
Antioxidant Bioactive Agents for Neuroprotection Against Perinatal Brain Injury

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
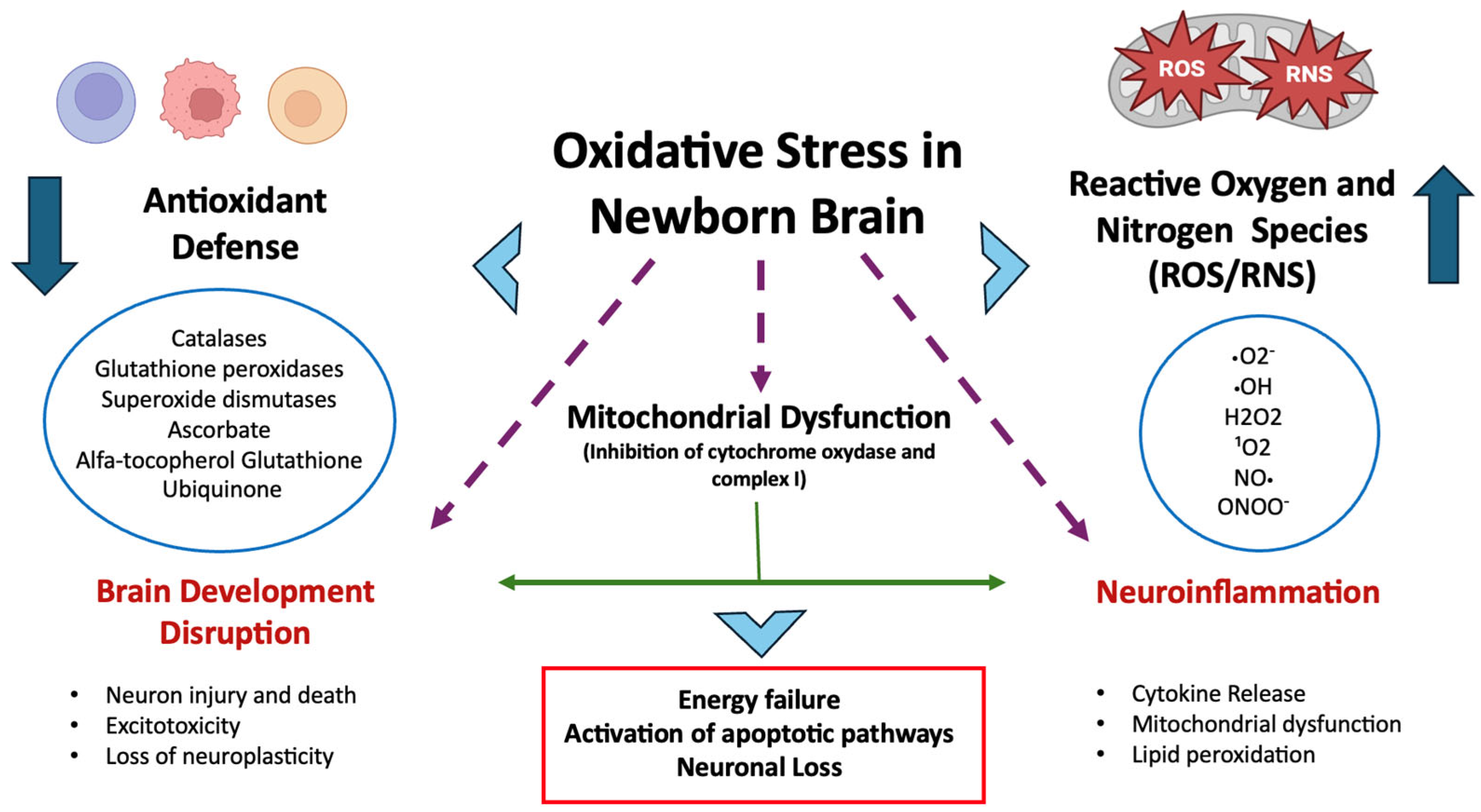
Oxidative Stress and Neonatal Brain Injury
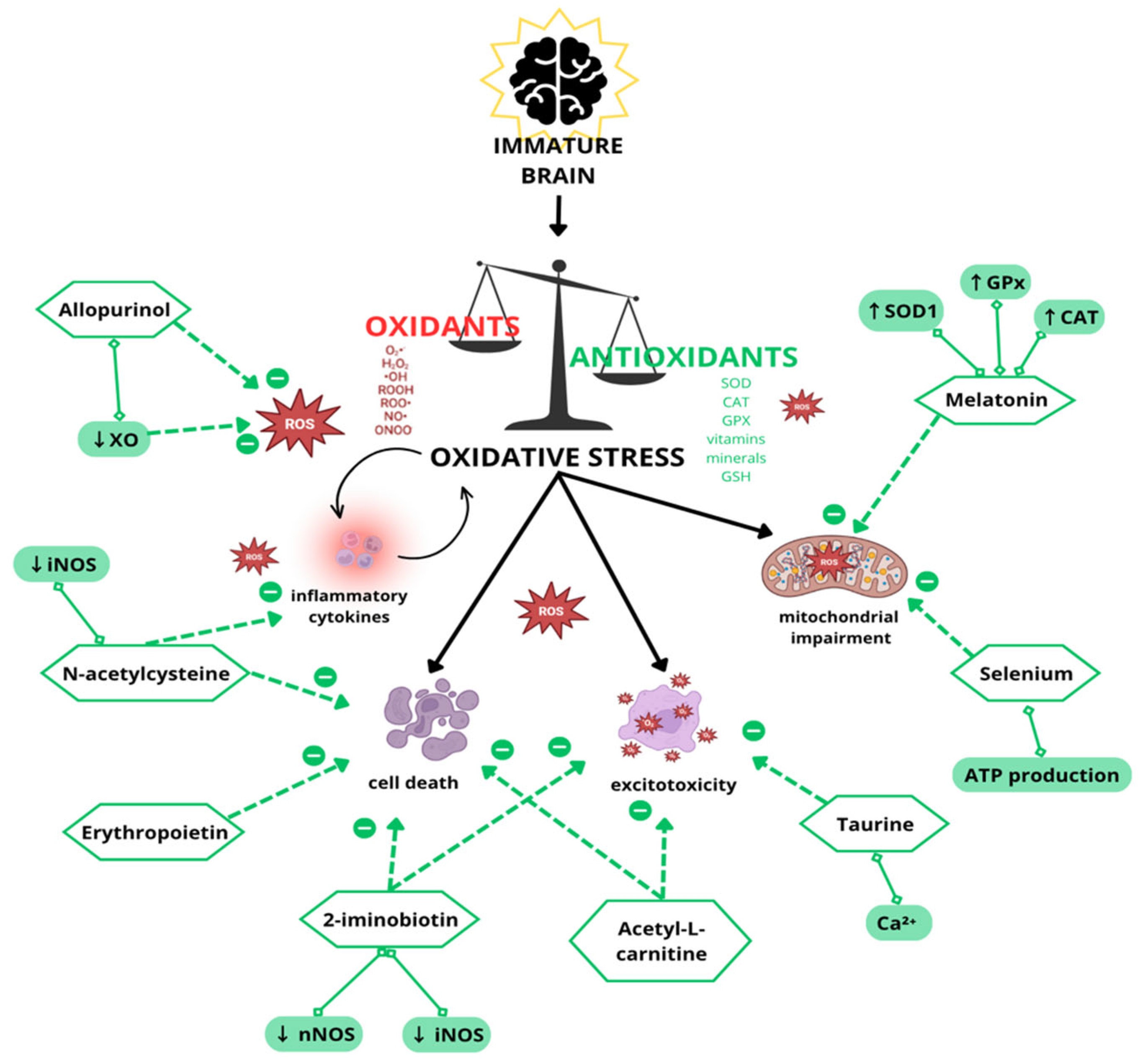
2. Antioxidant/Neuroprotective Strategies in Experimental and Clinical Studies
2.1. Erythropoietin
2.2. Melatonin
2.3. Allopurinol
2.4. N-Acetylcysteine
2.5. Acetyl-L-Carnitine
2.6. Selenium
2.7. Taurine
2.8. Iminobiotin
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [PubMed]
- Buonocore, G.; Perrone, S.; Tataranno, M.L. Oxygen toxicity: Chemistry and biology of reactive oxygen species. Semin. Fetal Neonatal Med. 2010, 15, 186–190. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Ostrakhovitch, E.A.; Semenikhin, O.A. The role of redox environment in neurogenic development. Arch. Biochem. Biophys. 2013, 534, 44–54. [Google Scholar] [CrossRef]
- Wu, F.; Tian, F.J.; Lin, Y.; Xu, W.M. Oxidative Stress: Placenta Function and Dysfunction. Am. J. Reprod. Immunol. 2016, 76, 258–271. [Google Scholar] [CrossRef]
- van den Tweel, E.R.; Nijboer, C.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; van Bel, F. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J. Neuroimmunol. 2005, 167, 64–71. [Google Scholar] [CrossRef]
- Buonocore, G.; Groenendaal, F. Anti-oxidant strategies. Semin. Fetal Neonatal Med. 2007, 12, 287–295. [Google Scholar] [CrossRef]
- Perrone, S.; Santacroce, A.; Longini, M.; Proietti, F.; Bazzini, F.; Buonocore, G. The Free Radical Diseases of Prematurity: From Cellular Mechanisms to Bedside. Oxidative Med. Cell. Longev. 2018, 2018, 7483062. [Google Scholar] [CrossRef]
- Tataranno, M.L.; Oei, J.L.; Perrone, S.; Wright, I.M.; Smyth, J.P.; Lui, K.; Tarnow-Mordi, W.O.; Longini, M.; Proietti, F.; Negro, S.; et al. Resuscitating preterm infants with 100% oxygen is associated with higher oxidative stress than room air. Acta Paediatr. 2015, 104, 759–765. [Google Scholar] [CrossRef]
- Dennery, P.A. Role of redox in fetal development and neonatal diseases. Antioxid. Redox Signal 2004, 6, 147–153. [Google Scholar] [CrossRef]
- Abad, C.; Chiarello, D.I.; Rojas, D.; Beretta, V.; Perrone, S.; Marín, R. Oxidative Stress in Preeclampsia and Preterm Newborn. In Biomarkers of Oxidative Stress; Andreescu, S., Henkel, R., Khelfi, A., Eds.; Springer: Cham, Switzerland, 2024. [Google Scholar]
- Perrone, S.; Tataranno, M.L.; Stazzoni, G.; Ramenghi, L.; Buonocore, G. Brain susceptibility to oxidative stress in the perinatal period. J. Matern. Neonatal Med. 2015, 28 (Suppl. S1), 2291–2295. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Abdull Razis, A.F.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative stress, free radicals and antioxidants: Potential crosstalk in the pathophysiology of human diseases. Front. Chem. 2023, 11, 1158198. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Longini, M.; Paffetti, P.; Vezzosi, P.; Gatti, M.G.; Bracci, R. Non protein bound iron as early predictive marker of neonatal brain damage. Brain 2003, 126 Pt 5, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, G.; Zani, S.; Perrone, S.; Caciotti, B.; Bracci, R. Intraerythrocyte nonprotein-bound iron and plasma malondialdehyde in the hypoxic newborn. Free Radic. Biol. Med. 1998, 25, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J. Polyunsaturated Fatty Acid Intake and Brain Health: Balance is the Key. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2022, 30, 774–776. [Google Scholar] [CrossRef]
- Sastry, P.S. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar]
- VanRollins, M.; Woltjer, R.L.; Yin, H.; Morrow, J.D.; Montine, T.J. F2-dihomo-isoprostanes arise from free radical attack on adrenic acid. J. Lipid Res. 2008, 49, 995–1005. [Google Scholar] [CrossRef]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Revuelta, M.; Urigüen, L.; Álvarez, A.; Montalvo, H.; Hilario, E. Pretreatment with Resveratrol Prevents Neuronal Injury and Cognitive Deficits Induced by Perinatal Hypoxia-Ischemia in Rats. PLoS ONE 2015, 10, e0142424. [Google Scholar] [CrossRef] [PubMed]
- Folkerth, R.D.; Haynes, R.L.; Borenstein, N.S.; Belliveau, R.A.; Trachtenberg, F.; Rosenberg, P.A.; Volpe, J.J.; Kinney, H.C. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J. Neuropathol. Exp. Neurol. 2004, 63, 990–999. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef]
- Menzel, A.; Samouda, H.; Dohet, F.; Loap, S.; Ellulu, M.S.; Bohn, T. Common and Novel Markers for Measuring Inflammation and Oxidative Stress Ex Vivo in Research and Clinical Practice-Which to Use Regarding Disease Outcomes? Antioxidants 2021, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.; Castellini, L.; Parladori, R.; Paoletti, V.; Aceti, A.; Corvaglia, L. Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives. Antioxidants 2021, 10, 2012. [Google Scholar] [CrossRef]
- Drury, P.P.; Bennet, L.; Gunn, A.J. Mechanisms of hypothermic neuroprotection. Semin. Fetal Neonatal Med. 2010, 15, 287–292. [Google Scholar] [CrossRef]
- Perrone, S.; Szabó, M.; Bellieni, C.V.; Longini, M.; Bangó, M.; Kelen, D.; Treszl, A.; Negro, S.; Tataranno, M.L.; Buonocore, G. Whole body hypothermia and oxidative stress in babies with hypoxic-ischemic brain injury. Pediatr. Neurol. 2010, 43, 236–240. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef]
- Vento, M. Oxygen supplementation in the neonatal period: Changing the paradigm. Neonatology 2014, 105, 323–331. [Google Scholar] [CrossRef]
- Perrone, S.; Manti, S.; Petrolini, C.; Dell’Orto, V.G.; Boscarino, G.; Ceccotti, C.; Bertini, M.; Buonocore, G.; Esposito, S.M.R.; Gitto, E. Oxygen for the Newborn: Friend or Foe? Children 2023, 10, 579. [Google Scholar] [CrossRef]
- Wyckoff, M.H.; Wyllie, J.; Aziz, K.; de Almeida, M.F.; Fabres, J.; Fawke, J.; Guinsburg, R.; Hosono, S.; Isayama, T.; Kapadia, V.S.; et al. Neonatal Life Support Collaborators Neonatal Life Support: 2020 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Circulation 2020, 142 (Suppl. S1), S185–S221. [Google Scholar] [CrossRef]
- Tataranno, M.L.; Perrone, S.; Longini, M.; Buonocore, G. New antioxidant drugs for neonatal brain injury. Oxidative Med. Cell. Longev. 2015, 2015, 108251. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, O.; Akar, M.; Uras, N.; Eras, Z.; Erdeve, O.; Oguz, S.S.; Dilmen, U. Total antioxidant capacity and total oxidant status in perinatal asphyxia in relation to neurological outcome. Neuropediatrics 2011, 42, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef] [PubMed]
- Traudt, C.M.; McPherson, R.J.; Bauer, L.A.; Richards, T.L.; Burbacher, T.M.; McAdams, R.M.; Juul, S.E. Concurrent erythropoietin and hypothermia treatment improve outcomes in a term nonhuman primate model of perinatal asphyxia. Dev. Neurosci. 2013, 35, 491–503. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Wang, Y.; Zhang, R.; Chopp, M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004, 35, 1732–1737. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Windsor, C.; Lee, B.S.; Arteaga Cabeza, O.; Ferriero, D.M. Erythropoietin Treatment Exacerbates Moderate Injury after Hypoxia-Ischemia in Neonatal Superoxide Dismutase Transgenic Mice. Dev. Neurosci. 2017, 39, 228–237. [Google Scholar] [CrossRef]
- Miller, S.L.; Yan, E.B.; Castillo-Meléndez, M.; Jenkin, G.; Walker, D.W. Melatonin provides neuroprotection in the late-gestation fetal sheep brain in response to umbilical cord occlusion. Dev. Neurosci. 2005, 27, 200–210. [Google Scholar] [CrossRef]
- Welin, A.K.; Svedin, P.; Lapatto, R.; Sultan, B.; Hagberg, H.; Gressens, P.; Kjellmer, I.; Mallard, C. Melatonin reduces inflammation and cell death in white matter in the mid-gestation fetal sheep following umbilical cord occlusion. Pediatr. Res. 2007, 61, 153–158. [Google Scholar] [CrossRef]
- Carloni, S.; Perrone, S.; Buonocore, G.; Longini, M.; Proietti, F.; Balduini, W. Melatonin protects from the long-term consequences of a neonatal hypoxic-ischemic brain injury in rats. J. Pineal Res. 2008, 44, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hutton, L.C.; Abbass, M.; Dickinson, H.; Ireland, Z.; Walker, D.W. Neuroprotective properties of melatonin in a model of birth asphyxia in the spiny mouse (Acomys cahirinus). Dev. Neurosci. 2009, 31, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; Ciccoli, L.; Leoncini, S.; Carloni, S.; Perrone, S.; Comporti, M.; Balduini, W.; Buonocore, G. Free iron, total F-isoprostanes and total F-neuroprostanes in a model of neonatal hypoxic-ischemic encephalopathy: Neuroprotective effect of melatonin. J. Pineal Res. 2009, 46, 148–154. [Google Scholar] [CrossRef]
- Lekic, T.; Manaenko, A.; Rolland, W.; Virbel, K.; Hartman, R.; Tang, J.; Zhang, J.H. Neuroprotection by melatonin after germinal matrix hemorrhage in neonatal rats. Acta Neurochir. Suppl. 2011, 111, 201–206. [Google Scholar] [PubMed]
- Balduini, W.; Carloni, S.; Perrone, S.; Bertrando, S.; Tataranno, M.L.; Negro, S.; Proietti, F.; Longini, M.; Buonocore, G. The use of melatonin in hypoxic-ischemic brain damage: An experimental study. J. Matern. Fetal Neonatal Med. 2012, 25 (Suppl. S1), 119–124. [Google Scholar] [CrossRef]
- Robertson, N.J.; Faulkner, S.; Fleiss, B.; Bainbridge, A.; Andorka, C.; Price, D.; Powell, E.; Lecky-Thompson, L.; Thei, L.; Chandrasekaran, M.; et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 2013, 136, 90–105. [Google Scholar] [CrossRef]
- Albertini, M.C.; Vanzolini, T.; Perrone, S.; Weiss, M.D.; Buonocore, G.; Dell’Orto, V.; Balduini, W.; Carloni, S. MiR-126 and miR-146a as Melatonin-Responsive Biomarkers for Neonatal Brain Ischemia. J. Mol. Neurosci. 2023, 73, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Vannucci, R.C.; Towfighi, J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr. Res. 1990, 27, 332–336. [Google Scholar] [CrossRef]
- Palmer, C.; Towfighi, J.; Roberts, R.L.; Heitjan, D.F. Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr. Res. 1993, 33, 405–411. [Google Scholar]
- Xu, S.; Waddell, J.; Zhu, W.; Shi, D.; Marshall, A.D.; McKenna, M.C.; Gullapalli, R.P. In vivo longitudinal proton magnetic resonance spectroscopy on neonatal hypoxic-ischemic rat brain injury: Neuroprotective effects of acetyl-L-carnitine. Magn. Reson. Med. 2015, 74, 1530–1542. [Google Scholar] [CrossRef]
- Mehta, S.L.; Kumari, S.; Mendelev, N.; Li, P.A. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012, 13, 79. [Google Scholar] [CrossRef]
- Chan, C.Y.; Sun, H.S.; Shah, S.M.; Agovic, M.S.; Friedman, E.; Banerjee, S.P. Modes of direct modulation by taurine of the glutamate NMDA receptor in rat cortex. Eur. J. Pharmacol. 2014, 728, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Groenendaal, F.; Kavelaars, A.; Hagberg, H.H.; van Bel, F.; Heijnen, C.J. Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent pathway. J. Cereb. Blood Flow. Metab. 2007, 27, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, S.T.; Ireland, Z.; Fan, X.; van der Wal, W.M.; Roes, K.C.; Colditz, P.B.; Peeters-Scholte, C.M. Short-term dose-response characteristics of 2-iminobiotin immediately postinsult in the neonatal piglet after hypoxia-ischemia. Stroke 2013, 44, 809–811. [Google Scholar] [CrossRef]
- Zhu, C.; Kang, W.; Xu, F.; Cheng, X.; Zhang, Z.; Jia, L.; Ji, L.; Guo, X.; Xiong, H.; Simbruner, G.; et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics 2009, 124, e218–e226. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Bauer, L.A.; Ballard, R.A.; Ferriero, D.M.; Glidden, D.V.; Mayock, D.E.; Chang, T.; Durand, D.J.; Song, D.; Bonifacio, S.L.; et al. Erythropoietin for neuroprotection in neonatal encephalopathy: Safety and pharmacokinetics. Pediatrics 2012, 130, 683–691. [Google Scholar] [CrossRef]
- Avasiloaiei, A.; Dimitriu, C.; Moscalu, M.; Paduraru, L.; Stamatin, M. High-dose phenobarbital or erythropoietin for the treatment of perinatal asphyxia in term newborns. Pediatr. Int. 2013, 55, 589–593. [Google Scholar] [CrossRef]
- El Shimi, M.S.; Awad, H.A.; Hassanein, S.M.; Gad, G.I.; Imam, S.S.; Shaaban, H.A.; El Maraghy, M.O. Single dose recombinant erythropoietin versus moderate hypothermia for neonatal hypoxic ischemic encephalopathy in low resource settings. J. Matern. Fetal Neonatal Med. 2014, 27, 1295–1300. [Google Scholar] [CrossRef]
- Rogers, E.E.; Bonifacio, S.L.; Glass, H.C.; Juul, S.E.; Chang, T.; Mayock, D.E.; Durand, D.J.; Song, D.; Barkovich, A.J.; Ballard, R.A.; et al. Erythropoietin and hypothermia for hypoxic-ischemic encephalopathy. Pediatr. Neurol. 2014, 51, 657–662. [Google Scholar] [CrossRef]
- Malla, R.R.; Asimi, R.; Teli, M.A.; Shaheen, F.; Bhat, M.A. Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: A randomized placebo-controlled trial. J. Perinatol. 2017, 37, 596–601. [Google Scholar] [CrossRef]
- Mulkey, S.B.; Ramakrishnaiah, R.H.; McKinstry, R.C.; Chang, T.; Mathur, A.M.; Mayock, D.E.; Van Meurs, K.P.; Schaefer, G.B.; Luo, C.; Bai, S.; et al. Erythropoietin and Brain Magnetic Resonance Imaging Findings in Hypoxic-Ischemic Encephalopathy: Volume of Acute Brain Injury and 1-Year Neurodevelopmental Outcome. J. Pediatr. 2017, 186, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.Y.; Wu, S.J.; Wang, Q.L.; Yang, L.H.; Ren, P.S.; Qiao, B.J.; Wang, Z.Y.; Li, J.H.; Gu, X.L.; Li, L.X. Effect of erythropoietin combined with hypothermia on serum tau protein levels and neurodevelopmental outcome in neonates with hypoxic-ischemic encephalopathy. Neural Regen. Res. 2017, 12, 1655–1663. [Google Scholar] [PubMed]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F.; et al. A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Aly, H.; Elmahdy, H.; El-Dib, M.; Rowisha, M.; Awny, M.; El-Gohary, T.; Elbatch, M.; Hamisa, M.; El-Mashad, A.R. Melatonin use for neuroprotection in perinatal asphyxia: A randomized controlled pilot study. J. Perinatol. 2015, 35, 186–191. [Google Scholar] [CrossRef]
- Marseglia, L.; Gitto, E.; Laschi, E.; Giordano, M.; Romeo, C.; Cannavò, L.; Toni, A.L.; Buonocore, G.; Perrone, S. Antioxidant Effect of Melatonin in Preterm Newborns. Oxidative Med. Cell. Longev. 2021, 2021, 6308255. [Google Scholar] [CrossRef]
- Russell, G.A.; Cooke, R.W. Randomised controlled trial of allopurinol prophylaxis in very preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 1995, 73, F27–F31. [Google Scholar] [CrossRef]
- Kaandorp, J.J.; Benders, M.J.; Schuit, E.; Rademaker, C.M.; Oudijk, M.A.; Porath, M.M.; Oetomo, S.B.; Wouters, M.G.; van Elburg, R.M.; Franssen, M.T.; et al. Maternal allopurinol administration during suspected fetal hypoxia: A novel neuroprotective intervention? A multicentre randomised placebo controlled trial. Arch. Dis. Child. Fetal Neonatal Ed. 2015, 100, F216–F223. [Google Scholar] [CrossRef]
- Chu, W.Y.; Annink, K.V.; Nijstad, A.L.; Maiwald, C.A.; Schroth, M.; Bakkali, L.E.; van Bel, F.; Benders, M.J.N.L.; van Weissenbruch, M.M.; Hagen, A.; et al. Pharmacokinetic/Pharmacodynamic Modelling of Allopurinol, its Active Metabolite Oxypurinol, and Biomarkers Hypoxanthine, Xanthine and Uric Acid in Hypoxic-Ischemic Encephalopathy Neonates. Clin. Pharmacokinet. 2022, 61, 321–333. [Google Scholar] [CrossRef]
- Wiest, D.B.; Jenkins, D.D. N-Acetylcysteine rapidly replenishes central nervous system glutathione measured via magnetic resonance spectroscopy in human neonates with hypoxic-ischemic encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 950–958. [Google Scholar]
- El-Mazary, A.A.; Abdel-Aziz, R.A.; Mahmoud, R.A.; El-Said, M.A.; Mohammed, N.R. Correlations between maternal and neonatal serum selenium levels in full term neonates with hypoxic ischemic encephalopathy. Ital. J. Pediatr. 2015, 41, 83. [Google Scholar] [CrossRef]
- Gücüyener, K.; Atalay, Y.; Aral, Y.Z.; Hasanoğlu, A.; Türkyilmaz, C.; Biberoglu, G. Excitatory amino acids and taurine levels in cerebrospinal fluid of hypoxic ischemic encephalopathy in newborn. Clin. Neurol. Neurosurg. 1999, 101, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Biselele, T.; Bambi, J.; Betukumesu, D.M.; Ndiyo, Y.; Tabu, G.; Kapinga, J.; Bola, V.; Makaya, P.; Tjabbes, H.; Vis, P.; et al. A Phase IIa Clinical Trial of 2-Iminobiotin for the Treatment of Birth Asphyxia in DR Congo, a Low-Income Country. Paediatr. Drugs 2020, 22, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.; Balsari, A.; Giallongo, T.; Ottolenghi, S.; Di Giulio, A.M.; Samaja, M.; Carelli, S. Erythropoietin as a Neuroprotective Molecule: An Overview of Its Therapeutic Potential in Neurodegenerative Diseases. ASN Neuro 2019, 11, 1759091419871420. [Google Scholar] [CrossRef]
- Perrone, S.; Lembo, C.; Gironi, F.; Petrolini, C.; Catalucci, T.; Corbo, G.; Buonocore, G.; Gitto, E.; Esposito, S.M.R. Erythropoietin as a Neuroprotective Drug for Newborn Infants: Ten Years after the First Use. Antioxidants 2022, 11, 652. [Google Scholar] [CrossRef]
- Ott, C.; Martens, H.; Hassouna, I.; Oliveira, B.; Erck, C.; Zafeiriou, M.-P.; Peteri, U.-K.; Hesse, D.; Gerhart, S.; Altas, B.; et al. Widespread Expression of Erythropoietin Receptor in Brain and Its Induction by Injury. Mol. Med. 2015, 21, 803–815. [Google Scholar] [CrossRef]
- Berger, H.R.; Brekke, E.; Widerøe, M.; Morken, T.S. Neuroprotective Treatments after Perinatal Hypoxic-Ischemic Brain Injury Evaluated with Magnetic Resonance Spectroscopy. Dev. Neurosci. 2017, 39, 36–48, Erratum in Dev. Neurosci. 2017, 39, 442. [Google Scholar] [CrossRef]
- Wu, Y.W.; Mathur, A.M.; Chang, T.; McKinstry, R.C.; Mulkey, S.B.; Mayock, D.E.; Van Meurs, K.P.; Rogers, E.E.; Gonzalez, F.F.; Comstock, B.A.; et al. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics 2016, 137, e20160191. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress. A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Furmaga-Jabłońska, W.; Januszewski, S.; Tarkowska, A. Melatonin: A Potential Candidate for the Treatment of Experimental and Clinical Perinatal Asphyxia. Molecules 2023, 28, 1105. [Google Scholar] [CrossRef]
- Fulia, F.; Gitto, E.; Cuzzocrea, S.; Reiter, R.J.; Dugo, L.; Gitto, P.; Barberi, S.; Cordaro, S.; Barberi, I. Increased levels of malondialdehyde and nitrite/nitrate in the blood of asphyxiated newborns: Reduction by melatonin. J. Pineal Res. 2001, 31, 343–349. [Google Scholar] [CrossRef]
- Weiss, M.D.; Carloni, S.; Vanzolini, T.; Coppari, S.; Balduini, W.; Buonocore, G.; Longini, M.; Perrone, S.; Sura, L.; Mohammadi, A.; et al. Human-rat integrated microRNAs profiling identified a new neonatal cerebral hypoxic-ischemic pathway melatonin-sensitive. J. Pineal Res. 2022, 73, e12818. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Álvarez, A.; Revuelta, M.; Santaolalla, F.; Urtasun, A.; Hilario, E. Role of Antioxidants in Neonatal Hypoxic-Ischemic Brain Injury: New Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 265. [Google Scholar] [CrossRef]
- Engel, C.; Rüdiger, M.; Benders, M.J.N.L.; van Bel, F.; Allegaert, K.; Naulaers, G.; Bassler, D.; Klebermaß-Schrehof, K.; Vento, M.; Vilan, A.; et al. Detailed statistical analysis plan for ALBINO: Effect of Allopurinol in addition to hypothermia for hypoxic-ischemic Brain Injury on Neurocognitive Outcome—A blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). Trials 2024, 25, 81. [Google Scholar]
- Liu, X.; Wang, L.; Cai, J.; Liu, K.; Liu, M.; Wang, H.; Zhang, H. N-acetylcysteine alleviates H2O2-induced damage via regulating the redox status of intracellular antioxidants in H9c2 cells. Int. J. Mol. Med. 2019, 43, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, S.A.; Terluk, M.R.; Kartha, R.V. N-acetylcysteine Pharmacology and Applications in Rare Diseases-Repurposing an Old Antioxidant. Antioxidants 2023, 12, 1316. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Cheung, P.Y.; Johnson, S.T.; Bigam, D.L.; Todd, K.G. Cerebral amino acid profiles after hypoxia-reoxygenation and N-acetylcysteine treatment in the newborn piglet. Neonatology 2010, 97, 195–203. [Google Scholar] [CrossRef]
- Kiuru, A.; Ahola, T.; Klenberg, L.; Tommiska, V.; Lano, A.; Kleemola, P.; Haavisto, A.; Fellman, V. Postnatal N-acetylcysteine does not provide neuroprotection in extremely low birth weight infants: A follow-up of a randomized controlled trial. Early Hum. Dev. 2019, 132, 13–17. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]
- Onofrj, M.; Ciccocioppo, F.; Varanese, S.; di Muzio, A.; Calvani, M.; Chiechio, S.; Osio, M.; Thomas, A. Acetyl-L-carnitine: From a biological curiosity to a drug for the peripheral nervous system and beyond. Expert. Rev. Neurother. 2013, 13, 925–936. [Google Scholar] [CrossRef]
- Tindell, R.; Tipple, T. Selenium: Implications for outcomes in extremely preterm infants. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2018, 38, 197–202. [Google Scholar] [CrossRef]
- Zhang, F.; Li, X.; Wei, Y. Selenium and Selenoproteins in Health. Biomolecules 2023, 13, 799. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Shanaida, M.; Lysiuk, R.; Antonyak, H.; Klishch, I.; Shanaida, V.; Peana, M. Selenium: An Antioxidant with a Critical Role in Anti-Aging. Molecules 2022, 27, 6613. [Google Scholar] [CrossRef]
- Algieri, C.; Oppedisano, F.; Trombetti, F.; Fabbri, M.; Palma, E.; Nesci, S. Selenite ameliorates the ATP hydrolysis of mitochondrial F1FO-ATPase by changing the redox state of thiol groups and impairs the ADP phosphorylation. Free Radic. Biol. Med. 2024, 210, 333–343. [Google Scholar] [CrossRef]
- Jakaria, M.; Azam, S.; Haque, M.E.; Jo, S.H.; Uddin, M.S.; Kim, I.S.; Choi, D.K. Taurine and its analogs in neurological disorders: Focus on therapeutic potential and molecular mechanisms. Redox Biol. 2019, 24, 101223. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, Z.; García-Serrano, A.M.; Duarte, J.M.N. Taurine Supplementation as a Neuroprotective Strategy upon Brain Dysfunction in Metabolic Syndrome and Diabetes. Nutrients 2022, 14, 1292. [Google Scholar] [CrossRef]
- Lima, L.; Obregón, F.; Roussó, T.; Quintal, M.; Benzo, Z.; Auladell, C. Content and concentration of taurine, hypotaurine, and zinc in the retina, the hippocampus, and the dentate gyrus of the rat at various postnatal days. Neurochem. Res. 2004, 29, 247–255. [Google Scholar] [CrossRef]
- Roldán, A.; Figueras-Aloy, J.; Deulofeu, R.; Jiménez, R. Glycine and other neurotransmitter amino acids in cerebrospinal fluid in perinatal asphyxia and neonatal hypoxic-ischaemic encephalopathy. Acta Paediatr. 1999, 88, 1137–1141. [Google Scholar] [CrossRef]
- Chen, W.Q.; Jin, H.; Nguyen, M.; Carr, J.; Lee, Y.J.; Hsu, C.C.; Faiman, M.D.; Schloss, J.V.; Wu, J.Y. Role of taurine in regulation of intracellular calcium level and neuroprotective function in cultured neurons. J. Neurosci. Res. 2001, 66, 612–619. [Google Scholar] [CrossRef]
- Wu, H.; Jin, Y.; Wei, J.; Jin, H.; Sha, D.; Wu, J.Y. Mode of action of taurine as a neuroprotector. Brain Res. 2005, 1038, 123–131. [Google Scholar] [CrossRef]
- Kulak, A.; Duarte, J.M.; Do, K.Q.; Gruetter, R. Neurochemical profile of the developing mouse cortex determined by in vivo 1H NMR spectroscopy at 14.1 T and the effect of recurrent anaesthesia. J. Neurochem. 2010, 115, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Santacroce, A.; Buonocore, G. 2-Iminobiotin for the treatment of perinatal asphyxia. Expert Opin. Orphan Drugs 2013, 1, 935–945. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Sup, J.; Green, B.G.; Grant, S.K. 2-Iminobiotin is an inhibitor of nitric oxide synthases. Biochem. Biophys. Res. Commun. 1994, 204, 962–968. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.; Stazzoni, G.; Buonocore, G. Oxidative stress and free radicals related diseases of the newborn. Adv. Biosci. Biotechnol. 2012, 3, 1043–1050. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, X.; Qiu, L.; Peeters-Scholte, C.; Hagberg, H.; Blomgren, K. Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor in neonatal rat cerebral hypoxia-ischaemia. J. Neurochem. 2004, 90, 462–471. [Google Scholar] [CrossRef]
- Favié, L.M.A.; Peeters-Scholte, C.M.P.C.D.; Bakker, A.; Tjabbes, H.; Egberts, T.C.G.; van Bel, F.; Rademaker, C.M.A.; Vis, P.; Groenendaal, F. Pharmacokinetics and short-term safety of the selective NOS inhibitor 2-iminobiotin in asphyxiated neonates treated with therapeutic hypothermia. Pediatr. Res. 2020, 87, 689–696. [Google Scholar] [CrossRef]
- Fernandes, L.F.; Bruch, G.E.; Massensini, A.R.; Frézard, F. Recent Advances in the Therapeutic and Diagnostic Use of Liposomes and Carbon Nanomaterials in Ischemic Stroke. Front. Neurosci. 2018, 12, 453. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, Y.A.; Kim, S.Y.; Su, L.; Liu, J.; Kannan, R.M.; Kannan, S. Systemic dendrimer-drug nanomedicines for long-term treatment of mild-moderate cerebral palsy in a rabbit model. J. Neuroinflamm. 2020, 17, 319. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Reference | Target Population | Intervention | Outcomes | Findings | |
|---|---|---|---|---|---|
| Erythropoietin (EPO) | Traudt et al., 2013 [37] | 35 Macaca nemestrina with UCO. GA: 168 ± 1 days. | Groups: (a) saline; (b) EPO only; (c) TH only; (d) TH + EPO; (e) ctrl. EPO: 3500 U/kg × 1 dose i.v., followed by 2500 U/kg × 3 doses (30 min, 24 h, 48 h, 7 d) or 1000 U/kg/day i.v. × 4 doses (30 min, 24 h, 48 h, 7 d). | Blood samples; Behavioral and motor assessment; MRI. | TH combined with 4 doses of EPO: ↓ risk of death or moderate-severe CP to 0%; Normal motor functions; Repeated 1000 U/kg i.v. EPO is a safe and effective dose. |
| Wang et al., 2004 [38] | 28 Male Wistar rats with MCAo. | rhEPO, i.p., 5000/10,000 units/kg daily for 7 days starting 24 h after MCAo. | VEGF and BDNF. Behavioral Tests. Infarct volume from 7 H&E-stained coronal sections. | rhEPO treatment: ↑ neurological outcome. ↑ VEGF that mediates rhEPO-induced angiogenesis. ↑BDNF levels. | |
| Sheldon et al., 2016 [39] | 10 transgenic mice overexpressing hSOD1-tg; 13 WT littermates (C57/Bl6) and 3 sham mice. PND9. | HI mouse model; Recombinant EPO (5 IU/g, R&D Systems) or vehicle (saline) was administered immediately, at 24 h, and 5 days after HI. | Histological analysis. | ↑ injury in SOD-tg mice than WT; No improvement from EPO treatment. | |
| Melatonin | Miller et al., 2005 [40] | 15 pregnant ewes with singleton fetuses; GA: 124–127 d. | 10 min UCO; Maternal melatonin administered i.v. (1 mg bolus + 1 mg/h for 2 h). | Detection of •OH; Fetal and maternal blood sampling for melatonin, blood gases, and PG; Histological analysis of OS and DNA damage. | Maternal melatonin infusion lead to ↓ •OH in the fetal brain; ↓ lipid peroxidation. |
| Welin et al., 2007 [41] | 15 pregnant Gotland sheep ewes. GA: 89–90 | Maternal aseptic surgery; Fetal administration of melatonin, i.v., 20 mg/kg/h for 6 h starting 10 min after reperfusion (n = 9); vehicle (n = 10). | FMAP and FHR; 8-Isoprostane; total and free thiol levels; Histological and neuropathological analysis. | Mel: ↓ 8-isoprostane levels at 6 h; ↓ microglia count and density in several brain regions. | |
| Carloni et al., 2008 [42] | 18 pregnant Sprague Dawley rats. PND7 | Groups: (n = 8): (a) HI-Mel 5 mg/kg-PRE; (b) HI-Mel 15 mg/kg-PRE; (c) HI-Mel 5 mg/kg × 3; (d) HI-Mel 15 mg/kg-POST; (e) HI group (n = 10) saline. Mel: diluted in saline 5% DMSO an i.p. injected. | Short- and long-term histological analysis and long-term behavioral assessment. | ↓ brain injury in a dose-dependent manner: 5 mg/kg pre-HI (−29%), 3 × 5 mg/kg (−45%), and 15 mg/kg pre-HI (−64%). | |
| Hutton et al., 2009 [43] | 31 pregnant spiny mice. GA: 29 days. | Groups: (a) saline + C-section (n = 4); (b) saline + birth asphyxia (n = 3); (c) Mel + C-section (n = 7); (d) Mel + birth asphyxia (n = 5). Subscapular implantation of osmotic minipump delivering Mel (0.1 mg/kg/day) or saline. | Mel concentrations; Immunohistochemical Staining and Analysis. | Mel treatment: ↓ asphyxia-related cell death in cortical gray matter and the corpus callosum. ↓ inflammatory cells in the cortical gray matter, dentate gyrus, and corpus callosum. | |
| Signorini et al., 2009 [44] | 9 neonatal Sprague Dawley rats. PND 7 | Groups: (a) HI; (b) HI + Mel; (c) sham controls. Mel: i.p., 15 mg/kg, single dose 30 min before HI (5% DMSO/saline). | Histological Assessment; Image analysis for intact brain volume; Biochemical Markers; Free iron levels. | HI-Mel: ↓ OS, F2-IsoPs, F4-NeuroPs, and DFO-chelatable free iron in the cerebral cortex; ↓ DHA oxidation; ↓lipid peroxidation. | |
| Lekic et al., 2011 [45] | 40 Timed pregnant Sprague Dawley rats. PND7 | Groups (n = 8): (a) sham-naïve; (b) needle-control; (c) GMH (collagenase-infusion); (d) GMH + 5 mg/kg melatonin i.p.; (e) GMH + 10 mg/kg melatonin i.p. | Cognitive function; sensorimotor ability; Cerebral, cardiac and splenic growths. | Systemic Mel treatment: ↓ long-term brain atrophy; near-normal levels of sensorimotor and cognitive function. | |
| Balduini et al., 2012 [46] | 56 Sprague Dawley rat pups. PND7 | Groups: (a) HI-Mel: melatonin i.p., 15 mg/kg, 5 min after HI (5% DMSO/saline). (b) V-HI: vehicle 5% DMSO in saline. | Lipid peroxidation; Western blot analyses; Immunohistochemistry. | HI-Mel: ↓ OS, IsoPs, NPs, and NFs after HI injury. ↓ inflammatory cell recruitment and glial cell activation. ↓ED1-positive cells. ↓ GFAP expression in the brain. | |
| Robertson et al., 2012 [47] | 17 male piglets. Age: >24 h. | Groups: (a) HT (n = 8); (b) TH + Mel (n = 9): melatonin i.v. 5 mg/kg over 6 h, starting 10 min after HI and repeated at 24 h. | Cerebral energetics and metabolite ratios; aEEG; Seizure activity; Histology and Biochemical markers; Plasma Mel levels. | TH + Mel: ↑ cerebral energy metabolism. ↑ ATP levels. ↓ cell death in several brain regions. ↓ microglial activation. ↓ proinflammatory markers. | |
| Albertini et al., 2023 [48] | 54 pup rats. PND7 | Groups (n = 18): (a) HI + Mel: melatonin, i.p. to 5 min after HI at the dose of 15 mg/kg; (b) CTRL: Sham-operated controls; (c) HI-injured animals. | Serum and brain samples; Quantitative Real-Time PCR. | Dysregulation of miR-126 and miR-146a in neonatal rats in the early phase of HI injury and restored effects after Mel treatment. | |
| Allopurinol | Palmer et al., 1990 [49] | 63 Wistar rat pups PND7 | Right hemisphere HI insult. Randomized to: (a) s.c. injection of allopurinol (0.2 mL); (b) s.c. injection normal saline (0.2 mL) | Water content; Gross neuropathology; Histopathology. | In allopurinol group: ↓ brain water content in the right hemisphere; ↓brain damage; |
| Palmer et al., 1993 [50] | 65 Wistar rat pups PND7 | Right hemisphere HI insult. Randomized to: (a) single s.c. injection of allopurinol 135 mg/kg; (b) equal volume s.c. injection (0.01 mL/g animal weight) of saline. | Morphologic analysis of the brain; Water content; Gross neuropathology; Histopathology. | In allopurinol group: ↓ Acute brain edema; ↓ HI long term brain damage. | |
| Acetyl-L-carnitine | Xu, et al., 2015 [51] | 12 Sprague Dawley rats. PND7 | Randomized to: (a) Control; (b) HI; (c) HI + ALCAR; 4 doses 100 mg/kg immedi-ately after HI, at 4 h, 24 h, and 48 h. | Lesion Characteristics on T2 -Weighted MRI; Metabolic Changes in Hippocampus and Cortex. | ↓ lactate (Lac) levels in the ipsilateral hippocampus of HI + ALCAR vs. HI rats at 24 h postinjury. |
| Selenium | Mehta et al., 2012 [52] | 43 male C57BL/6J mice. | Sodium selenite, i.p., 0.2 mg/kg for 7 days before ischemia; MCAO for 1 h, followed by 5 or 24 h reperfusion. | Infarct volume, neurodegeneration, oxidative DNA damage, and protein expression. | Selenium administration: ↓ ischemia-induced brain damage; limiting the activation of autophagy; ↓ levels of Beclin 1 and LC3-II. ↓ mitochondrial fragmentation. |
| Taurine | Chan et al., 2014 [53] | Male Sprague Dawley rats. Age: 4–6 weeks. | No intervention. | Field potential recordings; Neurochemistry; | Inhibition of the NMDA receptor complex by taurine. |
| Iminobiotin | Nijboer et al., 2007 [54] | Timed Wistar rats pups. | s.c. 2-IB (10 mg/kg) or vehicle (10 mL/kg) at 0, 12, and 24 h post-hypoxia (Rice-Vannucci); | Histology; Nitrite and Nitrate measurements; Western blot. | 2-IB prevent the increase in cytosolic cytochrome c and activation of caspase 3 only in females. |
| Bjorkman et al., 2013 [55] | HI insult in 47 term newborn piglets. | Groups: (a): vehicle; (b) 2-IB (0.1 mg/kg, 0.2 mg/kg, or 1.0 mg/kg) | Tissue analyzed for caspase-3 activity, tyrosine nitration, and histology. | Greater survival with a normal aEEG at 48 h and ↓ tyrosine nitration in 2-IB treated group. |
| Reference | Target Population | Intervention | Outcomes | Findings | |
|---|---|---|---|---|---|
| Erythropoietin (EPO) | Zhu et al., 2009 [56] | 167 term infants with perinatal HIE. GA: 37 weeks. | Groups: (a) EPO (n = 83); (b) CTRL (n = 84). rhEPO, 300/500 U/kg: 1st dose s.c., then i.v. for 2 weeks. | Blood and CSF sampled pre-dose and at 3 h, 8 h, and 24 h after. Daily neuro exam × 7 days. Follow-up every 6 m until 18 m. | EPO: ↑ neurologic outcomes, for patients with moderate but not severe HIE. |
| Wu et al., 2012 [57] | 24 newborns with HIE. GA: ≥36 weeks | 6 EPO doses i.v.: 250 (n = 3), 500 (n = 6), 1000 (n = 7), 2500 (n = 8) U/kg. Standard HT. | Pharmacokinetic Analysis in plasma and CSF. Laboratory data at 1, 3, 5, and 14 days. | EPO 1000 U/kg per dose intravenously given in conjunction with hypothermia is well tolerated. | |
| Avasiloaiei et al., 2013 [58] | 67 term neonates with PA. | Randomized to: (a) supportive care; (b) PB: 40 mg/kg i.v. within 4 h of birth + supportive treatment; (c) EPO: 1000 UI/kg/day s.c. for 3 days + supportive treatment. | Total antioxidant TAS and MDA. Samples at 4, 24, 48, 72 h, 7 d. Neuro exam at birth and periodically, aEEG. | ↑ TAS in EPO; ↓ MDA in EPO; ↓ incidence of sequelae in PB and EPO. | |
| El Shimi et al., 2014 [59] | 45 full-term neonates. GA: ≥36 weeks. | Randomized: (a) no- intervention (n = 10); (b) HT (n = 10); (c) rEPO (n = 10; 1500 U/kg s.c. on day 1); (d) CTRL (n = 15 healthy, GA/sex-matched). | Brain MRI at 21–28 days. Neuro exam at 3 m + short-term neurodevelopmental outcome. | ↑ survival in whole body HT; Better MRI and NMS scores at 3 mo in HT vs. rEPO. | |
| Rogers et al., 2014 [60] | 24 newborns with HIE. GA: ≥37 weeks. | EPO doses i.v.: 250 (n = 3), 500 (n = 6), 1000 (n = 7), 2500 U/kg (n = 8). Standard 72 h HT (whole body: n = 21, head: n = 3). | MRI after HT. Neurodevelopmental follow up at 24 months. | ↓ rate of death or moderate to severe disability. High-dose Epo also appears to be safe. | |
| Malla et al., 2017 [61] | 100 neonates with HIE. GA: ≥37 weeks. | Randomized: (a) EPO: 5 doses EPO 500 U/kg i.v. every 48 h starting within 6 h of birth; (b) Placebo: 2 mL saline. | Clinical and neurodevelopmental outcomes at 19 m. | ↓ death or mod-severe encephalopathy. ↓NO, ↑antioxidants, ↓ glutamate toxicity, ↓ lipid peroxidation, ↓ inflammation. | |
| Mulkey et al., 2017 [62] | 50 term newborns with moderate to severe HIE. GA: ≥37 weeks. | Randomized: (a) EPO: EPO 1000 U/kg; (b) placebo: saline. Standard HT. | Brain MRI post-hypothermia. Development and motor assessment at 12 months. | ↓ acute brain injury volume in EPO vs. placebo. ↑ injury volume linked to worse 12-mo outcomes in placebo. | |
| Lv et al., 2017 [63] | 41 neonates with moderate/severe HIE. GA: mean 39 weeks | Randomized: (a): Control (HT alone, n = 20); (b): EPO (rhEPO + HT, n = 21) rhEPO 200 U/kg i.v., in 10% glucose, once daily × 10 days. | Serum tau protein measurement. Neonatal behavioral neurological assessment. Neurodevelopmental outcomes. | ↓ Serum tau protein at 8 and 12 days in EPO vs. Control. ↑ NBNA at 7, 14, and 28 days in EPO vs. Control. | |
| Juul et al., 2020 [64] | 941 preterm infants. GA: >24 + 0 and <27 + 6 | Randomized: (a) EPO 1000 U/kg IV q48h × 6 doses + 400 U/kg SC 3×/week until 32 + 6 weeks PMA (n = 477); (b) placebo (n = 464). | Head ultrasound on days 7–9 and at 37 weeks PMA. Outcome at 22–26 mo PMA. MRI at 36 PMA. | No reduction in death risk or improved neurodevelopment with high-dose EPO in extremely preterm infants vs. placebo. | |
| Melatonin | Aly et al., 2015 [65] | 45 newborns. GA: 38 to 42 weeks. | Randomized: (a) HIE − HT (n = 15); (b) Mel + HT (n = 15), 5 daily oral doses of Mel 10 mg kg−1; (c): Healthy control (n = 15). | Laboratory evaluations; EEG; MRI; Neurologic and developmental outcomes; | ↓ SOD and NO from baseline to follow-up in Mel + HT. ↓ seizure activity in Mel + HT. ↑ survival without abnormalities at 6 months in Mel + HT. |
| Marseglia et al., 2021 [66] | 36 preterm newborns GA: <37 weeks. | Randomized: (a): MEL group, 0.5 mg/kg/day orally, first week (n = 21); (b) placebo, 0.5 mL 5% glucose (n = 15). | Melatonin and OS biomarker (AOPP, F2-IsoPs, and NPBI) concentrations. | ↓ F2-Isopr at 48 h in MEL vs. placebo. No significant effect on NPBI or AOPP at 24/48 h. | |
| Allopurinol | Russell et al., 1995 [67] | 400 babies GA: 24–32 weeks. | Randomized: (a) allopurinol (20 mg/kg/day via gastric tube × 7 days); (b) placebo. | Adverse outcome; Biochemical Analysis. | Treated patients: Inhibition of xanthine oxidase; ↓ significant in uric acid; ↑ hypoxanthine; |
| Kaandorp et al., 2015 [68] | 222 pregnant women. GA: ≥36 weeks | Randomized: (a) Single antenatal i.v. dose of 500 mg allopurinol; (b) antenatal i.v. placebo. | Biochemical markers in cord blood; Neonatal outcome; | ↓ S100β and neuroketal values in the newborn female of treatment group. | |
| Chu et al., 2022 [69] | 46 term and near-term infants with severe PA from the ALBINO study. GA: ≥36 weeks. | Allopurinol i.v. 20 mg/kg first dose to all; second dose of i.v. 20 mg/kg in 13 and 10 mg/kg in other 13; TH (n = 13) | PK/PD modeling. | In the final PK/PD model, the combined allopurinol and oxypurinol concentration at the half maximal XO inhibition was 0.36 mg/L (95% CI 0.31–0.42). | |
| N-Acetylcysteine | Moss et al., 2018 [70] | 24 term neonates with HIE after HT. age: 5–6 day. | Neonates received daily i.v. infusions of NAC (25–40 mg/kg every 12 h) and calcitriol (0.03–0.1 mg/kg/day) from 6 h of life until day 10 or discharge. | Magnetic resonance imaging (MRI); Magnetic resonance spectroscopy (MRS); Quantification of GSH. | ↑ GSH in basal ganglia within 12–30 min after NAC infusion; No difference in GSH changes between the NAC only (n = 9) and NAC plus calcitriol (n = 14) groups. |
| Selenium | El-Mazary et al., 2015 [71] | 60 full term neonates with HIE; 20 healthy term. GA: ≥37 weeks. | No intervention. | Serum selenium levels, clinical chemistry values for early assessment of neonatal status. | ↓ serum selenium levels in HIE vs. Controls; Selenium levels showed a negative correlation with HIE severity based on the Sarnat and Sarnat staging system. |
| Taurine | Gücüyener et al., 1999 [72] | 22 infants (10 term, 12 preterm) with PA; 10 Ctrl. GA: 30–39 weeks. | No intervention. | Measurement of amino acids in CSF using high-performance liquid chromatography. | ↑ ASP, GLU, and TAU levels in CSF of PA vs. Ctrl. |
| Iminobiotin | Biselele et al., 2020 [73] | 7 near-term neonates treated with HIE. GA: ≥36 weeks. | 6 i.v. infusions of 2-IB (0.16 mg/kg) every 4 h via umbilical catheter (0.75 mg/mL solution); first dose within 6 h after birth. | Pharmacokinetic analysis. | No adverse effects that could be attributed to the use of 2-IB. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beretta, V.; Scarpa, E.; Carloni, S.; Petrolini, C.; Dell’Orto, V.; Ravenda, S.; Perrone, S. Antioxidant Bioactive Agents for Neuroprotection Against Perinatal Brain Injury. Cells 2025, 14, 818. https://doi.org/10.3390/cells14110818
Beretta V, Scarpa E, Carloni S, Petrolini C, Dell’Orto V, Ravenda S, Perrone S. Antioxidant Bioactive Agents for Neuroprotection Against Perinatal Brain Injury. Cells. 2025; 14(11):818. https://doi.org/10.3390/cells14110818
Chicago/Turabian StyleBeretta, Virginia, Elena Scarpa, Silvia Carloni, Chiara Petrolini, Valentina Dell’Orto, Sebastiano Ravenda, and Serafina Perrone. 2025. "Antioxidant Bioactive Agents for Neuroprotection Against Perinatal Brain Injury" Cells 14, no. 11: 818. https://doi.org/10.3390/cells14110818
APA StyleBeretta, V., Scarpa, E., Carloni, S., Petrolini, C., Dell’Orto, V., Ravenda, S., & Perrone, S. (2025). Antioxidant Bioactive Agents for Neuroprotection Against Perinatal Brain Injury. Cells, 14(11), 818. https://doi.org/10.3390/cells14110818