
The Development and Characterisation of A Porcine Large Intestinal Biological Scaffold by Perfusion Decellularisation

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Intestinal Retrieval and Bench Preparation
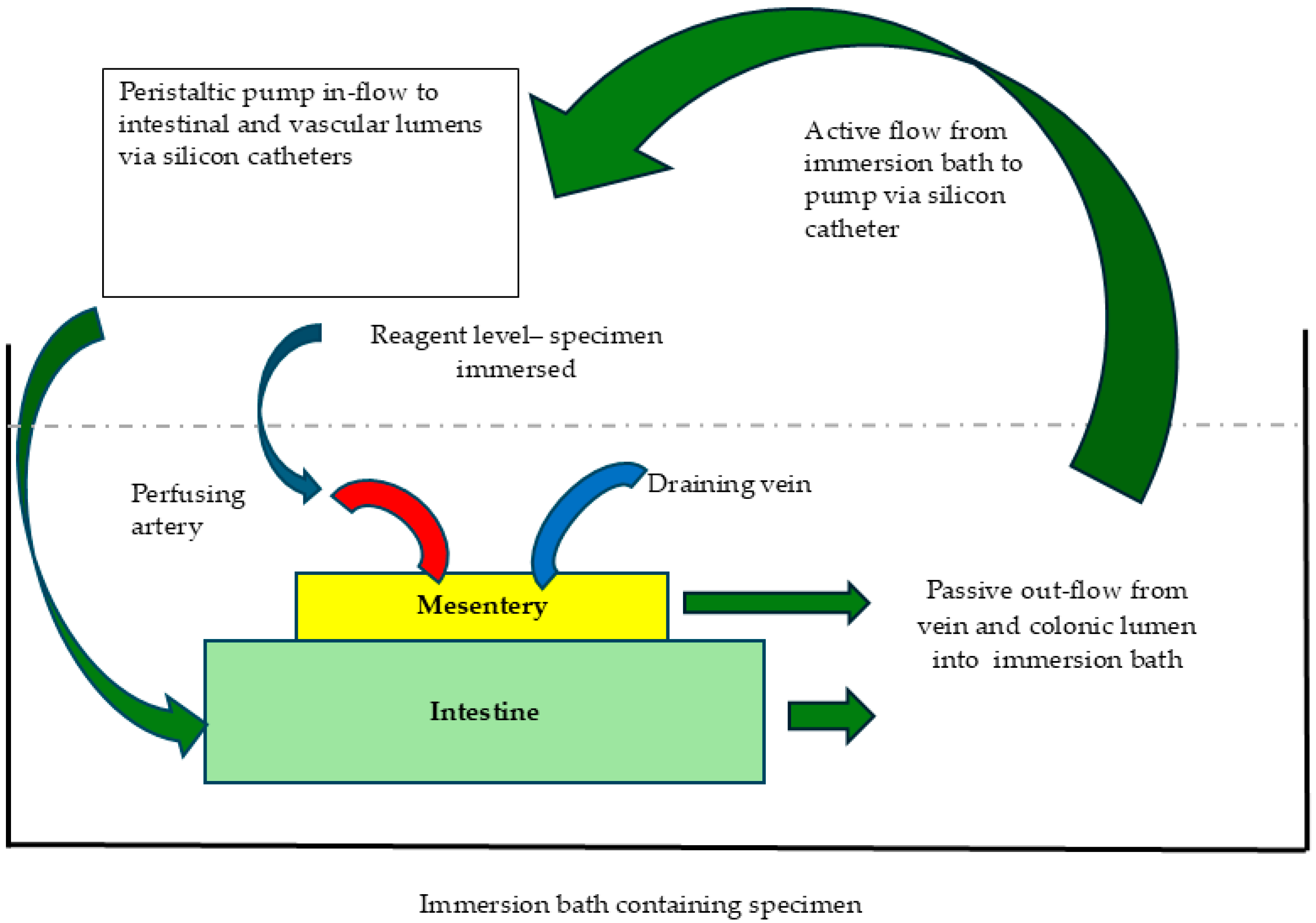
2.2. Perfusion Decellularisation
2.3. Histological Analysis
2.4. Immunohistochemical Analysis
2.5. Molecular Analysis
2.5.1. DNA Quantification
2.5.2. Glycosaminoglycan (GAG) Quantification
2.5.3. Transmission Electron Microscopy (TEM)
2.6. In Vivo Biocompatibility
2.7. Perfusion Studies
2.7.1. Intraluminal Patency
2.7.2. Vascular Dye Injection Studies
2.7.3. Vascular Transplantation
2.7.4. Computerised Tomographic (CT) Angiography
3. Results
3.1. Specimen Retrieval and Assessment of Decellularisation
3.2. Assessment of ECM Architecture
3.3. Scaffold Immunogenicity and Biocompatibility
3.4. Vascular Transplantation and CT Angiographic Evaluation
3.5. Perfusion Studies and Vascular Integrity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DNA | Deoxyribose Nucleic Acid |
| ITE | Intestinal Tissue Engineering |
| TELI | Tissue engineered large intestine |
| ECM | Extracellular Matrix |
| H&E | Haematoxylin and Eoisin |
| TEM | Transmission Electron Microscopy |
| CTA | computerised tomographic angiography |
| SDS | sodium dodecylsulfate |
| PME | Picro-sirius red with Millers elastin |
| NBF | Neutral buffered formalin |
| GAG | glycosaminoglycan |
| APTS | Aminopropyltriethoxysilane |
| MHC | Major Histocompatibility Complex |
References
- Tullie, L.; Jones, B.C.; De Coppi, P.; Li, V.S.W. Building gut from scratch—progress and update of intestinal tissue engineering. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.A.; Reuther, M.S.; Briggs, K.K.; Schumacher, B.L.; Williams, G.M.; Corr, M.; Sah, R.L.; Watson, D. In vivo implantation of tissue engineered human nasal septal neocartilage constructs: A pilot study. Otolaryngol. Head Neck Surg. 2012, 146, 46. [Google Scholar] [CrossRef] [PubMed]
- Olausson, M.; Patil, P.B.; Kuna, V.K.; Chougule, P.; Hernandez, N.; Methe, K.; Kullberg-Lindh, C.; Borg, H.; Ejnell, H.; Sumitran-Holgersson, S. Transplantation of an allogeneic vein bioengineered with autologous stem cells: A proof-of-concept study. Lancet 2012, 380, 230–237. [Google Scholar] [CrossRef]
- Elliott, M.J.; De Coppi, P.; Speggiorin, S.; Roebuck, D.; Butler, C.R.; Samuel, E.; Crowley, C.; McLaren, C.; Fierens, A.; Vondrys, D.; et al. Stem-cell-based, tissue engineered tracheal replacement in a child: A 2-year follow-up study. Lancet 2012, 380, 994–1000. [Google Scholar] [CrossRef]
- Esdaille, C.J.; Washington, K.S.; Laurencin, C.T. Regenerative engineering: A review of recent advances and future directions. Regen. Med. 2021, 16, 495. [Google Scholar] [CrossRef]
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e3. [Google Scholar] [CrossRef]
- Qi, D.; Shi, W.; Black, A.R.; Kuss, M.A.; Pang, X.; He, Y.; Liu, B.; Duan, B. Repair and regeneration of small intestine: A review of current engineering approaches. Biomaterials 2020, 240, 119832. [Google Scholar] [CrossRef] [PubMed]
- Collier, C.A.; Mendiondo, C.; Raghavan, S. Tissue engineering of the gastrointestinal tract: The historic path to translation. J. Biol. Eng. 2022, 16, 9. [Google Scholar] [CrossRef]
- Gupta, A.; Dixit, A.; Sales, K.M.; Winslet, M.C.; Seifalian, A.M. Tissue engineering of small intestine—Current status. Biomacromolecules 2006, 7, 2701–2709. [Google Scholar] [CrossRef]
- Meran, L.; Tullie, L.; Eaton, S.; De Coppi, P.; Li, V.S.W. Bioengineering human intestinal mucosal grafts using patient-derived organoids, fibroblasts and scaffolds. Nat. Protoc. 2023, 18, 108–135. [Google Scholar] [CrossRef]
- Kitano, K.; Schwartz, D.M.; Zhou, H.; Gilpin, S.E.; Wojtkiewicz, G.R.; Ren, X.; Sommer, C.A.; Capilla, A.V.; Mathisen, D.J.; Goldstein, A.M.; et al. Bioengineering of functional human induced pluripotent stem cell-derived intestinal grafts. Nat. Commun. 2017, 8, 765. [Google Scholar] [CrossRef] [PubMed]
- Bitar, K.N.; Raghavan, S. Intestinal Tissue Engineering: Current Concepts and Future Vision of Regenerative Medicine in the Gut. Neurogastroenterol. Motil. 2012, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Roh, T.T.; Chen, Y.; Paul, H.T.; Guo, C.; Kaplan, D.L. 3D Bioengineered Tissue Model of the Large Intestine to Study Inflammatory Bowel Disease. Biomaterials 2019, 225, 119517. [Google Scholar] [CrossRef]
- Grikscheit, T.C.; Ochoa, E.R.; Ramsanahie, A.; Alsberg, E.; Mooney, D.; Whang, E.E.; Vacanti, J.P. Tissue-Engineered Large Intestine Resembles Native Colon With Appropriate In Vitro Physiology and Architecture. Ann. Surg. 2003, 238, 35–41. [Google Scholar] [CrossRef]
- Ansari, T.; Southgate, A.; Obiri-Yeboa, I.; Jones, L.G.; Greco, K.; Olayanju, A.; Mbundi, L.; Somasundaram, M.; Davidson, B.; Sibbons, P.D. Development and Characterization of a Porcine Liver Scaffold. Stem Cells Dev. 2020, 29, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Guyette, J.P.; Gilpin, S.E.; Gonzalez, G.; Vacanti, J.P.; Ott, H.C. Regeneration and Experimental Orthotopic Transplantation of a Bioengineered Kidney. Nat. Med. 2013, 19, 646–651. [Google Scholar] [CrossRef]
- Ott, H.C.; Clippinger, B.; Conrad, C.; Schuetz, C.; Pomerantseva, I.; Ikonomou, L.; Kotton, D.; Vacanti, J.P. Regeneration and orthotopic transplantation of a bioartificial lung. Nat. Med. 2010, 16, 927–933. [Google Scholar] [CrossRef]
- Ott, H.C.; Matthiesen, T.S.; Goh, S.-K.; Black, L.D.; Kren, S.M.; Netoff, T.I.; Taylor, D.A. Perfusion-decellularized matrix: Using nature’s platform to engineer a bioartificial heart. Nat. Med. Vol. 2008, 14, 213–221. [Google Scholar] [CrossRef]
- Urciuolo, A.; De Coppi, P. Decellularized Tissue for Muscle Regeneration. Int. J. Mol. Sci. 2018, 19, 2392. [Google Scholar] [CrossRef]
- Moffat, D.; Ye, K.; Jin, S. Decellularization for the retention of tissue niches. J. Tissue Eng. 2022, 13, 20417314221101151. [Google Scholar] [CrossRef]
- Badylak, S.F.; Taylor, D.; Uygun, K. Whole-Organ Tissue Engineering: Decellularization and Recellularization of Three-Dimensional Matrix Scaffolds. Annu. Rev. Biomed. Eng. 2011, 13, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Speer, A.L.; Ren, X.; McNeill, E.P.; Aziz, J.M.; Muir, S.M.; Marino, D.I.; Dadhich, P.; Sawant, K.; Ciccocioppo, R.; Asthana, A.; et al. Bioengineering of the digestive tract: Approaching the clinic. Cytotherapy 2021, 23, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Korossis, S.; Iop, L. Editorial: Tissue and organ decellularization strategies in regenerative medicine; recent advances, current translational challenges, and future directions. Front. Bioeng. Biotechnol. 2023, 11, 1201041. [Google Scholar] [CrossRef] [PubMed]
- Nowocin, A.K.; Southgate, A.; Shurey, S.; Sibbons, P.; Gabe, S.M.; Ansari, T. The development and implantation of a biologically derived allograft scaffold. J. Tissue Eng. Regen. Med. 2016, 10, 140–148. [Google Scholar] [CrossRef]
- Lozanovski, V.J.; Döhler, B.; Weiss, K.H.; Mehrabi, A.; Süsal, C. The Differential Influence of Cold Ischemia Time on Outcome After Liver Transplantation for Different Indications-Who Is at Risk? A Collaborative Transplant Study Report. Front. Immunol. 2020, 11, 892. [Google Scholar] [CrossRef]
- Totsuka, E.; Fung, J.J.; Lee, M.C.; Ishii, T.; Umehara, M.; Makino, Y.; Chang, T.H.; Toyoki, Y.; Narumi, S.; Hakamada, K.; et al. Influence of Cold Ischemia Time and Graft Transport Distance on Postoperative Outcome in Human Liver Transplantation. Surg. Today 2002, 32, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, C.E.; Montgomery, R.A.; Hawxby, A.M.; Locke, J.E.; Gentry, S.E.; Warren, D.S.; Segev, D.L. Cold ischemia time and allograft outcomes in live donor renal transplantation: Is live donor organ transport feasible? Am. J. Transplant. 2007, 7, 99–107. [Google Scholar] [CrossRef]
- Wang, X.; Cui, J.; Zhang, B.Q.; Zhang, H.; Bi, Y.; Kang, Q.; Wang, N.; Bie, P.; Yang, Z.; Wang, H.; et al. Decellularized liver scaffolds effectively support the proliferation and differentiation of mouse fetal hepatic progenitors. J. Biomed. Mater. Res. A 2014, 102, 1017. [Google Scholar] [CrossRef]
- Macchiarini, P.; Jungebluth, P.; Go, T.; Asnaghi, M.A.; Rees, L.E.; Cogan, T.A.; Dodson, A.; Martorell, J.; Bellini, S.; Parnigotto, P.P.; et al. Clinical transplantation of a tissue-engineered airway. Lancet 2008, 372, 2023–2030. [Google Scholar] [CrossRef]
- Guyette, J.P.; Gilpin, S.E.; Charest, J.M.; Tapias, L.F.; Ren, X.; Ott, H.C. Perfusion decellularization of whole organs. Nat. Protoc. 2014, 9, 1451–1468. [Google Scholar] [CrossRef]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233. [Google Scholar] [CrossRef] [PubMed]
- Narciso, M.; Ulldemolins, A.; Júnior, C.; Otero, J.; Navajas, D.; Farré, R.; Gavara, N.; Almendros, I. Novel Decellularization Method for Tissue Slices. Front. Bioeng. Biotechnol. 2022, 10, 832178. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Griffiths, L.G. Immunogenicity in xenogeneic scaffold generation: Antigen removal versus decellularization. Acta Biomater. 2014, 10, 1806. [Google Scholar] [CrossRef]
- Gerecht-Nir, S.; Radisic, M.; Park, H.; Cannizzaro, C.; Boublik, J.; Langer, R.; Vunjak-Novakovic, G. Biophysical regulation during cardiac development and application to tissue engineering. Int. J. Dev. Biol. 2003, 50, 233–243. [Google Scholar] [CrossRef]
- Casarin, M.; Fortunato, T.M.; Imran, S.; Todesco, M.; Sandrin, D.; Borile, G.; Toniolo, I.; Marchesan, M.; Gerosa, G.; Bagno, A.; et al. Porcine Small Intestinal Submucosa (SIS) as a Suitable Scaffold for the Creation of a Tissue-Engineered Urinary Conduit: Decellularization, Biomechanical and Biocompatibility Characterization Using New Approaches. Int. J. Mol. Sci. 2022, 23, 2826. [Google Scholar] [CrossRef]
- Totonelli, G.; Maghsoudlou, P.; Garriboli, M.; Riegler, J.; Orlando, G.; Burns, A.J.; Sebire, N.J.; Smith, V.V.; Fishman, J.M.; Ghionzoli, M.; et al. A rat decellularized small bowel scaffold that preserves villus-crypt architecture for intestinal regeneration. Biomaterials 2012, 33, 3401. [Google Scholar] [CrossRef] [PubMed]
- Williams, D. Revisiting the definition of biocompatibility. Med. Device Technol. 2003, 14, 10–13. [Google Scholar]
- Spurrier, R.G.; Grikscheit, T.C. Tissue engineering the small intestine. Clin. Gastroenterol. Hepatol. 2013, 11, 354–358. [Google Scholar] [CrossRef]
- Guo, Y.; Wu, C.; Xu, L.; Xu, Y.; Xiaohong, L.; Hui, Z.; Jingjing, L.; Lu, Y.; Wang, Z. Vascularization of pancreatic decellularized scaffold with endothelial progenitor cells. J. Artif. Organs 2018, 21, 230–237. [Google Scholar] [CrossRef]
- Hansmann, J.; Groeber, F.; Kahlig, A.; Kleinhans, C.; Walles, H. Bioreactors in tissue engineering—Principles, applications and commercial constraints. Biotechnol. J. 2013, 8, 298–307. [Google Scholar] [CrossRef]
- Jin, G.; Yang, G.H.; Kim, G. Tissue engineering bioreactor systems for applying physical and electrical stimulations to cells. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 103, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.F. Biocompatibility Pathways in Tissue-Engineering Templates. Engineering 2018, 4, 286–290. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Agent (Concentration) | Duration | RPM | Solution Volume |
|---|---|---|---|---|
| 1 | Trypsin (0.05%) and EDTA (0.05%) | 30 min | 30 | 1 L |
| 2 | Distilled Water Wash | 15 min | 30 | 1 L |
| (Wash repeated three times) | ||||
| 3 | DNase I (2KU) | 30 min | 30 | 1 L |
| 4 | Distilled Water Wash | 15 min | 30 | 1 L |
| (Wash repeated three times) | ||||
| 5 | SDS (0.05%) | 30 min | 30 | 1 L |
| 6 | Distilled Water Wash | 15 min | 30 | 1 L |
| (Wash repeated three times) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somasundaram, M.; Greco, K.V.; Bhatnagar, G.; Gabe, S.; Sibbons, P.; Friend, P.; Ansari, T. The Development and Characterisation of A Porcine Large Intestinal Biological Scaffold by Perfusion Decellularisation. Cells 2025, 14, 817. https://doi.org/10.3390/cells14110817
Somasundaram M, Greco KV, Bhatnagar G, Gabe S, Sibbons P, Friend P, Ansari T. The Development and Characterisation of A Porcine Large Intestinal Biological Scaffold by Perfusion Decellularisation. Cells. 2025; 14(11):817. https://doi.org/10.3390/cells14110817
Chicago/Turabian StyleSomasundaram, Murali, Karin V. Greco, Gauraang Bhatnagar, Simon Gabe, Paul Sibbons, Peter Friend, and Tahera Ansari. 2025. "The Development and Characterisation of A Porcine Large Intestinal Biological Scaffold by Perfusion Decellularisation" Cells 14, no. 11: 817. https://doi.org/10.3390/cells14110817
APA StyleSomasundaram, M., Greco, K. V., Bhatnagar, G., Gabe, S., Sibbons, P., Friend, P., & Ansari, T. (2025). The Development and Characterisation of A Porcine Large Intestinal Biological Scaffold by Perfusion Decellularisation. Cells, 14(11), 817. https://doi.org/10.3390/cells14110817