5. [Google Scholar] [CrossRef]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef]
- Yao, X.; Kang, J.H.; Kim, K.P.; Shin, H.; Jin, Z.L.; Guo, H.; Xu, Y.N.; Li, Y.H.; Hali, S.; Kwon, J.; et al. Production of Highly Uniform Midbrain Organoids from Human Pluripotent Stem Cells. Stem Cells Int. 2023, 2023, 3320211. [Google Scholar] [CrossRef]
- Renner, H.; Grabos, M.; Becker, K.J.; Kagermeier, T.E.; Wu, J.; Otto, M.; Peischard, S.; Zeuschner, D.; TsyTsyura, Y.; Disse, P.; et al. A fully automated high-throughput workflow for 3D-based chemical screening in human midbrain organoids. Elife 2020, 9, e52904. [Google Scholar] [CrossRef]
- Chen, M.; Niclis, J.C.; Denham, M. Protocol for generating reproducible miniaturized controlled midbrain organoids. STAR Protoc. 2023, 4, 102451. [Google Scholar] [CrossRef]
- Pavlinov, I.; Tambe, M.; Abbott, J.; Nguyen, H.N.; Xu, M.; Pradhan, M.; Farkhondeh, A.; Zheng, W. In depth characterization of midbrain organoids derived from wild type iPSC lines. PLoS ONE 2023, 18, e0292926. [Google Scholar] [CrossRef]
- Berger, E.; Magliaro, C.; Paczia, N.; Monzel, A.S.; Antony, P.; Linster, C.L.; Bolognin, S.; Ahluwalia, A.; Schwamborn, J.C. Millifluidic culture improves human midbrain organoid vitality and differentiation. Lab Chip 2018, 18, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Melka, N.; Pszczolinska, A.; Klejbor, I.; Morys, J. The cerebellum: The ‘little’ brain and its big role. Folia Morphol. 2024, 83, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Kloth, A.D.; Badura, A. The cerebellum, sensitive periods, and autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, A.M.; Crocetti, D.; Mostofsky, S.H.; Stoodley, C.J. Cerebellar gray matter and lobular volumes correlate with core autism symptoms. Neuroimage Clin. 2015, 7, 631–639. [Google Scholar] [CrossRef]
- Weinschutz Mendes, H.; Neelakantan, U.; Liu, Y.; Fitzpatrick, S.E.; Chen, T.; Wu, W.; Pruitt, A.; Jin, D.S.; Jamadagni, P.; Carlson, M.; et al. High-throughput functional analysis of autism genes in zebrafish identifies convergence in dopaminergic and neuroimmune pathways. Cell Rep. 2023, 42, 112243. [Google Scholar] [CrossRef]
- Ahmadi, A.; Saadatmand, M.; Wallois, F. Evaluation of potential alterations related to ADHD in the effective connectivity between the default mode network and cerebellum, hippocampus, thalamus, and primary visual cortex. Cereb. Cortex 2024, 34, bhae335. [Google Scholar] [CrossRef]
- Parkkinen, S.; Radua, J.; Andrews, D.S.; Murphy, D.; Dell’Acqua, F.; Parlatini, V. Cerebellar network alterations in adult attention-deficit/hyperactivity disorder. J. Psychiatry Neurosci. 2024, 49, E233–E241. [Google Scholar] [CrossRef]
- Tse, N.Y.; Chen, Y.; Irish, M.; Cordato, N.J.; Landin-Romero, R.; Hodges, J.R.; Piguet, O.; Ahmed, R.M. Cerebellar contributions to cognition in corticobasal syndrome and progressive supranuclear palsy. Brain Commun. 2020, 2, fcaa194. [Google Scholar] [CrossRef]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child. Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef]
- Haldipur, P.; Millen, K.J.; Aldinger, K.A. Human Cerebellar Development and Transcriptomics: Implications for Neurodevelopmental Disorders. Annu. Rev. Neurosci. 2022, 45, 515–531. [Google Scholar] [CrossRef]
- Stoodley, C.J. The Cerebellum and Neurodevelopmental Disorders. Cerebellum 2016, 15, 34–37. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.E.; Gill, J.S.; Sillitoe, R.V. Abnormal Cerebellar Development in Autism Spectrum Disorders. Dev. Neurosci. 2021, 43, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum 2018, 17, 419–427. [Google Scholar] [CrossRef]
- Silva, T.P.; Sousa-Luis, R.; Fernandes, T.G.; Bekman, E.P.; Rodrigues, C.A.V.; Vaz, S.H.; Moreira, L.M.; Hashimura, Y.; Jung, S.; Lee, B.; et al. Transcriptome profiling of human pluripotent stem cell-derived cerebellar organoids reveals faster commitment under dynamic conditions. Biotechnol. Bioeng. 2021, 118, 2781–2803. [Google Scholar] [CrossRef]
- Nayler, S.; Agarwal, D.; Curion, F.; Bowden, R.; Becker, E.B.E. High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep. 2021, 11, 12959. [Google Scholar] [CrossRef]
- Hua, T.T.; Bejoy, J.; Song, L.; Wang, Z.; Zeng, Z.; Zhou, Y.; Li, Y.; Sang, Q.A. Cerebellar Differentiation from Human Stem Cells Through Retinoid, Wnt, and Sonic Hedgehog Pathways. Tissue Eng. Part A 2021, 27, 881–893. [Google Scholar] [CrossRef]
- Chen, Y.; Bury, L.A.; Chen, F.; Aldinger, K.A.; Miranda, H.C.; Wynshaw-Boris, A. Generation of advanced cerebellar organoids for neurogenesis and neuronal network development. Hum. Mol. Genet. 2023, 32, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Atamian, A.; Birtele, M.; Hosseini, N.; Quadrato, G. Generation and long-term culture of human cerebellar organoids from pluripotent stem cells. Nat. Protoc. 2024. [Google Scholar] [CrossRef]
- Makrygianni, E.A.; Chrousos, G.P. From Brain Organoids to Networking Assembloids: Implications for Neuroendocrinology and Stress Medicine. Front. Physiol. 2021, 12, 621970. [Google Scholar] [CrossRef]
- Marton, R.M.; Pasca, S.P. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020, 30, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Kanton, S.; Pasca, S.P. Human assembloids. Development 2022, 149, dev201120. [Google Scholar] [CrossRef] [PubMed]
- Pașca, S.P.; Arlotta, P.; Bateup, H.S.; Camp, J.G.; Cappello, S.; Gage, F.H.; Knoblich, J.A.; Kriegstein, A.R.; Lancaster, M.A.; Ming, G.L.; et al. A framework for neural organoids, assembloids and transplantation studies. Nature 2025, 639, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Cho, H. Human brain organoids in Alzheimer’s disease. Organoid 2021, 1, e5. [Google Scholar] [CrossRef]
- Holloway, P.M.; Willaime-Morawek, S.; Siow, R.; Barber, M.; Owens, R.M.; Sharma, A.D.; Rowan, W.; Hill, E.; Zagnoni, M. Advances in microfluidic in vitro systems for neurological disease modeling. J. Neurosci. Res. 2021, 99, 1276–1307. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Castiglione, H.; Vigneron, P.A.; Baquerre, C.; Yates, F.; Rontard, J.; Honegger, T. Human Brain Organoids-on-Chip: Advances, Challenges, and Perspectives for Preclinical Applications. Pharmaceutics 2022, 14, 2301. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Li, Z.; Wang, H.; Li, N.; Deng, Y. Microfluidic Brain-on-a-Chip: From Key Technology to System Integration and Application. Small 2023, 19, e2304427. [Google Scholar] [CrossRef]
- Soubéran, A.; Jiguet-Jiglaire, C.; Toutain, S.; Morando, P.; Baeza-Kallee, N.; Appay, R.; Boucard, C.; Graillon, T.; Meyer, M.; Farah, K.; et al. Brain tumoroids: Treatment prediction and drug development for brain tumors with fast, reproducible, and easy-to-use personalized models. Neuro Oncol. 2025, 27, 415–429. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, D.; Liu, A.; Wu, K. Tumor organoids: Applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 2022, 15, 58. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Soubéran, A.; Tchoghandjian, A. Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation. Cancers 2020, 12, 2347. [Google Scholar] [CrossRef]
- Poli, D.; Magliaro, C.; Ahluwalia, A. Experimental and Computational Methods for the Study of Cerebral Organoids: A Review. Front. Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef]
- Passaro, A.P.; Stice, S.L. Electrophysiological Analysis of Brain Organoids: Current Approaches and Advancements. Front. Neurosci. 2020, 14, 622137. [Google Scholar] [CrossRef]
- Quadrato, G.; Brown, J.; Arlotta, P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med. 2016, 22, 1220–1228+1078–8956. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Ozaki, Y.; Ashida, T.; Matsubara, T.; Oishi, N.; Kihara, S.; Takahashi, J. Self-Organized Synchronous Calcium Transients in a Cultured Human Neural Network Derived from Cerebral Organoids. Stem Cell Rep. 2019, 13, 458–473. [Google Scholar] [CrossRef]
- Romoser, V.A.; Hinkle, P.M.; Persechini, A. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 1997, 272, 13270–13274. [Google Scholar] [CrossRef]
- Shafer, T.J.; Brown, J.P.; Lynch, B.; Davila-Montero, S.; Wallace, K.; Friedman, K.P. Evaluation of Chemical Effects on Network Formation in Cortical Neurons Grown on Microelectrode Arrays. Toxicol. Sci. 2019, 169, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.L.; Brown, J.P.; Wallace, K.; Mundy, W.R.; Shafer, T.J. From the Cover: Developmental Neurotoxicants Disrupt Activity in Cortical Networks on Microelectrode Arrays: Results of Screening 86 Compounds During Neural Network Formation. Toxicol. Sci. 2017, 160, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, E.; Hall, D.; Wallace, K.; Mundy, W.R.; Eglen, S.J.; Shafer, T.J. Characterization of Early Cortical Neural Network Development in Multiwell Microelectrode Array Plates. J. Biomol. Screen. 2016, 21, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Poli, D.; Wheeler, B.C.; Demarse, T.B.; Brewer, G.J. Pattern separation and completion of distinct axonal inputs transmitted via micro-tunnels between co-cultured hippocampal dentate, CA3, CA1 and entorhinal cortex networks. J. Neural Eng. 2018, 15, 046009. [Google Scholar] [CrossRef]
- Berdondini, L.; Imfeld, K.; Maccione, A.; Tedesco, M.; Neukom, S.; Koudelka-Hep, M.; Martinoia, S. Active pixel sensor array for high spatio-temporal resolution electrophysiological recordings from single cell to large scale neuronal networks. Lab Chip 2009, 9, 2644–2651. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef]
- Soscia, D.A.; Lam, D.; Tooker, A.C.; Enright, H.A.; Triplett, M.; Karande, P.; Peters, S.K.G.; Sales, A.P.; Wheeler, E.K.; Fischer, N.O. A flexible 3-dimensional microelectrode array for in vitro brain models. Lab Chip 2020, 20, 901–911. [Google Scholar] [CrossRef]
- Shin, H.; Jeong, S.; Lee, J.H.; Sun, W.; Choi, N.; Cho, I.J. 3D high-density microelectrode array with optical stimulation and drug delivery for investigating neural circuit dynamics. Nat. Commun. 2021, 12, 492. [Google Scholar] [CrossRef]
- Shiri, Z.; Simorgh, S.; Naderi, S.; Baharvand, H. Optogenetics in the Era of Cerebral Organoids. Trends Biotechnol. 2019, 37, 1282–1294. [Google Scholar] [CrossRef]
- Hochbaum, D.R.; Zhao, Y.; Farhi, S.L.; Klapoetke, N.; Werley, C.A.; Kapoor, V.; Zou, P.; Kralj, J.M.; Maclaurin, D.; Smedemark-Margulies, N.; et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 2014, 11, 825–833. [Google Scholar] [CrossRef]
- Werley, C.A.; Brookings, T.; Upadhyay, H.; Williams, L.A.; McManus, O.B.; Dempsey, G.T. All-Optical Electrophysiology for Disease Modeling and Pharmacological Characterization of Neurons. Curr. Protoc. Pharmacol. 2017, 11, 825–833. [Google Scholar] [CrossRef]
- Kiskinis, E.; Kralj, J.M.; Zou, P.; Weinstein, E.N.; Zhang, H.; Tsioras, K.; Wiskow, O.; Ortega, J.A.; Eggan, K.; Cohen, A.E. All-Optical Electrophysiology for High-Throughput Functional Characterization of a Human iPSC-Derived Motor Neuron Model of ALS. Stem Cell Rep. 2018, 10, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Versace, A.; Hitchens, T.K.; Wallace, C.T.; Watkins, S.C.; D’Aiuto, L. 11.7T Diffusion Magnetic Resonance Imaging and Tractography to Probe Human Brain Organoid Microstructure. Biol. Psychiatry Glob. Open Sci. 2024, 4, 100344. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.A.; Muotri, A.R. Brain Organoids and the Study of Neurodevelopment. Trends Mol. Med. 2018, 24, 982–990. [Google Scholar] [CrossRef]
- Gordon, A.; Geschwind, D.H. Human in vitro models for understanding mechanisms of autism spectrum disorder. Mol. Autism 2020, 11, 26. [Google Scholar] [CrossRef]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef]
- Gusyatiner, O.; Hegi, M.E. Glioma epigenetics: From subclassification to novel treatment options. Semin. Cancer Biol. 2018, 51, 50–58. [Google Scholar] [CrossRef]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Bergthold, G.; London, W.B.; Goumnerova, L.C.; Morales La Madrid, A.; Marcus, K.J.; Guo, D.; Ullrich, N.J.; Robison, N.J.; Chi, S.N.; et al. Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: An analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr. Blood Cancer 2014, 61, 1173–1179. [Google Scholar] [CrossRef]
- Qaddoumi, I.; Sultan, I.; Gajjar, A. Outcome and prognostic features in pediatric gliomas: A review of 6212 cases from the Surveillance, Epidemiology, and End Results database. Cancer 2009, 115, 5761–5770. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed]
- Rybin, M.J.; Ivan, M.E.; Ayad, N.G.; Zeier, Z. Organoid Models of Glioblastoma and Their Role in Drug Discovery. Front. Cell Neurosci. 2021, 15, 605255. [Google Scholar] [CrossRef]
- Ishahak, M.; Han, R.H.; Annamalai, D.; Woodiwiss, T.; McCornack, C.; Cleary, R.T.; DeSouza, P.A.; Qu, X.; Dahiya, S.; Kim, A.H.; et al. Genetically Engineered Brain Organoids Recapitulate Spatial and Developmental States of Glioblastoma Progression. Adv. Sci. 2025, 12, e2410110. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, J.; Wang, S.; Guo, P.; Liao, K.; Shi, Z.; Zhao, J.; Lin, S.; Yang, M.; Cai, G.; et al. Generation and banking of patient-derived glioblastoma organoid and its application in cancer neuroscience. Am. J. Cancer Res. 2024, 14, 5000–5010. [Google Scholar] [CrossRef]
- Sarnow, K.; Majercak, E.; Qurbonov, Q.; Cruzeiro, G.A.V.; Jeong, D.; Haque, I.A.; Khalil, A.; Baird, L.C.; Filbin, M.G.; Tang, X. Neuroimmune-competent human brain organoid model of diffuse midline glioma. Neuro Oncol. 2025, 27, 369–382. [Google Scholar] [CrossRef] [PubMed]
- van Essen, M.J.; Apsley, E.J.; Riepsaame, J.; Xu, R.; Northcott, P.A.; Cowley, S.A.; Jacob, J.; Becker, E.B.E. PTCH1-mutant human cerebellar organoids exhibit altered neural development and recapitulate early medulloblastoma tumorigenesis. Dis. Model. Mech. 2024, 17, dmm050323. [Google Scholar] [CrossRef]
- van Essen, M.J.; Nicheperovich, A.; Schuster-Bockler, B.; Becker, E.B.E.; Jacob, J. Sonic hedgehog medulloblastoma cells in co-culture with cerebellar organoids converge towards in vivo malignant cell states. Neurooncol. Adv. 2025, 7, vdae218. [Google Scholar] [CrossRef]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 583. [Google Scholar] [CrossRef]
- Han, X.; He, Y.; Wang, Y.; Hu, W.; Chu, C.; Huang, L.; Hong, Y.; Han, L.; Zhang, X.; Gao, Y.; et al. Deficiency of FABP7 Triggers Premature Neural Differentiation in Idiopathic Normocephalic Autism Organoids. Adv. Sci. 2025, 12, e2406849. [Google Scholar] [CrossRef]
- Chalkiadaki, K.; Statoulla, E.; Zafeiri, M.; Voudouri, G.; Amvrosiadis, T.; Typou, A.; Theodoridou, N.; Moschovas, D.; Avgeropoulos, A.; Samiotaki, M.; et al. GABA/Glutamate Neuron Differentiation Imbalance and Increased AKT/mTOR Signaling in CNTNAP2(-/-) Cerebral Organoids. Biol. Psychiatry Glob. Open Sci. 2025, 5, 100413. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Saito, Y.; Takasaki, A.; Nakano, K.; Yamamoto, S.; Suzuki, C.; Kawamura, N.; Hattori, A.; Oikawa, M.; Nagashima, S.; et al. Role of immature choroid plexus in the pathology of model mice and human iPSC-derived organoids with autism spectrum disorder. Cell Rep. 2025, 44, 115133. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.X.; Mao, Y.; Han, X.; Qian, H.Y.; Chu, K.K. EGR1 Regulates SHANK3 Transcription at Different Stages of Brain Development. Neuroscience 2024, 540, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, J.; Chen, X.; Wettschurack, K.; Que, Z.; Deming, B.A.; Olivero-Acosta, M.I.; Cui, N.; Eaton, M.; Zhao, Y.; et al. Microglial over-pruning of synapses during development in autism-associated SCN2A-deficient mice and human cerebral organoids. Mol. Psychiatry 2024, 29, 2424–2437. [Google Scholar] [CrossRef]
- Kang, S.C.; Sarn, N.B.; Venegas, J.; Tan, Z.; Hitomi, M.; Eng, C. Germline PTEN genotype-dependent phenotypic divergence during the early neural developmental process of forebrain organoids. Mol. Psychiatry 2024, 29, 1767–1781. [Google Scholar] [CrossRef]
- Villa, C.E.; Cheroni, C.; Dotter, C.P.; Lopez-Tobon, A.; Oliveira, B.; Sacco, R.; Yahya, A.C.; Morandell, J.; Gabriele, M.; Tavakoli, M.R.; et al. CHD8 haploinsufficiency links autism to transient alterations in excitatory and inhibitory trajectories. Cell Rep. 2022, 39, 110615. [Google Scholar] [CrossRef]
- Jourdon, A.; Wu, F.; Mariani, J.; Capauto, D.; Norton, S.; Tomasini, L.; Amiri, A.; Suvakov, M.; Schreiner, J.D.; Jang, Y.; et al. Modeling idiopathic autism in forebrain organoids reveals an imbalance of excitatory cortical neuron subtypes during early neurogenesis. Nat. Neurosci. 2023, 26, 1505–1515. [Google Scholar] [CrossRef]
- Birtele, M.; Del Dosso, A.; Xu, T.; Nguyen, T.; Wilkinson, B.; Hosseini, N.; Nguyen, S.; Urenda, J.P.; Knight, G.; Rojas, C.; et al. Non-synaptic function of the autism spectrum disorder-associated gene SYNGAP1 in cortical neurogenesis. Nat. Neurosci. 2023, 26, 2090–2103. [Google Scholar] [CrossRef]
- Lu, R.; Xu, Y.; Li, H.; Xiong, M.; Zhou, W.; Feng, W.; Zhao, R. Identifying the Pathogenicity of a Novel NPRL3 Missense Mutation Using Personalized Cortical Organoid Model of Focal Cortical Dysplasia. J. Mol. Neurosci. 2024, 75, 3. [Google Scholar] [CrossRef]
- Niu, W.; Deng, L.; Mojica-Perez, S.P.; Tidball, A.M.; Sudyk, R.; Stokes, K.; Parent, J.M. Abnormal cell sorting and altered early neurogenesis in a human cortical organoid model of Protocadherin-19 clustering epilepsy. Front. Cell Neurosci. 2024, 18, 1339345. [Google Scholar] [CrossRef]
- Muller, Y.; Lengacher, L.; Friscourt, F.; Quairiaux, C.; Stoppini, L.; Magistretti, P.J.; Lengacher, S.; Finsterwald, C. Epileptiform activity in brain organoids derived from patient with Glucose Transporter 1 Deficiency Syndrome. Front. Neurosci. 2024, 18, 1498801. [Google Scholar] [CrossRef] [PubMed]
- Eichmuller, O.L.; Corsini, N.S.; Vertesy, A.; Morassut, I.; Scholl, T.; Gruber, V.E.; Peer, A.M.; Chu, J.; Novatchkova, M.; Hainfellner, J.A.; et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 2022, 375, eabf5546. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Guo, R.; Ye, X.; Tang, S.; Chen, M.; Zhou, P.; Yang, M.; Liao, C.; Li, H.; Lin, B.; et al. Wybutosine hypomodification of tRNAphe activates HERVK and impairs neuronal differentiation. iScience 2024, 27, 109748. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, E.M.; Means, R.E.; Michaud, M.; Madri, J.A.; Katz, S.G. Minocycline mitigates the effect of neonatal hypoxic insult on human brain organoids. Cell Death Dis. 2019, 10, 325. [Google Scholar] [CrossRef]
- Zhang, D.; Eguchi, N.; Okazaki, S.; Sora, I.; Hishimoto, A. Telencephalon Organoids Derived from an Individual with ADHD Show Altered Neurodevelopment of Early Cortical Layer Structure. Stem Cell Rev. Rep. 2023, 19, 1482–1491. [Google Scholar] [CrossRef]
- Miura, Y.; Li, M.Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef]
- Wu, S.; Hong, Y.; Chu, C.; Gan, Y.; Li, X.; Tao, M.; Wang, D.; Hu, H.; Zheng, Z.; Zhu, Q.; et al. Construction of human 3D striato-nigral assembloids to recapitulate medium spiny neuronal projection defects in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2024, 121, e2316176121. [Google Scholar] [CrossRef]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef]
- Meng, X.; Yao, D.; Imaizumi, K.; Chen, X.; Kelley, K.W.; Reis, N.; Thete, M.V.; Arjun McKinney, A.; Kulkarni, S.; Panagiotakos, G.; et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature 2023, 622, 359–366. [Google Scholar] [CrossRef]
- Kalpana, K.; Rao, C.; Semrau, S.; Zhang, B.; Noggle, S.; Fossati, V. Generating Neuroimmune Assembloids Using Human Induced Pluripotent Stem Cell (iPSC)-Derived Cortical Organoids and Microglia. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2024. [Google Scholar] [CrossRef]
- Barmpa, K.; Saraiva, C.; Lopez-Pigozzi, D.; Gomez-Giro, G.; Gabassi, E.; Spitz, S.; Brandauer, K.; Rodriguez Gatica, J.E.; Antony, P.; Robertson, G.; et al. Modeling early phenotypes of Parkinson’s disease by age-induced midbrain-striatum assembloids. Commun. Biol. 2024, 7, 1561. [Google Scholar] [CrossRef]
- Patton, M.H.; Thomas, K.T.; Bayazitov, I.T.; Newman, K.D.; Kurtz, N.B.; Robinson, C.G.; Ramirez, C.A.; Trevisan, A.J.; Bikoff, J.B.; Peters, S.T.; et al. Synaptic plasticity in human thalamocortical assembloids. Cell Rep. 2024, 43, 114503. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Miura, Y.; Li, M.Y.; Revah, O.; Selvaraj, S.; Birey, F.; Meng, X.; Thete, M.V.; Pavlov, S.D.; Andersen, J.; et al. Human assembloids reveal the consequences of CACNA1G gene variants in the thalamocortical pathway. Neuron 2024, 112, 4048–4059.e4047. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Park, K.H.; Kim, D.H.; Kim, N.G.; Lee, S.E.; Shin, N.; Kook, M.G.; Kim, Y.B.; Kang, K.S. Cortical-blood vessel assembloids exhibit Alzheimer’s disease phenotypes by activating glia after SARS-CoV-2 infection. Cell Death Discov. 2023, 9, 32. [Google Scholar] [CrossRef]
- Kim, J.I.; Imaizumi, K.; Jurjut, O.; Kelley, K.W.; Wang, D.; Thete, M.V.; Hudacova, Z.; Amin, N.D.; Levy, R.J.; Scherrer, G.; et al. Human assembloid model of the ascending neural sensory pathway. Nature 2025. [Google Scholar] [CrossRef]
- Correia, C.D.; Calado, S.M.; Matos, A.; Esteves, F.; De Sousa-Coelho, A.L.; Campinho, M.A.; Fernandes, M.T. Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models. Cells 2025, 14, 292. [Google Scholar] [CrossRef] [PubMed]
- Khamis, Z.I.; Sarker, D.B.; Xue, Y.; Al-Akkary, N.; James, V.D.; Zeng, C.; Li, Y.; Sang, Q.A. Modeling Human Brain Tumors and the Microenvironment Using Induced Pluripotent Stem Cells. Cancers 2023, 15, 1253. [Google Scholar] [CrossRef]
- Deligne, C.; Tourbez, A.; Bénard, F.; Meyer, S.; Curt, A.; Gianesello, M.; Hamadou, M.; Clavier, L.; Coquet, C.; Bocquet, C.; et al. Establishing a living biobank of pediatric high-grade glioma and ependymoma suitable for cancer pharmacology. Neuro Oncol. 2025. [Google Scholar] [CrossRef]
- Rutkowski, S.; von Hoff, K.; Emser, A.; Zwiener, I.; Pietsch, T.; Figarella-Branger, D.; Giangaspero, F.; Ellison, D.W.; Garre, M.L.; Biassoni, V.; et al. Survival and prognostic factors of early childhood medulloblastoma: An international meta-analysis. J. Clin. Oncol. 2010, 28, 4961–4968. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef]
- Eyring, K.W.; Geschwind, D.H. Three decades of ASD genetics: Building a foundation for neurobiological understanding and treatment. Hum. Mol. Genet. 2021, 30, R236–R244. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Lamanna, J.; Meldolesi, J. Autism Spectrum Disorder: Brain Areas Involved, Neurobiological Mechanisms, Diagnoses and Therapies. Int. J. Mol. Sci. 2024, 25, 2423. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.R.; Szoko, N.; Barnard, J.; Rubin, R.A.; Schlatzer, D.; Lundberg, K.; Li, X.; Natowicz, M.R. Proteomic Investigations of Autism Brain Identify Known and Novel Pathogenetic Processes. Sci. Rep. 2019, 9, 13118. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Le, T.L.; Malesys, S.; Cliquet, F.; Tabet, A.C.; Delorme, R.; Rolland, T.; Bourgeron, T. Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol. Cell Neurosci. 2021, 113, 103623. [Google Scholar] [CrossRef]
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Puffenberger, E.G.; Huentelman, M.J.; Gottlieb, S.; Dobrin, S.E.; Parod, J.M.; Stephan, D.A.; Morton, D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006, 354, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Shimogori, T.; Ohtsuka, T.; Kageyama, R. Hes genes and neurogenin regulate non-neural versus neural fate specification in the dorsal telencephalic midline. Development 2008, 135, 2531–2541. [Google Scholar] [CrossRef]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Moller, R.S.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.A.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef]
- Santos, J.L.S.; Araújo, C.A.; Rocha, C.A.G.; Costa-Ferro, Z.S.M.; Souza, B.S.F. Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids. Biomolecules 2023, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes 2023, 14, 677. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Chan, Y.; Dawes, P.; Guo, X.; Erdin, S.; Tai, D.J.C.; Liu, S.; Reichert, J.M.; Burns, M.J.; Chan, Y.K.; et al. Orgo-Seq integrates single-cell and bulk transcriptomic data to identify cell type specific-driver genes associated with autism spectrum disorder. Nat. Commun. 2022, 13, 3243. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhang, W.; Wang, X.; Jiao, C.; Xu, S.; Liu, C.; Tang, B.; Chen, C. Human forebrain organoids reveal connections between valproic acid exposure and autism risk. Transl. Psychiatry 2022, 12, 130. [Google Scholar] [CrossRef]
- Modafferi, S.; Zhong, X.; Kleensang, A.; Murata, Y.; Fagiani, F.; Pamies, D.; Hogberg, H.T.; Calabrese, V.; Lachman, H.; Hartung, T.; et al. Gene-Environment Interactions in Developmental Neurotoxicity: A Case Study of Synergy between Chlorpyrifos and CHD8 Knockout in Human BrainSpheres. Environ. Health Perspect. 2021, 129, 77001. [Google Scholar] [CrossRef]
- Pearson, G.; Song, C.; Hohmann, S.; Prokhorova, T.; Sheldrick-Michel, T.M.; Knöpfel, T. DNA Methylation Profiles of GAD1 in Human Cerebral Organoids of Autism Indicate Disrupted Epigenetic Regulation during Early Development. Int. J. Mol. Sci. 2022, 23, 9188. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP2-KO neurons and cortical organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Urresti, J.; Zhang, P.; Moran-Losada, P.; Yu, N.K.; Negraes, P.D.; Trujillo, C.A.; Antaki, D.; Amar, M.; Chau, K.; Pramod, A.B.; et al. Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 2021, 26, 7560–7580. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chiola, S.; Yang, G.; Russell, C.; Armstrong, C.J.; Wu, Y.; Spampanato, J.; Tarboton, P.; Ullah, H.M.A.; Edgar, N.U.; et al. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun. 2022, 13, 5688. [Google Scholar] [CrossRef] [PubMed]
- Wegscheid, M.L.; Anastasaki, C.; Hartigan, K.A.; Cobb, O.M.; Papke, J.B.; Traber, J.N.; Morris, S.M.; Gutmann, D.H. Patient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis. Cell Rep. 2021, 36, 109315. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.F. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef]
- Murray, C.J.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef]
- Nolan, D.; Fink, J. Genetics of epilepsy. Handb. Clin. Neurol. 2018, 148, 467–491. [Google Scholar] [CrossRef]
- Brodie, M.J.; Barry, S.J.; Bamagous, G.A.; Norrie, J.D.; Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012, 78, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.L. Effect of Seizures on the Developing Brain and Cognition. Semin. Pediatr. Neurol. 2016, 23, 120–126. [Google Scholar] [CrossRef]
- Miller, D.J.; Bhaduri, A.; Sestan, N.; Kriegstein, A. Shared and derived features of cellular diversity in the human cerebral cortex. Curr. Opin. Neurobiol. 2019, 56, 117–124. [Google Scholar] [CrossRef]
- Zeng, H.; Shen, E.H.; Hohmann, J.G.; Oh, S.W.; Bernard, A.; Royall, J.J.; Glattfelder, K.J.; Sunkin, S.M.; Morris, J.A.; Guillozet-Bongaarts, A.L.; et al. Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell 2012, 149, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef]
- Semah, F.; Picot, M.C.; Adam, C.; Broglin, D.; Arzimanoglou, A.; Bazin, B.; Cavalcanti, D.; Baulac, M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Gopalappa, R.; Kim, S.H.; Ramakrishna, S.; Lee, M.; Kim, W.I.; Kim, J.; Park, S.M.; Lee, J.; Oh, J.H.; et al. Somatic Mutations in TSC1 and TSC2 Cause Focal Cortical Dysplasia. Am. J. Hum. Genet. 2017, 100, 454–472. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Saitsu, H.; Takei, N.; Tohyama, J.; Kato, M.; Kitaura, H.; Shiina, M.; Shirozu, H.; Masuda, H.; Watanabe, K.; et al. Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann. Neurol. 2015, 78, 375–386. [Google Scholar] [CrossRef]
- Ribierre, T.; Deleuze, C.; Bacq, A.; Baldassari, S.; Marsan, E.; Chipaux, M.; Muraca, G.; Roussel, D.; Navarro, V.; Leguern, E.; et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Investig. 2018, 128, 2452–2458. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Tarpey, P.S.; Hynes, K.; Bayly, M.A.; Scheffer, I.E.; Smith, R.; Bomar, J.; Sutton, E.; Vandeleur, L.; Shoubridge, C.; et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 2008, 40, 776–781. [Google Scholar] [CrossRef]
- Leen, W.G.; Klepper, J.; Verbeek, M.M.; Leferink, M.; Hofste, T.; van Engelen, B.G.; Wevers, R.A.; Arthur, T.; Bahi-Buisson, N.; Ballhausen, D.; et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar] [CrossRef]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 342. [Google Scholar] [CrossRef]
- Thiele, E.A. Managing and understanding epilepsy in tuberous sclerosis complex. Epilepsia 2010, 51 (Suppl. S1), 90–91. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yao, H.; Negraes, P.D.; Wang, J.; Trujillo, C.A.; de Souza, J.S.; Muotri, A.R.; Haddad, G.G. Neuronal hyperexcitability and ion channel dysfunction in CDKL5-deficiency patient iPSC-derived cortical organoids. Neurobiol. Dis. 2022, 174, 105882. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’Agostino, G.A.; Tran, H.D.; Itahana, Y.; et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366, 1486–1492. [Google Scholar] [CrossRef]
- Repudi, S.; Steinberg, D.J.; Elazar, N.; Breton, V.L.; Aquilino, M.S.; Saleem, A.; Abu-Swai, S.; Vainshtein, A.; Eshed-Eisenbach, Y.; Vijayaragavan, B.; et al. Neuronal deletion of Wwox, associated with WOREE syndrome, causes epilepsy and myelin defects. Brain 2021, 144, 3061–3077. [Google Scholar] [CrossRef] [PubMed]
- Schiariti, V.; Shierk, A.; Stashinko, E.E.; Sukal-Moulton, T.; Feldman, R.S.; Aman, C.; Mendoza-Puccini, M.C.; Brandenburg, J.E.; National Institute of Neurological Disorders; Stroke Cerebral Palsy Common Data Elements Oversight Committee. Cerebral palsy pain instruments: Recommended tools for clinical research studies by the National Institute of Neurological Disorders and Stroke Cerebral Palsy Common Data Elements project. Dev. Med. Child. Neurol. 2024, 66, 610–622. [Google Scholar] [CrossRef]
- Vitrikas, K.; Dalton, H.; Breish, D. Cerebral Palsy: An Overview. Am. Fam. Physician 2020, 101, 213–220. [Google Scholar]
- Sadowska, M.; Sarecka-Hujar, B.; Kopyta, I. Cerebral Palsy: Current Opinions on Definition, Epidemiology, Risk Factors, Classification and Treatment Options. Neuropsychiatr. Dis. Treat. 2020, 16, 1505–1518. [Google Scholar] [CrossRef]
- Li, N.; Zhou, P.; Tang, H.; He, L.; Fang, X.; Zhao, J.; Wang, X.; Qi, Y.; Sun, C.; Lin, Y.; et al. In-depth analysis reveals complex molecular aetiology in a cohort of idiopathic cerebral palsy. Brain 2022, 145, 119–141. [Google Scholar] [CrossRef]
- Harnett, D.; Ambrozkiewicz, M.C.; Zinnall, U.; Rusanova, A.; Borisova, E.; Drescher, A.N.; Couce-Iglesias, M.; Villamil, G.; Dannenberg, R.; Imami, K.; et al. A critical period of translational control during brain development at codon resolution. Nat. Struct. Mol. Biol. 2022, 29, 1277–1290. [Google Scholar] [CrossRef]
- Curristin, S.M.; Cao, A.; Stewart, W.B.; Zhang, H.; Madri, J.A.; Morrow, J.S.; Ment, L.R. Disrupted synaptic development in the hypoxic newborn brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15729–15734. [Google Scholar] [CrossRef]
- Ment, L.R.; Vohr, B.; Allan, W.; Katz, K.H.; Schneider, K.C.; Westerveld, M.; Duncan, C.C.; Makuch, R.W. Change in cognitive function over time in very low-birth-weight infants. JAMA 2003, 289, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, B.; Yang, M.; Guo, R.; Yuan, S.; Wang, J.; Hu, H. Generation of GPAM knockout human embryonic stem cell line SYSUe-008-A using CRISPR/Cas9. Stem Cell Res. 2021, 53, 102303. [Google Scholar] [CrossRef] [PubMed]
- Agnew-Blais, J.C.; Polanczyk, G.V.; Danese, A.; Wertz, J.; Moffitt, T.E.; Arseneault, L. Evaluation of the Persistence, Remission, and Emergence of Attention-Deficit/Hyperactivity Disorder in Young Adulthood. JAMA Psychiatry 2016, 73, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, S.P. Attention Deficit Hyperactivity Disorder (ADHD): Controversy, Developmental Mechanisms, and Multiple Levels of Analysis. Annu. Rev. Clin. Psychol. 2018, 14, 291–316. [Google Scholar] [CrossRef]
- Thapar, A. Discoveries on the Genetics of ADHD in the 21st Century: New Findings and Their Implications. Am. J. Psychiatry 2018, 175, 943–950. [Google Scholar] [CrossRef]
- Lee, P.H.; Anttila, V.; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.G.; Grotzinger, A.D.; Posthuma, D.; et al. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179, 1469–1482.e11. [Google Scholar] [CrossRef]
- Shepherd, G.M. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013, 14, 278–291. [Google Scholar] [CrossRef]
- Milad, M.R.; Rauch, S.L. Obsessive-compulsive disorder: Beyond segregated cortico-striatal pathways. Trends Cogn. Sci. 2012, 16, 43–51. [Google Scholar] [CrossRef]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Welch, J.M.; Lu, J.; Rodriguiz, R.M.; Trotta, N.C.; Peca, J.; Ding, J.D.; Feliciano, C.; Chen, M.; Adams, J.P.; Luo, J.; et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 2007, 448, 894–900. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Li, M.Y.; Gordon, A.; Thete, M.V.; Valencia, A.M.; Revah, O.; Pasca, A.M.; Geschwind, D.H.; Pasca, S.P. Dissecting the molecular basis of human interneuron migration in forebrain assembloids from Timothy syndrome. Cell Stem Cell 2022, 29, 248–264.e247. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.; Coelho, L.; Mo, G.; Adang, L.A.; Patne, M.; Chen, Z.; Garcia-Bassets, I.; Mesci, P.; Muotri, A.R. TREX1 is required for microglial cholesterol homeostasis and oligodendrocyte terminal differentiation in human neural assembloids. Mol. Psychiatry 2024, 29, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, S.; Cho, B.; An, S.; Kim, Y.; Kim, J. Parkinson’s Disease Modeling Using Directly Converted 3D Induced Dopaminergic Neuron Organoids and Assembloids. Adv. Sci. 2025, 12, e2412548. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Jandrey, E.H.F.; Granha, I.; Endo, A.K.; Ferreira, R.O.; Araujo, B.H.S.; Zatz, M.; Okamoto, O.K. Differential Replication and Oncolytic Effects of Zika Virus in Aggressive CNS Tumor Cells: Insights from Organoid and Tumoroid Models. Viruses 2024, 16, 1764. [Google Scholar] [CrossRef]
- Costamagna, G.; Comi, G.P.; Corti, S. Advancing Drug Discovery for Neurological Disorders Using iPSC-Derived Neural Organoids. Int. J. Mol. Sci. 2021, 22, 2659. [Google Scholar] [CrossRef]
- Li, C.; Fleck, J.S.; Martins-Costa, C.; Burkard, T.R.; Themann, J.; Stuempflen, M.; Peer, A.M.; Vertesy, A.; Littleboy, J.B.; Esk, C.; et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature 2023, 621, 373–380. [Google Scholar] [CrossRef]
- Park, J.C.; Jang, S.Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef]
- Langhans, S.A. Using 3D in vitro cell culture models in anti-cancer drug discovery. Expert. Opin. Drug Discov. 2021, 16, 841–850. [Google Scholar] [CrossRef]
- Durens, M.; Nestor, J.; Williams, M.; Herold, K.; Niescier, R.F.; Lunden, J.W.; Phillips, A.W.; Lin, Y.C.; Dykxhoorn, D.M.; Nestor, M.W. High-throughput screening of human induced pluripotent stem cell-derived brain organoids. J. Neurosci. Methods 2020, 335, 108627. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Shrestha, S.; Joshi, P.; Choi, N.Y.; Lekkala, V.K.R.; Kang, S.Y.; Ni, G.; Lee, M.Y. Dynamic culture of cerebral organoids using a pillar/perfusion plate for the assessment of developmental neurotoxicity. Biofabrication 2024, 17, 015001. [Google Scholar] [CrossRef]
- Acharya, P.; Joshi, P.; Shrestha, S.; Choi, N.Y.; Jeong, S.; Lee, M.Y. Uniform cerebral organoid culture on a pillar plate by simple and reproducible spheroid transfer from an ultralow attachment well plate. Biofabrication 2024, 16, 025005. [Google Scholar] [CrossRef]
- Ramani, A.; Pasquini, G.; Gerkau, N.J.; Jadhav, V.; Vinchure, O.S.; Altinisik, N.; Windoffer, H.; Muller, S.; Rothenaigner, I.; Lin, S.; et al. Reliability of high-quantity human brain organoids for modeling microcephaly, glioma invasion and drug screening. Nat. Commun. 2024, 15, 10703. [Google Scholar] [CrossRef] [PubMed]
- Abedellatif, S.E.; Hosni, R.; Waha, A.; Gielen, G.H.; Banat, M.; Hamed, M.; Guresir, E.; Frohlich, A.; Sirokay, J.; Wulf, A.L.; et al. Melanoma Brain Metastases Patient-Derived Organoids: An In Vitro Platform for Drug Screening. Pharmaceutics 2024, 16, 1042. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A. Chimeric brain organoids capture human genetic diversity. Nature 2024, 631, 32–33. [Google Scholar] [CrossRef]
- Shalita, R.; Amit, I. The industrial genomic revolution: A new era in neuroimmunology. Neuron 2022, 110, 3429–3443. [Google Scholar] [CrossRef]
- Simões-Abade, M.B.C.; Patterer, M.; Nicaise, A.M.; Pluchino, S. Brain organoid methodologies to explore mechanisms of disease in progressive multiple sclerosis. Front. Cell Neurosci. 2024, 18, 1488691. [Google Scholar] [CrossRef]
- Li, K.; Gu, L.; Cai, H.; Lu, H.C.; Mackie, K.; Guo, F. Human brain organoids for understanding substance use disorders. Drug Metab. Pharmacokinet. 2025, 60, 101036. [Google Scholar] [CrossRef]
- Villanueva, R. Advances in the knowledge and therapeutics of schizophrenia, major depression disorder, and bipolar disorder from human brain organoid research. Front. Psychiatry 2023, 14, 1178494. [Google Scholar] [CrossRef]
- Cheng, K.; Kshirsagar, A.; Nixon, J.; Lau, J.; Yang, K.; Sawa, A.; Kathuria, A. Model systems for emulating human tissue and physiology in psychiatric research. Front. Neurosci. 2025, 19, 1527826. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, Á.; Silva, T.P.; Ledesma, M.D. Models to study basic and applied aspects of lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 190, 114532. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Organoid Type | Disease Modeled | Aim/Methods | Main Findings | Ref. |
|---|---|---|---|---|
| GBM brain organoid | Glioblastoma | HiPSCs were mutated with genes from the proneural and mesenchymal subtypes. | Mutations linked to GBM disrupted neural development within brain organoids. | [135] |
| GBM brain organoid | Glioblastoma | Patient-derived cells. | Organoids maintained characteristics of parental GBM. | [136] |
| GBM brain organoid | Diffuse midline glioma (DMG) | Produced a neuroimmune-competent brain organoid by fusing microglia organoids with DMG organoids. | Replicated DMG infiltration pattern, with microglia mobility and interaction with tumor cells influenced by external factors and microenvironment. | [137] |
| Cerebellar | Medulloblastoma (MB) | Produced an MB organoid by the CRISPR/Cas9 gene editing of the PTCH1 gene. | Loss of PTCH1 was associated with disrupted neuronal growth and MB. | [138] |
| Cerebellar | Medulloblastoma (MB) | Generated an SHH (Sonic Hedgehog)-MB cell line organoid model by co-culturing SHH-MB lines with nonmalignant cerebellar organoids to replicate the tumor microenvironment (TME). | SHH-MB cells in cerebellar organoid co-culture mimicked in vivo malignant states, with organoid microenvironment regulating SHH-MB cell states. | [139] |
| Cerebellar | Medulloblastoma (MB) | Modeled Group 3 medulloblastoma by overexpressing Otx2 and c-Myc genes. | Organoids exhibited DNA methylation like Group 3 MB, and treatment with Tazemetostat diminished Otx2/c-MYC tumorigenesis. | [140] |
| Cerebral | Autism Spectrum Disorder (ASD) | Identified the FABP7/MEK axis as a potential drug target in idiopathic ASD using ASD patient-derived iPSCs. | ASD organoids showed premature differentiation with impaired expression of FABP7. | [141] |
| Cerebral | ASD | Studied the effects of the elimination of the CNTNAP2 gene in ASD, using organoids generated from CRISPR-edited iPSCs. | CNTNAP2 plays vital role in development of brain during early stages and may be linked to mechanisms underlying ASD. | [142] |
| Cerebral | ASD | Investigated the impact of an underdeveloped choroid plexus on the onset of ASD, using ASD patient-derived iPSCs. | Undeveloped choroid plexus is prevalent characteristic in ASD pathology. | [143] |
| Cerebral | ASD | Investigated the relationship between EGR1 and SHANK3, using cerebral organoids at different developmental stages. | EGR1 expression influenced SHANK3 transcription. | [144] |
| Cerebral | ASD | Explored the role of microglial phagocytosis in synapses, using a microglia-SCN2A-mutated cerebral organoid. | Role of microglia in eliminating surplus synapses is evolutionarily conserved. | [145] |
| Forebrain | ASD | Analyzed alterations in the PTEN gene, using CRISPR-edited iPSCs. | PTEN regulated several crucial neurodevelopmental processes in early and late stages of development. | [146] |
| Cortical | ASD | Generated hESCs with CHD8 haploinsufficiency and CHD8 mutations and used these cells to recapitulate a macrocephaly-like phenotype in cerebral organoids. | CHD8 mutations affected proliferation and differentiation dynamics with accelerated and delayed generation of inhibitory and excitatory neurons, respectively. CHD8 haploinsufficiency also disrupted neurodevelopmental trajectories. | [147] |
| Cortical | ASD | Explored the genetic etiology of ASD, using organoids derived from sons with idiopathic ASD and unaffected fathers. | Macrocephalic and normocephalic probands showed different expression of transcription factors during early cortical development. | [148] |
| Cortical | ASD | Analyzed the alteration of SYNGAP1, using patient-derived organoids. | SYNGAP1 haploinsufficiency led to abnormal cytoskeleton dynamics, disrupting and dividing orientation of radial cells. | [149] |
| Cortical | Epilepsy (Focal cortical dysplasia II) | Investigated the role of GATOR1 subunit NPRL3, using iPSCs derived from an FCD II patient. | Produced novel organoid model to investigate NPRL3-related epilepsy. Findings aligned with hyperactivation of mTOR pathway observed in FCD. | [150] |
| Cortical | Epilepsy (PCDH19-clustering epilepsy) | Studied the role of PCDH19, using organoids produced from CRISPR-edited female iPSCs. | PCDH19 is crucial for organizing radial glial cells and early cortical development in human ventricular zone. | [151] |
| Cerebral | Epilepsy (GLUT1-Deficiency syndrome; GLUT1-DS) | Investigated the role of the GLUT1 gene, using patient-derived organoids. | GLUT1-DS organoids showed lower cell density and smaller dimensions, exhibiting epileptiform activity and increased glucose sensitivity deprivation. | [152] |
| Cerebral | Epilepsy (Tuberous sclerosis; TSC) | Demonstrated how human-specific developmental processes contribute to malformations of cortical development, utilizing patient-derived iPSCs with TSC2 mutations. | Identified caudal late interneuron progenitor (CLIP) cells in TSC organoids that exhibit excessive proliferation, resulting in overabundance of interneurons, brain tumors, and cortical malformations. | [153] |
| Cerebral | Cerebral Palsy | Analyzed the effects of the loss of the TYW1 gene, using organoids produced from CRISPR-edited iPSCs. | Loss of TYW1 led to impaired differentiation of neurons. | [154] |
| Cerebral | Cerebral palsy | Evaluated the effectiveness of minocycline on neonatal hypoxic insult (NHI) using an organoid model of hypoxia. | Minocycline reduced impact of NHI on organoids. | [155] |
| Telencephalic | ADHD | Explored the emergence of ADHD in the early cerebral cortex through patient-derived organoids. | In ADHD organoids, cortical maturation was delayed, along with alterations in qualities of neural stem cells and development of layered structures. | [156] |
| Assembloids (Cortico-striatal) | Developed a human in vitro model that recapitulates the dopaminergic innervation of the striatum and cortex. | Assembloids comprised ventral midbrain–striatum–cortical organoids, which can be used to study dopaminergic neuron maturation, innervation, and functions. | [157] | |
| Assembloids (Striato-nigral) | Generated Str-SN assembloids by fusing striatum-like and midbrain SN-like organoids. | Model showed significant medium spiny neuron (MSN) projections from striatum to SN. | [158] | |
| Assembloids (Cortico-motor) | Developed cortico-motor assembloids by fusing cerebral/hindbrain/spinal and skeletal muscle organoids. | Model demonstrated that corticofugal neurons project and establish connections with spinal organoids. Optogenetic activation of cortical spheroids elicited significant contractions in 3D muscle. | [159] | |
| Assembloids (Forebrain) | Combined CRISPR screening with assembloids to explore 425 neurodevelopmental disorder (NDD) genes related to interneuron development. | Discovered 33 candidate genes from interneuron migration screen using more than 1000 forebrain assembloids. Emphasized efficacy of CRISPR-assembloid platform in studying NDDs and human development. | [160] | |
| Assembloids (Neuroimmune) | Created neuroimmune assembloids using cortical organoids and microglia. | Model serves as tool for exploring interactions between neural and immune cells. | [161] | |
| Assembloids (Neuron–astrocyte) | Developed a model that more accurately mimics the aging brain and Parkinson’s disease (PD)-related pathologies by incorporating astrocytes. | Neuron–astrocyte assembloids highlighted role of glial cells in neurodegeneration processes. | [162] | |
| Assembloids (Thalamocortical) | Created thalamocortical assembloids by fusing thalamus and cortical organoids. | scRNA analysis showed that over 80% of thalamic organoid cells were glutamatergic neurons. Assembloids formed reciprocal projections and synapses, demonstrating short-term plasticity. Valuable model for studying synaptic plasticity in human neural system circuits. | [163] | |
| Assembloids (Thalamocortical) | Investigated the contribution of genetic variants in T-type calcium channels in the early development of the human thalamocortical pathway. | M15331V CACNA1G variant led to changes in T-type currents in thalamic neurons, which resulted in hyperactivity of thalamic and cortical neurons and abnormal thalamocortical connectivity. | [164] | |
| Assembloids (Cortical–blood vessel) | Developed assembloids by merging cortical and blood vessel organoids. | Observed notable increase in microglia and astrocytes in brain organoids, providing platform to study neurotrophic diseases such as COVID-19 and its correlation with AD. | [165] | |
| Assembloids (Somatosensory) | Developed human ascending somatosensory assembloids (hASAs). | Four-part assembloids integrating somatosensory, spinal, thalamic, and cortical organoids model spinothalamic pathway. Transcriptomic profiling confirmed key cell types, and chemical stimulation showed coordinated response. | [166] |
| Organoid | Disease | Aim | Strategy | Main Findings | Ref. |
|---|---|---|---|---|---|
| Midbrain | Created a scalable and automated platform for generating and analyzing organoids. | Automated high-throughput workflow, high-content imaging (HCI). | Organoids with minimal variability in size, morphology, and cellular composition within and across batches. RNA sequencing and HCI confirmed reproducibility. | [69] | |
| Cerebral | Alzheimer’s disease (AD) | Developed a drug screening model for AD and evaluated FDA-approved drugs that can penetrate the blood–brain barrier. | Mathematical modeling, high-content screening. | Generated a network-based computational model of AD and identified FDA-approved compounds that alleviate AD-related phenotypes. | [240] |
| Cortical | Aimed to establish a scalable and automated platform for high-throughput screening (HTS) using iPSC-derived brain organoids. | High-content imaging (HCI), multi-electrode arrays (MEAs), and single cell-calcium imaging. | Demonstrated the feasibility of using iPSC-derived brain organoids in an automated HTS platform, offering a promising tool for drug discovery and neurotoxicity. | [243] | |
| Cerebral | Developmental neurotoxicity (DNT) | Aimed to enhance the evaluation of DNT by employing a dynamic culture system. | Pillar/perfusion plate platform. | A dynamic culture approach offers a high-throughput platform for assessing the DNT of various compounds. | [244] |
| Cerebral | Aimed to develop a simple and reproducible method to enhance scalability and constituency in organoid production. | Pillar plate/ultra-low attachment well plate. | Uniform cerebral organoids offer a scalable and efficient platform for high-throughput organoid-based assay. | [245] | |
| Cerebral | Glioma | Described a method that reproducibly generates thousands of organoids across multiple iPSC lines. | High-quantity (Hi-Q) brain organoid approach. | Enabled a medium-throughput drug screen that identified selumetinib and fulvestrant as inhibitors of glioma invasion. | [246] |
| Patient-derived organoids (PDO) | Melanoma brain metastases | Created patient-derived organoids from melanoma brain metastases (MBM-PDOs) and evaluated the practicality of utilizing them as a model for testing targeted therapy drugs in vitro. | Cell viability assessment after MBM-PDOs were exposed to drugs. | Utilizing FDA-approved inhibitors for BRAF and MEK, demonstrated the practicality of employing MBM-PDOs for identifying targeted therapeutics. | [247] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajongbolo, A.O.; Langhans, S.A. Brain Organoids and Assembloids—From Disease Modeling to Drug Discovery. Cells 2025, 14, 842. https://doi.org/10.3390/cells14110842
Ajongbolo AO, Langhans SA. Brain Organoids and Assembloids—From Disease Modeling to Drug Discovery. Cells. 2025; 14(11):842. https://doi.org/10.3390/cells14110842
Chicago/Turabian StyleAjongbolo, Aderonke O., and Sigrid A. Langhans. 2025. "Brain Organoids and Assembloids—From Disease Modeling to Drug Discovery" Cells 14, no. 11: 842. https://doi.org/10.3390/cells14110842
APA StyleAjongbolo, A. O., & Langhans, S. A. (2025). Brain Organoids and Assembloids—From Disease Modeling to Drug Discovery. Cells, 14(11), 842. https://doi.org/10.3390/cells14110842