
Congenital Heart Diseases: Recent Insights into Epigenetic Mechanisms

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
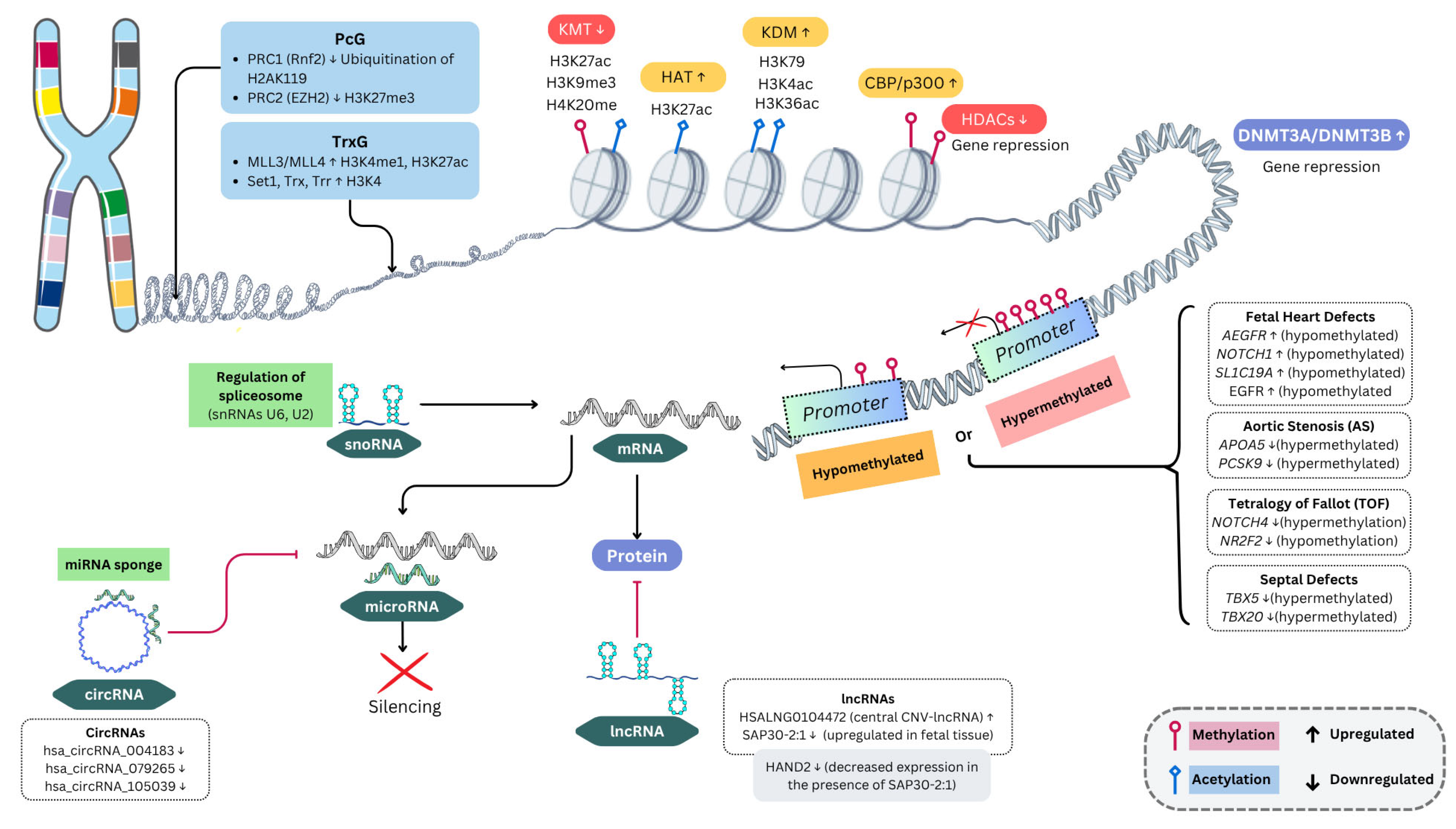
2. Epigenetics
3. Congenital Heart Diseases and the Environment
4. Cardiac Development
5. DNA Methylation
6. Histone Modification
7. ATP-Dependent Chromatin Remodeling
8. Polycomb and Trithorax Complex Proteins
9. Non-Coding RNAs
9.1. MicroRNAs (miRNAs) and CHD
9.2. CHD and Other ncRNAs
10. Limitations
11. Perspectives
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peixoto, P.; Cartron, P.-F.; Serandour, A.A.; Hervouet, E. From 1957 to Nowadays: A Brief History of Epigenetics. Int. J. Mol. Sci. 2020, 21, 7571. [Google Scholar] [CrossRef]
- Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Zühlke, L.; Babu-Narayan, S.V.; Black, G.C.; Choy, M.; Li, N.; Keavney, B.D. Global Prevalence of Congenital Heart Disease in School-Age Children: A Meta-Analysis and Systematic Review. BMC Cardiovasc. Disord. 2020, 20, 488. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Zühlke, L.; Black, G.C.; Choy, M.-K.; Li, N.; Keavney, B.D. Global Birth Prevalence of Congenital Heart Defects 1970–2017: Updated Systematic Review and Meta-Analysis of 260 Studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef]
- Stout, K.K.; Daniels, C.J.; Aboulhosn, J.A.; Bozkurt, B.; Broberg, C.S.; Colman, J.M.; Crumb, S.R.; Dearani, J.A.; Fuller, S.; Gurvitz, M.; et al. 2018 AHA/ACC Guideline for the Management of Adults with Congenital Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e81–e192. [Google Scholar] [CrossRef] [PubMed]
- NHLBI; NIH. Congenital Heart Defects. Available online: https://www.nhlbi.nih.gov/health/congenital-heart-defects/types (accessed on 8 March 2025).
- Rohit, M.; Shrivastava, S. Acyanotic and Cyanotic Congenital Heart Diseases. Indian J. Pediatr. 2018, 85, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Sheng, W.; Ma, D.; Huang, G.; Liu, F. DNA Methylation Status of TBX20 in Patients with Tetralogy of Fallot. BMC Med. Genom. 2019, 12, 75. [Google Scholar] [CrossRef]
- Yang, X.; Kong, Q.; Li, Z.; Xu, M.; Cai, Z.; Zhao, C. Association Between the Promoter Methylation of the TBX20 Gene and Tetralogy of Fallot. Scand. Cardiovasc. J. 2018, 52, 287–291. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, X.; Zhang, Y.; Zheng, J.; Yang, R. Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases. World J. Pediatr. Surg. 2021, 4, e000196. [Google Scholar] [CrossRef]
- Nagy, O.; Baráth, S.; Ujfalusi, A. The Role of microRNAs in Congenital Heart Disease. EJIFCC 2019, 30, 165–178. [Google Scholar]
- Tafazoli, A.; Hemmati, M.; Rafigh, M.; Alimardani, M.; Khaghani, F.; Korostyński, M.; Karnes, J.H. Leveraging long-read sequencing technologies for pharmacogenomic testing: Applications, analytical strategies, challenges, and future perspectives. Front. Genet. 2025, 16, 1435416. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Kumar, K.R.; Zech, M. Long-Read Sequencing: The Third Generation of Diagnostic Testing for Dystonia. Mov. Disord. 2025. Early View. [Google Scholar] [CrossRef]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C. A Brief History of Epigenetics. Immunol. Lett. 2022, 249, 1–4. [Google Scholar] [CrossRef]
- Jarrell, D.K.; Lennon, M.L.; Jacot, J.G. Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Rdu, E. Epigenética: Candados y Llaves Durante la Lectura del ADN. RDU UNAM. Available online: https://www.revista.unam.mx/2020v21n6/epigenetica_candados_y_llaves_durante_la_lectura_del_adn/ (accessed on 5 March 2025).
- Thiagalingam, S. Epigenetic Memory in Development and Disease: Unraveling the Mechanism. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188349. [Google Scholar] [CrossRef]
- D’Adamo, G.L.; Widdop, J.T.; Giles, E.M. The Future Is Now? Clinical and Translational Aspects of “Omics” Technologies. Immunol. Cell Biol. 2021, 99, 168–176. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861. [Google Scholar] [CrossRef]
- Cheng, Y.; Yin, J.; Yang, L.; Xu, M.; Lu, X.; Huang, W.; Dai, G.; Sun, G. Ambient Air Pollutants in the First Trimester of Pregnancy and Birth Defects: An Observational Study. BMJ Open 2023, 13, e063712. [Google Scholar] [CrossRef]
- Linglart, L.; Bonnet, D. Epigenetics and Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Eckhardt, C.M.; Baccarelli, A.A. Molecular Mechanisms of Environmental Exposures and Human Disease. Nat. Rev. Genet. 2023, 24, 332–344. [Google Scholar] [CrossRef]
- Patel, J.; Nembhard, W.N.; Politis, M.D.; Rocheleau, C.M.; Langlois, P.H.; Shaw, G.M.; Romitti, P.A.; Gilboa, S.M.; Desrosiers, T.A.; Insaf, T.; et al. Maternal Occupational Exposure to Polycyclic Aromatic Hydrocarbons and the Risk of Isolated Congenital Heart Defects Among Offspring. Environ. Res. 2020, 186, 109550. [Google Scholar] [CrossRef]
- Ma, Z.; Cao, X.; Chang, Y.; Li, W.; Chen, X.; Tang, N.J. Association Between Gestational Exposure and Risk of Congenital Heart Disease: A Systematic Review and Meta-Analysis. Environ. Res. 2021, 197, 111014. [Google Scholar] [CrossRef]
- Liu, F.; Li, X.; Chen, J.; Huang, Y.; Dang, S. Maternal Pesticide Exposure and Risk of Birth Defects: A Population-Based Cross-Sectional Study in China. Front. Public Health 2024, 12, 1489365. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.L.; Yang, W.; Roberts, E.; Kegley, S.E.; Padula, A.M.; English, P.B.; Lammer, E.J.; Shaw, G.M. Residential Agricultural Pesticide Exposures and Risk of Selected Congenital Heart Defects among Offspring in the San Joaquin Valley of California. Environ. Res. 2014, 135, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.-Y.; Zhou, K.-Y.; Wang, T.; Zhang, Y.; Li, Y.-F.; Hua, Y.-M.; Wang, C. Disruption of Planar Cell Polarity Pathway Attributable to Valproic Acid-Induced Congenital Heart Disease through Hdac3 Participation in Mice. Chin. Med. J. 2018, 131, 2080–2088. [Google Scholar] [CrossRef]
- Desai, P.H.; Yagnik, P.J.; Ross Ascuitto, N.; Prajapati, P.; Sernich, S. Risk of Congenital Heart Disease in Newborns with Prenatal Exposure to Anti-Depressant Medications. Cureus 2019, 11, e4673. [Google Scholar] [CrossRef]
- Howley, M.M.; Papadopoulos, E.A.; Van Bennekom, C.M.; Van Zutphen, A.R.; Carmichael, S.L.; Munsie, J.W.; Herdt, M.L.; Browne, M.L. National Birth Defects Prevention Study. Asthma Medication Use and Risk of Birth Defects: National Birth Defects Prevention Study, 1997–2011. J. Allergy Clin. Immunol. Pract. 2020, 8, 3490–3499. [Google Scholar] [CrossRef]
- Patorno, E.; Huybrechts, K.F.; Hernandez-Diaz, S. Lithium Use in Pregnancy and the Risk of Cardiac Malformations. N. Engl. J. Med. 2017, 377, 893–894. [Google Scholar]
- Patel, S.S.; Burns, T.L. Nongenetic Risk Factors and Congenital Heart Defects. Pediatr. Cardiol. 2013, 34, 1535–1555. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, L.; Yang, T.; Chen, L.; Wang, T.; Chen, L.; Zhao, L.; Zhang, S.; Zheng, Z.; Luo, L.; et al. Maternal Viral Infection and Risk of Fetal Congenital Heart Diseases: A Meta-Analysis of Observational Studies. J. Am. Heart Assoc. 2019, 8, e011264. [Google Scholar] [CrossRef] [PubMed]
- Forrest, J.M.; Turnbull, F.M.; Sholler, G.F.; Hawker, R.E.; Martin, F.J.; Doran, T.T.; Burgess, M.A. Gregg’s Congenital Rubella Patients 60 Years Later. Med. J. Aust. 2002, 177, 664–667. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Guo, L.; Peng, X.; Liu, Y. Prenatal Alcohol Exposure Induced Congenital Heart Diseases: From Bench to Bedside. Birth Defects Res. 2021, 113, 521–534. [Google Scholar] [CrossRef]
- Borjali, M.; Amini-Rarani, M.; Nosratabadi, M. Nonmedical Determinants of Congenital Heart Diseases in Children from the Perspective of Mothers: A Qualitative Study in Iran. Cardiol. Res. Pract. 2021, 2021, 6647260. [Google Scholar] [CrossRef]
- Spinder, N.; Bergman, J.E.; Kromhout, H.; Vermeulen, R.; Corsten-Janssen, N.; Boezen, H.M.; du Marchie Sarvaas, G.J.; de Walle, H.E. Maternal Occupational Exposure and Congenital Heart Defects in Offspring. Scand. J. Work Environ. Health 2020, 46, 599–608. [Google Scholar] [CrossRef]
- Al-Qattan, M.M.; Abou Al-Shaar, H. Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene. 2015, 560, 129–136. [Google Scholar] [CrossRef] [PubMed]
- De Gannes, M.; Ko, C.I.; Zhang, X.; Biesiada, J.; Niu, L.; Koch, S.E.; Medvedovic, M.; Rubinstein, J.; Puga, A. Dioxin Disrupts Dynamic DNA Methylation Patterns in Genes that Govern Cardiomyocyte Maturation. Toxicol. Sci. 2020, 178, 325–337. [Google Scholar] [CrossRef]
- Rossi, G.; Broguiere, N.; Miyamoto, M.; Boni, A.; Guiet, R.; Girgin, M.; Kelly, R.G.; Kwon, C.; Lutolf, M.P. Capturing Cardiogenesis in Gastruloids. Cell Stem Cell 2021, 28, 230–240. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D. Molecular Mechanisms of Cardiac Development and Disease. Int. J. Mol. Sci. 2023, 24, 8784. [Google Scholar] [CrossRef]
- Heallen, T.R.; Kadow, Z.A.; Wang, J.; Martin, J.F. Determinants of Cardiac Growth and Size. Cold Spring Harb. Perspect. Biol. 2020, 12, a037150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, H.M.; Wang, F.; Zhao, C.M.; Huang, R.T.; Xue, S.; Li, R.G.; Qiu, X.B.; Xu, Y.J.; Liu, X.Y.; et al. A New ISL1 Loss-of-Function Mutation Predisposes to Congenital Double Outlet Right Ventricle. Int. Heart J. 2019, 60, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Nim, H.T.; Dang, L.; Thiyagarajah, H.; Bakopoulos, D.; See, M.; Charitakis, N.; Sibbritt, T.; Eichenlaub, M.P.; Archer, S.K.; Fossat, N.; et al. A Cis-Regulatory-Directed Pipeline for the Identification of Genes Involved in Cardiac Development and Disease. Genome Biol. 2021, 22, 335. [Google Scholar] [CrossRef]
- Buijtendijk, M.F.J.; Barnett, P.; van den Hoff, M.J.B. Development of the Human Heart. Am. J. Med. Genet. Semin. Med. Genet. 2020, 184, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Nzomvuama, A.; Nader, J.; Remadi, J.-P. Ectopia Cordis: An Uncommon Fatal Disease. Ann. Thorac. Surg. 2021, 112, e201. [Google Scholar] [CrossRef]
- Anderson, R.H.; Spicer, D.E.; Brown, N.A.; Mohun, T.J. The Development of Septation in the Four-Chambered Heart. Anat. Rec. 2014, 297, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-Box Genes and Retinoic Acid Signaling Regulate the Segregation of Arterial and Venous Pole Progenitor Cells in the Murine Second Heart Field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef]
- Ortega-Zhindón, D.B.; Flores-Sarria, I.P.; Minakata-Quiróga, M.A.; Angulo-Cruzado, S.T.; Romero-Montalvo, L.A.; Cervantes-Salazar, J.L. Isomorfismo cardiaco: Una perspectiva multidisciplinaria. Arch. Cardiol. Mex. 2021, 91, 470–479. [Google Scholar] [CrossRef]
- Ortega-Zhindón, D.B.; Calderón-Colmenero, J.; García-Montes, J.A.; Sandoval, J.P.; Minakata-Quiroga, M.A.; Cervantes-Salazar, J.L. Cardiac Surgery in Patients with Atrial Isomerism: Long-Term Results and Outcomes. J. Card. Surg. 2021, 36, 4476–4484. [Google Scholar] [CrossRef]
- Ortega-Zhindón, D.B.; Pérez-Hernández, N.; Rodríguez-Pérez, J.M.; García-Montes, J.A.; Calderón-Colmenero, J.; Rivera-Buendía, F.; Cervantes-Salazar, J.L. Cardiac Laterality: Surgical Results of Right Atrial Isomerism. Diseases 2023, 11, 170. [Google Scholar] [CrossRef]
- Desgrange, A.; Le Garrec, J.-F.; Meilhac, S.M. Left-Right Asymmetry in Heart Development and Disease: Forming the Right Loop. Development 2018, 145, dev162776. [Google Scholar] [CrossRef] [PubMed]
- Uludag Alkaya, D.; Ozturk, B.; Yuksel Ulker, A.; Bozlak, S.; Ozturk, E.; Dedeoglu, R.; Eroglu, A.G.; Oztunc, F.; Tuysuz, B. Congenital Heart Defects and Outcome in a Large Cohort of Down Syndrome: A Single-Center Experience from Turkey. Turk. Arch. Pediatr. 2023, 58, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Courtney, J.; Troja, W.; Owens, K.J.; Brockway, H.M.; Hinton, A.C.; Hinton, R.B.; Cnota, J.F.; Jones, H.N. Abnormalities of Placental Development and Function Are Associated with the Different Fetal Growth Patterns of Hypoplastic Left Heart Syndrome and Transposition of the Great Arteries. Placenta 2020, 101, 57–65. [Google Scholar] [CrossRef]
- Firulli, B.A.; George, R.M.; Harkin, J.; Toolan, K.P.; Gao, H.; Liu, Y.; Zhang, W.; Field, L.J.; Liu, Y.; Shou, W.; et al. HAND1 Loss-of-Function Within the Embryonic Myocardium Reveals Survivable Congenital Cardiac Defects and Adult Heart Failure. Cardiovasc. Res. 2020, 116, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Saw, S.N.; Dai, Y.; Yap, C.H. A Review of Biomechanics Analysis of the Umbilical–Placenta System with Regards to Diseases. Front. Physiol. 2021, 12, 587635. [Google Scholar] [CrossRef]
- Moumne, O.; Chowdhurry, R.; Doll, C.; Pereira, N.; Hashimi, M.; Grindrod, T.; Dollar, J.J.; Riva, A.; Kasahara, H. Mechanism Sharing Between Genetic and Gestational Hypoxia-Induced Cardiac Anomalies. Front. Cardiovasc. Med. 2018, 5, 100. [Google Scholar] [CrossRef]
- Moreau, J.L.M.; Kesteven, S.; Martin, E.M.M.A.; Lau, K.S.; Yam, M.X.; O’Reilly, V.C.; Del Monte-Nieto, G.; Baldini, A.; Feneley, M.P.; Moon, A.M.; et al. Gene-Environment Interaction Impacts on Heart Development and Embryo Survival. Development 2019, 146, dev172957. [Google Scholar] [CrossRef]
- Kelsey, G. Imprints in the History of Epigenetics. Nat. Rev. Mol. Cell Biol. 2020, 21, 566–567. [Google Scholar] [CrossRef]
- Wang, G.; Wang, B.; Yang, P. Epigenetics in Congenital Heart Disease. J. Am. Heart Assoc. 2022, 11, e025163. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA Methylation: Implications for Gene Regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar]
- Cao, J.; Wu, Q.; Huang, Y.; Wang, L.; Su, Z.; Ye, H. The Role of DNA Methylation in Syndromic and Non-Syndromic Congenital Heart Disease. Clin. Epigenetics 2021, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.O.; Kukshal, P.; Chellappan, S.; Guhathakurta, S. The Study of Expression Levels of DNA Methylation Regulators in Patients Affected with Congenital Heart Defects (CHDs). Birth Defects Res. 2022, 114, 228–237. [Google Scholar] [CrossRef]
- Radhakrishna, U.; Albayrak, S.; Alpay-Savasan, Z.; Zeb, A.; Turkoglu, O.; Sobolewski, P.; Bahado-Singh, R.O. Genome-Wide DNA Methylation Analysis and Epigenetic Variations Associated with Congenital Aortic Valve Stenosis (AVS). PLoS ONE 2016, 11, e0154010. [Google Scholar] [CrossRef]
- Forman, J.; Beech, R.; Slugantz, L.; Donnellan, A. A Review of Tetralogy of Fallot and Postoperative Management. Crit. Care Nurs. Clin. N. Am. 2019, 31, 315–328. [Google Scholar] [CrossRef]
- Wise-Faberowski, L.; Asija, R.; McElhinney, D.B. Tetralogy of Fallot: Everything You Wanted to Know but were Afraid to Ask. Paediatr. Anaesth. 2019, 29, 475–482. [Google Scholar] [CrossRef]
- Zhu, Y.; Ye, M.; Xu, H.; Gu, R.; Ma, X.; Chen, M.; Li, X.; Sheng, W.; Huang, G. Methylation Status of CpG Sites in the NOTCH4 Promoter Region Regulates NOTCH4 Expression in Patients with Tetralogy of Fallot. Mol. Med. Rep. 2020, 22, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef]
- Xiaodi, L.; Ming, Y.; Hongfei, X.; Yanjie, Z.; Ruoyi, G.; Ma, X.; Wei, S.; Guoying, H. DNA Methylation at CpG Island Shore and RXRα Regulate NR2F2 in Heart Tissues of Tetralogy of Fallot Patients. Biochem. Biophys. Res. Commun. 2020, 529, 1209–1215. [Google Scholar] [CrossRef]
- Zhou, J.; Xiong, Y.; Dong, X.; Wang, H.; Qian, Y.; Ma, D.; Li, X. Genome-Wide Methylation Analysis Reveals Differentially Methylated CpG Sites and Altered Expression of Heart Development-Associated Genes in Fetuses with Cardiac Defects. Exp. Ther. Med. 2021, 22, 1032. [Google Scholar] [CrossRef]
- Wilson, V.; Conlon, F.L. The T-Box Family. Genome Biol. 2002, 3, reviews3008. [Google Scholar] [CrossRef] [PubMed]
- García-Flores, E.; Rodríguez-Pérez, J.M.; Borgonio-Cuadra, V.M.; Vargas-Alarcón, G.; Calderón-Colmenero, J.; Sandoval, J.P.; García-Montes, J.A.; Espinoza-Gutiérrez, V.M.; Reyes-García, J.G.; Cazarín-Santos, B.G.; et al. DNA Methylation Levels of the TBX5 Gene Promoter Are Associated with Congenital Septal Defects in Mexican Paediatric Patients. Biology 2022, 11, 96. [Google Scholar] [CrossRef]
- Mouat, J.S.; Li, S.; Myint, S.S.; Laufer, B.I.; Lupo, P.J.; Schraw, J.M.; Woodhouse, J.P.; de Smith, A.J.; LaSalle, J.M. Epigenomic Signature of Major Congenital Heart Defects in Newborns with Down Syndrome. Hum. Genom. 2023, 17, 92. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, Y.; Xin, Y.; Wang, S.; Luo, Y.; Wang, L.; Zhang, H.; Li, J. DNA Methylation Abnormalities of Imprinted Genes in Congenital Heart Disease: A Pilot Study. BMC Med. Genom. 2021, 14, 4. [Google Scholar] [CrossRef]
- He, K.; Cao, X.; Deng, X. Histone Methylation in Epigenetic Regulation and Temperature Responses. Current Opin. Plant Biol. 2021, 61, 102001. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Atilla, H.; Kannu, P.; Schneider, A.; Thompson, S.; Bardakjian, T.; Semina, E.V. Distinct Roles of Histone Lysine Demethylases and Methyltransferases in Developmental Eye Disease. Genes 2023, 14, 216. [Google Scholar] [CrossRef]
- Zhou, W.; Jiang, D.; Tian, J.; Liu, L.; Lu, T.; Huang, X.; Sun, H. Acetylation of H3K4, H3K9, and H3K27 Mediated by P300 Regulates the Expression of GATA4 in Cardiocytes. Genes Dis. 2018, 6, 318–325. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar]
- Huang, W.; Zhu, J.-Y.; Fu, Y.; van de Leemput, J.; Han, Z. Lpt, Trr, and Hcf Regulate Histone Mono- and Dimethylation that Are Essential for Drosophila Heart Development. Dev. Biol. 2022, 490, 53–65. [Google Scholar] [CrossRef]
- Matsushita, N. Dysregulated Histone Acetylation Causes Congenital Diseases. Gene Rep. 2023, 31, 101778. [Google Scholar] [CrossRef]
- Zhou, P.; VanDusen, N.J.; Zhang, Y.; Cao, Y.; Sethi, I.; Hu, R.; Zhang, S.; Wang, G.; Ye, L.; Mazumdar, N.; et al. Dynamic Changes in P300 Enhancers and Enhancer-Promoter Contacts Control Mouse Cardiomyocyte Maturation. Dev. Cell 2023, 58, 898–914.e7. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K. p300 in Cardiac Development and Accelerated Cardiac Aging. Aging Dis. 2020, 11, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Leigh, R.S.; Välimäki, M.J.; Kaynak, B.L.; Ruskoaho, H.J. TAF1 Bromodomain Inhibition as a Candidate Epigenetic Driver of Congenital Heart Disease. Biochim. Biophys. Acta. Mol. Basis. Dis. 2023, 1869, 166689. [Google Scholar] [CrossRef] [PubMed]
- Crombie, E.M.; Cleverley, K.; Timmers, H.T.M.; Fisher, E.M.C. The roles of TAF1 in neuroscience and beyond. R. Soc. Open Sci. 2024, 11, 240790. [Google Scholar] [CrossRef]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H. Genome-Wide Histone Methylation Profile for Heart Failure. Genes Cells 2009, 14, 69–77. [Google Scholar] [CrossRef]
- Blakeslee, W.W.; Demos-Davies, K.M.; Lemon, D.D.; Lutter, K.M.; Cavasin, M.A.; Payne, S.; Nunley, K.; Long, C.S.; McKinsey, T.A.; Miyamoto, S.D. Histone Deacetylase Adaptation in Single Ventricle Heart Disease and a Young Animal Model of Right Ventricular Hypertrophy. Pediatr. Res. 2017, 82, 642–649. [Google Scholar] [CrossRef]
- Huang, Z.; Song, S.; Zhang, X.; Zeng, L.; Sun, A.; Ge, J. Metabolic Substrates, Histone Modifications, and Heart Failure. Biochim. Biophys. Acta Gene Regul. Mech. 2023, 1866, 194898. [Google Scholar] [CrossRef]
- Hou, Y.S.; Wang, J.Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.X.; Xu, C.; Li, F.F.; Wang, G.Y.; Liu, S.L. Identification of Epigenetic Factor KAT2B Gene Variants for Possible Roles in Congenital Heart Diseases. Biosci. Rep. 2020, 40, BSR20191779. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Aparicio-Sánchez, J.J.; Buxton, S.; Ketley, A.; Mohamed, T.; Rutland, C.S.; Loughna, S.; Brook, J.D. Acetylation of TBX5 by KAT2B and KAT2A Regulates Heart and Limb Development. J. Mol. Cell Cardiol. 2018, 114, 185–198. [Google Scholar] [CrossRef]
- Campeau, P.M.; Lu, J.T.; Dawson, B.C.; Fokkema, I.F.; Robertson, S.P.; Gibbs, R.A.; Lee, B.H. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum. Mutat. 2012, 33, 1520–1525. [Google Scholar] [CrossRef]
- Yodh, J. ATP-Dependent Chromatin Remodeling. Adv. Exp. Med. Biol. 2013, 767, 263–295. [Google Scholar] [PubMed]
- Basson, M.A.; van Ravenswaaij-Arts, C. Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome. Trends Genet. 2015, 31, 600–611. [Google Scholar] [CrossRef]
- Al-Awar, A.; Hussain, S. Interplay of Reactive Oxygen Species (ROS) and Epigenetic Remodelling in Cardiovascular Diseases Pathogenesis: A Contemporary Perspective. Front. Biosci. 2024, 29, 398. [Google Scholar] [CrossRef] [PubMed]
- Chohra, I.; Chung, K.; Giri, S.; Malgrange, B. ATP-Dependent Chromatin Remodellers in Inner Ear Development. Cells 2023, 12, 532. [Google Scholar] [CrossRef]
- Meisner, J.K.; Martin, D.M. Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes. Am. J. Med. Genet. Semin. Med. Genet. 2020, 184, 81–89. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Epigenetic Regulation of Cellular Memory by the Polycomb and Trithorax Group Proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Occupying Chromatin: Polycomb Mechanisms for Getting to Genomic Targets, Stopping Transcriptional Traffic, and Staying Put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, C.B.; Nishiyama, A.; Ryan, J.; Nakamura, R.; Yiğit, M.; Glück, I.M.; Trummer, C.; Qin, W.; Bartoschek, M.D.; Traube, F.R.; et al. Recent Evolution of a TET-Controlled and DPPA3/STELLA-Driven Pathway of Passive DNA Demethylation in Mammals. Nat. Commun. 2020, 11, 5972. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Loh, C.H.; Veenstra, G.J.C. The Role of Polycomb Proteins in Cell Lineage Commitment and Embryonic Development. Epigenomes 2022, 6, 23. [Google Scholar] [CrossRef]
- Chetverina, D.A.; Lomaev, D.V.; Erokhin, M.M. Polycomb and Trithorax Group Proteins: The Long Road from Mutations in Drosophila to Use in Medicine. Acta Naturae 2020, 12, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, I.; Ishtiaq, A.; Ali, T.; Jan, M.I.; Murtaza, I. An Overview of Non-Coding RNAs and Cardiovascular System. Adv. Exp. Med. Biol. 2020, 1229, 3–45. [Google Scholar]
- Kan, Z.; Yan, W.; Wang, N.; Fang, Y.; Gao, H.; Song, Y. Identification of circRNA-miRNA-mRNA Regulatory Network and Crucial Signaling Pathway Axis Involved in Tetralogy of Fallot. Front. Genet. 2022, 13, 917454. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E.; Kibiryeva, N.; Zhou, X.-G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding RNA Expression in Myocardium from Infants with Tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar] [CrossRef]
- Ma, J.; Chen, S.; Hao, L.; Sheng, W.; Chen, W.; Ma, X.; Zhang, B.; Ma, D.; Huang, G. Long Non-Coding RNA SAP30-2:1 Is Downregulated in Congenital Heart Disease and Regulates Cell Proliferation by Targeting HAND2. Front. Med. 2021, 15, 91–100. [Google Scholar] [CrossRef]
- Lu, Y.; Fang, Q.; Qi, M.; Li, X.; Zhang, X.; Lin, Y.; Xiang, Y.; Fu, Q.; Wang, B. Copy Number Variation-Associated lncRNAs May Contribute to the Etiologies of Congenital Heart Disease. Commun. Biol. 2023, 6, 189. [Google Scholar] [CrossRef]
- Yan, J.; Dutta, B.; Hee, Y.T.; Chng, W.-J. Towards Understanding of PRC2 Binding to RNA. RNA Biol. 2019, 16, 176–184. [Google Scholar] [CrossRef]
- Wang, G.; Ye, H.; Wang, X.; Liu, B. Polycomb Repressive Complex 2 Controls Cardiac Cell Fate Decision via Interacting with RNA: Promiscuously or Well-Ordered. Front. Genet. 2022, 13, 1011228. [Google Scholar] [CrossRef] [PubMed]
- Hanafiah, A.; Geng, Z.; Liu, T.; Tai, Y.T.; Cai, W.; Wang, Q.; Christensen, N.; Liu, Y.; Yue, F.; Gao, Z. PRC1 and CTCF-Mediated Transition from Poised to Active Chromatin Loops Drives Bivalent Gene Activation. bioRxiv 2024. [Google Scholar] [CrossRef]
- He, A.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb Repressive Complex 2 Regulates Normal Development of the Mouse Heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef]
- Chrispijn, N.D.; Elurbe, D.M.; Mickoleit, M.; Aben, M.; de Bakker, D.E.M.; Andralojc, K.M.; Huisken, J.; Bakkers, J.; Kamminga, L.M. Loss of the Polycomb Group Protein Rnf2 Results in Derepression of Tbx-Transcription Factors and Defects in Embryonic and Cardiac Development. Sci. Rep. 2019, 9, 4327. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Nakka, K.; Zhu, J.; Dilworth, F.J. Polycomb/Trithorax Antagonism: Cellular Memory in Stem Cell Fate and Function. Cell Stem Cell 2019, 24, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, D.; Boileau, R.M.; Lenshoek, K.; Blelloch, R. MLL3/MLL4 Enzymatic Activity Shapes DNA Replication Timing. bioRxiv 2023. [Google Scholar] [CrossRef]
- Boileau, R.M.; Chen, K.X.; Blelloch, R. Loss of MLL3/4 Decouples Enhancer H3K4 Monomethylation, H3K27 Acetylation, and Gene Activation During Embryonic Stem Cell Differentiation. Genome Biol. 2023, 24, 41. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Lee, H.; Huang, X.; van de Leemput, J.; Han, Z. Distinct Roles for COMPASS Core Subunits Set1, Trx, and Trr in the Epigenetic Regulation of Drosophila Heart Development. Int. J. Mol. Sci. 2023, 24, 17314. [Google Scholar] [CrossRef]
- Loganathan, T.; Doss, C.G.P. Non-Coding RNAs in Human Health and Disease: Potential Function as Biomarkers and Therapeutic Targets. Funct. Integr. Genom. 2023, 23, 33. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, A.F.; Costa, M.C.; Enguita, F.J. Interactions Among Regulatory Non-Coding RNAs Involved in Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1229, 79–104. [Google Scholar]
- Smith, T.; Rajakaruna, C.; Caputo, M.; Emanueli, C. MicroRNAs in Congenital Heart Disease. Ann. Transl. Med. 2015, 3, 333. [Google Scholar]
- Wajahat, M.; Bracken, C.P.; Orang, A. Emerging Functions for snoRNAs and snoRNA-Derived Fragments. Int. J. Mol. Sci. 2021, 22, 10193. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, W.; Yu, W.; Zhang, Y.; Ao, X.; Wang, J. Long Non-Coding RNAs: Biogenesis, Functions, and Clinical Significance in Gastric Cancer. Mol. Ther. Oncolytics 2021, 23, 458–476. [Google Scholar] [CrossRef]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-Coding RNA Regulatory Networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ni, Y.Q.; Xu, H.; Xiang, Q.Y.; Zhao, Y.; Zhan, J.K.; He, J.Y.; Li, S.; Liu, Y.S. Roles and Mechanisms of Exosomal Non-Coding RNAs in Human Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Catalucci, D.; Condorelli, G. MicroRNA and Cardiac Pathologies. Physiol. Genom. 2008, 34, 239–242. [Google Scholar] [CrossRef]
- Si, H.; Zhang, N.; Shi, C.; Luo, Z.; Hou, S. Tumor-Suppressive miR-29c Binds to MAPK1 Inhibiting the ERK/MAPK Pathway in Pancreatic Cancer. Clin. Transl. Oncol. 2023, 25, 803–816. [Google Scholar] [CrossRef]
- Sayed, D.; Hong, C.; Chen, I.-Y.; Lypowy, J.; Abdellatif, M. MicroRNAs Play an Essential Role in the Development of Cardiac Hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA Expression in Hypoplastic Left Heart Syndrome. J. Card. Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef] [PubMed]
- AmiRsardari, Z.; Gholipour, A.; Khajali, Z.; Maleki, M.; Malakootian, M. Exploring the Role of Non-Coding RNAs in Atrial Septal Defect Pathogenesis: A Systematic Review. PLoS ONE 2024, 19, e0306576. [Google Scholar] [CrossRef]
- Rivas, D.A.; Peng, F.; Benard, T.; Ramos da Silva, A.S.; Fielding, R.A.; Margolis, L.M. miR-19b-3p Is Associated with a Diametric Response to Resistance Exercise in Older Adults and Regulates Skeletal Muscle Anabolism via PTEN Inhibition. Am. J. Physiol. Cell Physiol. 2021, 321, C977–C991. [Google Scholar] [CrossRef]
- Wang, F.; Liu, D.; Zhang, R.-R.; Yu, L.-W.; Zhao, J.-Y.; Yang, X.-Y.; Jiang, S.-S.; Ma, D.; Qiao, B.; Zhang, F.; et al. A TBX5 3′UTR Variant Increases the Risk of Congenital Heart Disease in the Han Chinese Population. Cell Discov. 2017, 3, 17026. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Zhou, Z.; Jiang, J.; Zhang, Z.; Ye, L.; Hong, H. A Gain-of-Function ACTC1 3′UTR Mutation That Introduces a miR-139-5p Target Site May Be Associated with a Dominant Familial Atrial Septal Defect. Sci. Rep. 2016, 6, 25404. [Google Scholar] [CrossRef]
- Yanagawa, B.; Lovren, F.; Pan, Y.; Garg, V.; Quan, A.; Tang, G.; Singh, K.K.; Shukla, P.C.; Kalra, N.P.; Peterson, M.D.; et al. miRNA-141 Is a Novel Regulator of BMP-2-Mediated Calcification in Aortic Stenosis. J. Thorac. Cardiovasc. Surg. 2012, 144, 256–262. [Google Scholar] [PubMed]
- Nigam, V.; Sievers, H.H.; Jensen, B.C.; Sier, H.A.; Simpson, P.C.; Srivastava, D.; Mohamed, S.A. Altered microRNAs in Bicuspid Aortic Valve: A Comparison between Stenotic and Insufficient Valves. J. Heart Valve Dis. 2010, 19, 459–465. [Google Scholar]
- Douvris, A.; Viñas, J.; Burns, K.D. miRNA-486-5p: Signaling Targets and Role in Non-Malignant Disease. Cell Mol. Life Sci. 2022, 79, 376. [Google Scholar] [CrossRef]
- NCBI. MIRLET7C microRNA let-7c [Homo sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/406885 (accessed on 9 March 2025).
- Yu, H.; Zhang, X.; Wang, X.; Chen, W.; Lao, W.; Chen, Y. MiR-99a-5p Inhibits the Proliferation and Migration of Human Retinal Microvascular Endothelial Cells by Targeting NOX4. Horm. Metab. Res. 2023, 55, 142–148. [Google Scholar] [PubMed]
- Ni, M.; Zhao, Y.; Zhang, W.J.; Jiang, Y.J.; Fu, H.; Huang, F.; Li, D.J.; Shen, F.M. microRNA-802 Accelerates Hepatocellular Carcinoma Growth by Targeting RUNX3. J. Cell Physiol. 2020, 235, 7128–7135. [Google Scholar] [CrossRef] [PubMed]
- Milan, K.L.; Jayasuriya, R.; Harithpriya, K.; Anuradha, M.; Ramkumar, K.M. MicroRNA-125b Regulates Vitamin D Resistance by Targeting CYP24A1 in the Progression of Gestational Diabetes Mellitus. J. Steroid Biochem. Mol. Biol. 2024, 239, 106475. [Google Scholar] [CrossRef]
- Cillo, F.; Coppola, E.; Habetswallner, F.; Cecere, F.; Pignata, L.; Toriello, E.; De Rosa, A.; Grilli, L.; Ammendola, A.; Salerno, P.; et al. Understanding the Variability of 22q11.2 Deletion Syndrome: The Role of Epigenetic Factors. Genes 2024, 15, 321. [Google Scholar] [CrossRef]
- Yap, X.L.; Chen, J.A. Elucidation of How the Mir-23-27-24 Cluster Regulates Development and Aging. Exp. Mol. Med. 2024, 56, 1263–1271. [Google Scholar]
- Si, W.; Wei, H.; Chen, W.; Chen, B.; Zhou, Y.; Zhang, H. Exosomal microRNA-363 Mediates the Destructive Effect of M1 Macrophages on Chondrocytes by Repressing G3BP2. Exp. Cell Res. 2024, 442, 114276. [Google Scholar] [CrossRef]
- Wang, L.; Shangguan, S.; Xin, Y.; Chang, S.; Wang, Z.; Lu, X.; Wu, L.; Niu, B.; Zhang, T. Folate Deficiency Disturbs Hsa-let-7 g Level through Methylation Regulation in Neural Tube Defects. J. Cell Mol. Med. 2017, 21, 3244–3253. [Google Scholar]
- Ramachandran, V.; Bhagavatheeswaran, S.; Shanmugam, S.; Vasudevan, M.; Ragunathan, M.; Cherian, K.M.; Munirajan, A.K.; Ravi, S.; Balakrishnan, A. Deep Sequencing Unveils Altered Cardiac miRNome in Congenital Heart Disease. Mol. Genet. Genom. 2022, 297, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, Y.; Chen, Q.; Li, S.; Yang, Y.; Lyu, G. Analyzing Exosomal miRNA Profiles in Tetralogy of Fallot Fetuses’ Amniotic Fluid. Sci. Rep. 2025, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Li, K.X.; Li, J.R.; Zuo, S.J.; Li, X.; Chen, X.T.; Xiao, P.Y.; Li, H.T.; Sun, L.; Qian, T.; Zhang, H.M.; et al. Identification of miR-20b-5p as an Inhibitory Regulator in Cardiac Differentiation via TET2 and DNA Hydroxymethylation. Clin. Epigenetics 2024, 16, 42. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, Y.; Zhang, X.; Zhang, X.; Xiang, Y.; Fu, Q.; Wang, B.; Xu, Z. FGD5-AS1 Is a Hub lncRNA ceRNA in Hearts with Tetralogy of Fallot Which Regulates Congenital Heart Disease Genes Transcriptionally and Epigenetically. Front. Cell Dev. Biol. 2021, 9, 630634. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Liu, H.; Yin, J.; Zhang, M.; Yu, Z.; Miao, H. Circulating Plasma Circular RNAs as Novel Diagnostic Biomarkers for Congenital Heart Disease in Children. J. Clin. Lab. Anal. 2019, 33, e22998. [Google Scholar] [CrossRef] [PubMed]
- García-Flores, E.; Calderón-Colmenero, J.; Borgonio-Cuadra, V.M.; Sandoval, J.P.; García-Montes, J.A.; Cazarín-Santos, B.G.; Miranda-Duarte, A.; Gamboa-Domínguez, A.; Rodríguez-Pérez, J.M.; Pérez-Hernández, N. Epigenetic Evaluation of the TBX20 Gene and Environmental Risk Factors in Mexican Paediatric Patients with Congenital Septal Defects. Cells 2023, 12, 586. [Google Scholar] [CrossRef]
- Tournoy, T.K.; Moons, P.; Daelman, B.; De Backer, J. Biological Age in Congenital Heart Disease-Exploring the Ticking Clock. J. Cardiovasc. Dev. Dis. 2023, 10, 492. [Google Scholar] [CrossRef]
- Crimmins, E.M.; Klopack, E.T.; Kim, J.K. Generations of epigenetic clocks and their links to socioeconomic status in the Health and Retirement Study. Epigenomics 2024, 16, 1031–1042. [Google Scholar] [CrossRef]
- Nicolini, H.; Genis-Mendoza, A.D. Genómica Psiquiátrica: Los Nuevos Retos, 1st ed.; APM Ediciones y Convenciones en Psiquiartía: Mexico City, Mexico, 2023; pp. 109–126. [Google Scholar]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef]
- Jang, J.; Song, G.; Pettit, S.M.; Li, Q.; Song, X.; Cai, C.L.; Kaushal, S.; Li, D. Epicardial HDAC3 Promotes Myocardial Growth Through a Novel MicroRNA Pathway. Circ. Res. 2022, 131, 151–164. [Google Scholar] [CrossRef]
- Griazeva, E.D.; Fedoseeva, D.M.; Radion, E.I.; Ershov, P.V.; Meshkov, I.O.; Semyanihina, A.V.; Makarova, A.S.; Makarov, V.V.; Yudin, V.S.; Keskinov, A.A.; et al. Current Approaches to Epigenetic Therapy. Epigenomes 2023, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Lempiäinen, J.K.; Garcia, B.A. Characterizing crosstalk in epigenetic signaling to understand disease physiology. Biochem. J. 2023, 480, 57–85. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Wang, H.; Kim, H.R.; Wenderski, W.; Fang, F.; Li, C.H.; Dewell, S.; Hughes, S.H.; Melnick, A.M.; Patel, D.J.; et al. Engineering of a Histone-Recognition Domain in Dnmt3a Alters the Epigenetic Landscape and Phenotypic Features of Mouse ESCs. Mol. Cell 2015, 59, 89–103. [Google Scholar] [CrossRef]
- Anderson, K.M.; Anderson, D.M.; McAnally, J.R.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2016, 539, 433–436. [Google Scholar] [CrossRef]

{kind=link}
| Gestational Environmental Exposure | Relevance | Associated Pathology | Reference |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons (PAHs) | Exposure to PAHs can interfere with proper cardiovascular development in fetuses. The combination of chemical compounds in tobacco fumes, vehicle exhausts, and certain industries’ emissions can induce oxidative stress and inflammation in fetal tissues. Since PAHs are lipophilic, they can cross through cell membranes as well as the placenta. In the fetus, they form intermediate reagents that bind covalently to DNA. | Conotruncal defects, obstruction of right ventricle outflow tract, ASD, TOF | [25] |
| Air pollutants (CO2, SO2) | Exposure to air pollutants can induce an inflammatory response and increase oxidative stress, which might alter placental circulation, affect fetal oxygenation, and disturb normal heart development. Interference with the development of fetal blood vessels affects heart perfusion, altering gene expression in heart cells. | ASD, CoA, TOF, PDA, PS | [26] |
| Pesticides | Pesticides are associated with fetal heart development by acting as endocrine disruptors, generating oxidative stress, altering gene expression (VEGF and NOTCH), and inducing maternal inflammation. They can interfere with cell migration from the neural crest, favoring conotruncal defects. They can also alter the equilibrium between the proliferation and apoptosis of key cardiac cells, generating alterations in angiogenesis and gene expression. | TOF, HLH, PS, VSD, ASD | [27,28] |
| Drugs | |||
| Valproic acid | Valproic acid causes an alteration in the expression of genes associated with cell polarity (Vangl2, Scrib) and dysfunction in the activities of histone deacetylases (HDAC1/2/3), which might interfere with the proper formation and closure of the interventricular septum. | VSD | [29] |
| Fluoxetine | Exposure to fluoxetine may induce an alteration of serotonin regulation, which affects heart and blood vessel development in fetuses. This can interfere with placental circulation, alter gene expression in heart cells, and disturb normal heart development. | Subaortic stenosis, secundum ASD, muscular VSD, CoA | [30] |
| Citalopram | Citalopram can affect serotonin regulation, altering fetal blood vessel development. This might interfere with adequate fetal heart perfusion, altering normal development and increasing the risk of septal defects. | Subaortic stenosis, secundum ASD, muscular VSD, CoA | [30] |
| Venlafaxine | Venlafaxine could induce changes in the regulation of fetal blood vessels, affecting placental circulation and causing alterations in fetal cardiac perfusion. This could interfere with heart development and increase the risk of cardiovascular defects like ductal constriction. | Subaortic stenosis, secundum ASD, muscular VSD, CoA | [30] |
| Escitalopram | Like citalopram, escitalopram can interfere with serotonin regulation. This can alter blood vessel development and placental circulation, increasing the risk of heart defects. | Subaortic stenosis, secundum ASD, muscular VSD, CoA | [30] |
| Albuterol | Bronchodilators, like albuterol, are drugs that act on the beta-adrenergic receptors in the lungs to dilate the respiratory tract. Excessive or early bronchodilator use may alter this signaling processes and result in defects of key heart structures like valves or septums. | Truncus Arteriosus, interauricular secondary communication | [31] |
| Lithium | Lithium can affect cellular calcium homeostasis, which is crucial for the development of cardiac and vascular structures. It has been suggested that it can interfere with G-protein-dependent signaling. Furthermore, lithium inhibits inositol monophosphatase and inositol polyphosphate 1 phosphatase. This cycle is crucial in cell signaling and growth and development regulation, as well as interfering with Wnt/beta-catenin signaling. | Obstruction of right ventricle outflow tract (RVOTO) and EA | [32] |
| Others | |||
| Viral infection | Rubella and cytomegalovirus are known human teratogens that can cause birth defects, including cardiac malformations. These infections induce a maternal inflammatory response that affects development, altering the formation of the heart and other organs. Moreover, they can interfere with the formation of fetal blood vessels or affect placental circulation, which contribute to fetal cardiac insufficiency. The drugs used to treat these infections, like antibiotics and analgesics–antipyretics, can also have teratogenic effects. | Conotruncal defects, PDA, peripheral PS | [33,34,35] |
| Alcohol | Alcohol has been associated with histone hypoacetylation, affecting the expression of development-related genes. Furthermore, it alters retinoic acid biosynthesis and signaling, as well as Wnt and BMP signaling. | TOF, VSD, atrioventricular channel malformation, dextro-transposition of great arteries (RTGA) | [36] |
| Pathology | Involved miRNAs | Target Genes | References |
|---|---|---|---|
| TOF | miR-27b, miR-421, miR-1275, miR-122, miR-1201, miR-22, miR-222, miR-375, miR-138, miR-421, miR-1, miR-206, miR-940, hsa-miR-148a, hsa-miR-221-3p, hsa-miR-218-5p, hsa-miR-873-5p, miR-19, let-7e-5p, miR-10a-5p, miR-181c, miR-940, miR-181, miR-130, miR-146b-5p, miR-29c, miR-720, miR-424, miR-660, miR-708, miR-363, miR-337-5p, miR-155, miR-154 | SOX4, BCL2L11, TBX5, CDK9, FN1, MAPK1 | [11,119,125,126,127] |
| HLHS | miR-30, miR-100, miR-378, miR-99a, miR-145a, miR-208, miR-204 | QKI, FOG2, CDK6, SOX11, BAZ2A | [11,128] |
| ASD | miR-20b-5p, hsa-miR-19b, hsa-miR-375, hsa-miR-29c, miR-139-5p, miR-196-a2, miR-9, miR-30a, hsa-let-7a, hsa-let-7b, hsa-miR-486, miR-29, miR-143/145, miR-17-92, miR-106b-25, miR-503/424 | ACTC1, TBX5, PTEN, VEGFR-1 | [129,130,131,132] |
| VSD | miR-1/2, miR-1/1, miR-181c, miR-92, let-7e-5p, miR-155-5p, miR-222-3p, miR-379-5p, miR-409-3p, miR-433, miR-487b, miR-498 | GJA1, SOX9, BMPR2, | [11,120] |
| BAV | miR-26a, miR-95, miR-30b, miR-141 | BMP2, ALPL, SMAD1, SMAD3 | [120,133,134] |
| TGA | has-let-7e, miR-16, miR-25, miR-18a, miR-93, miR-106a, miR-451, miR-486-3p, miR-486-5p | ATM, PTEN, BCL11A, FOXO1, MMP19, IGF1, HAT1, SMAD1 | [11,135] |
| Down Syndrome | miR-99a, has-let-7c, miR-125b2, miR-155, miR-802 | IL10, NOX4, RUNX3, CYP24A1 | [120,125,136,137,138,139,140] |
| DiGeorge Syndrome | miR-23, miR-363, let-7g, miR-361-5p, miR-324-5p, miR-194, miR-720, miR-150, miR-15b-3p, miR-185 | SOX17, AFP, G3BP2 | [141,142,143,144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Pérez, J.M.; Ortega-Zhindón, D.B.; Villamil-Castañeda, C.; Lara-Ortiz, J.S.; Borgonio-Cuadra, V.M.; Cervantes-Salazar, J.L.; Calderón-Colmenero, J.; Escalante-Ruiz, Z.N.; Retama-Méndez, E.; Hernández-García, Y.C.; et al. Congenital Heart Diseases: Recent Insights into Epigenetic Mechanisms. Cells 2025, 14, 820. https://doi.org/10.3390/cells14110820
Rodríguez-Pérez JM, Ortega-Zhindón DB, Villamil-Castañeda C, Lara-Ortiz JS, Borgonio-Cuadra VM, Cervantes-Salazar JL, Calderón-Colmenero J, Escalante-Ruiz ZN, Retama-Méndez E, Hernández-García YC, et al. Congenital Heart Diseases: Recent Insights into Epigenetic Mechanisms. Cells. 2025; 14(11):820. https://doi.org/10.3390/cells14110820
Chicago/Turabian StyleRodríguez-Pérez, José Manuel, Diego B. Ortega-Zhindón, Clara Villamil-Castañeda, Javier Santiago Lara-Ortiz, Verónica Marusa Borgonio-Cuadra, Jorge L. Cervantes-Salazar, Juan Calderón-Colmenero, Zeomara Nathali Escalante-Ruiz, Eduardo Retama-Méndez, Yessica C. Hernández-García, and et al. 2025. "Congenital Heart Diseases: Recent Insights into Epigenetic Mechanisms" Cells 14, no. 11: 820. https://doi.org/10.3390/cells14110820
APA StyleRodríguez-Pérez, J. M., Ortega-Zhindón, D. B., Villamil-Castañeda, C., Lara-Ortiz, J. S., Borgonio-Cuadra, V. M., Cervantes-Salazar, J. L., Calderón-Colmenero, J., Escalante-Ruiz, Z. N., Retama-Méndez, E., Hernández-García, Y. C., & Pérez-Hernández, N. (2025). Congenital Heart Diseases: Recent Insights into Epigenetic Mechanisms. Cells, 14(11), 820. https://doi.org/10.3390/cells14110820