.png)
Mitochondrial Dysfunction: The Silent Catalyst of Kidney Disease Progression


 ,
,  and
and 
Abstract
1. Introduction
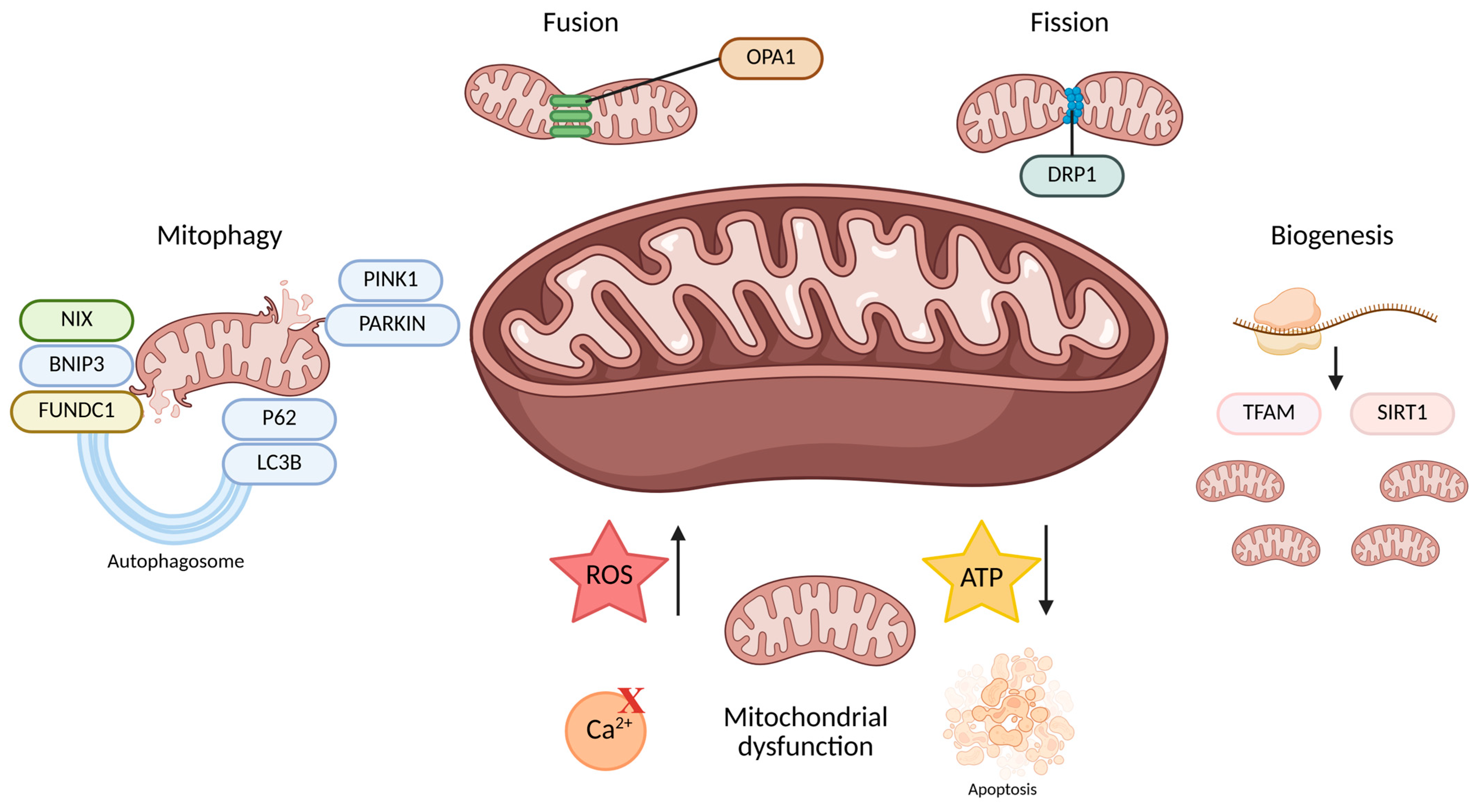
2. Mitochondria in Kidney Health and Disease
2.1. AKI
2.2. CKD
2.3. CAKUT
3. How Does Epigenetics Shape Mitochondrial Function in Renal Pathology?
4. Mitophagy in Kidney Disease
5. Feeding the Mitochondria: Can Diet Slow Kidney Disease?
6. Hormonal Guardians of the Mitochondria: Estrogen and Thyroid Hormones in Kidney Disease Regulation
7. Therapeutic Approaches for Kidney Disease
8. Future Perspective
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Disha, B.; Mathew, R.P.; Dalal, A.B.; Mahato, A.K.; Satyamoorthy, K.; Singh, K.K.; Thangaraj, K.; Govindaraj, P. Mitochondria in biology and medicine—2023. Mitochondrion 2024, 76, 101853. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, R.M.; Seli, E. Mitochondria as therapeutic targets in assisted reproduction. Hum. Reprod. 2024, 39, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.B.; Jeffery, G. Light stimulation of mitochondria reduces blood glucose levels. J. Biophotonics 2024, 17, e202300521. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Mechanisms and applications of the anti-inflammatory effects of photobiomodulation. AIMS Biophys. 2017, 4, 337–361. [Google Scholar] [CrossRef]
- Kaynezhad, P.; Tachtsidis, I.; Jeffery, G. Optical monitoring of retinal respiration in real time: 670 nm light increases the redox state of mitochondria. Exp. Eye Res. 2016, 152, 88–93. [Google Scholar] [CrossRef]
- Begum, R.; Calaza, K.; Kam, J.H.; Salt, T.E.; Hogg, C.; Jeffery, G. Near-infrared light increases ATP, extends lifespan and improves mobility in aged Drosophila melanogaster. Biol. Lett. 2015, 11, 20150073. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, R.M.; Seli, E. The role of mitochondrial dynamics in oocyte and early embryo development. Semin. Cell Dev. Biol. 2024, 159–160, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Morgenstern, M.; Peikert, C.D.; Lübbert, P.; Suppanz, I.; Klemm, C.; Alka, O.; Steiert, C.; Naumenko, N.; Schendzielorz, A.; Melchionda, L.; et al. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 2021, 33, 2464–2483.e18. [Google Scholar] [CrossRef]
- Suomalainen, A.; Nunnari, J. Mitochondria at the crossroads of health and disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Rizk, D.; Chapman, A.B. Cystic and inherited kidney diseases. Am. J. Kidney Dis. 2003, 42, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Narang, J.; Chhillar, A.K.; Rana, J.S.; Siddique, M.U.M.; Kenawy, E.-R.; Alkahtani, S.; Ahsan, M.N.; Nayak, A.K.; Hasnain, S. Diagnostic methods employing kidney biomarkers clinching biosensors as promising tools. Sensors Int. 2024, 5, 100253. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Mao, J.; Li, C.; Wu, F.; Wang, Y.; Zhu, J.; Wen, C. The relationship between kidney disease and mitochondria: A bibliometric study. Ren. Fail. 2024, 46, 2302963. [Google Scholar] [CrossRef]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Zelnick, L.R.; Weiss, N.S.; Kestenbaum, B.R.; Robinson-Cohen, C.; Heagerty, P.J.; Tuttle, K.; Hall, Y.N.; Hirsch, I.B.; de Boer, I.H. Diabetes and CKD in the United States Population, 2009–2014. Clin. J. Am. Soc. Nephrol. 2017, 12, 1984–1990. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Bleyer, A.J.; Molnar, M.Z.; Ma, J.Z.; Sim, J.J.; Cushman, W.C.; Darryl Quarles, L.; Kalantar-Zadeh, K. Blood pressure and mortality in U.S. Veterans with chronic kidney disease: A cohort study. Ann. Intern Med. 2013, 159, 4. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N. Temporal trends in the burden of chronic kidney disease in the United States. Curr. Opin. Nephrol. Hypertens. 2010, 19, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.J.; Gilbertson, D.T.; Snyder, J.J.; Chen, S.; Foley, R.N. Chronic kidney disease awareness, screening and prevention: Rationale for the design of a public education program. Nephrology 2010, 15, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Mizdrak, M.; Kumrić, M.; Kurir, T.T.; Božić, J. Emerging Biomarkers for Early Detection of Chronic Kidney Disease. J. Pers. Med. 2022, 12, 548. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11, 565023. [Google Scholar] [CrossRef]
- Ho, H.-J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Kanasaki, K.; Goodwin, J.E. Loss of Mitochondrial Control Impacts Renal Health. Front. Pharmacol. 2020, 11, 543973. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.-M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Miao, M.; Xu, X.; Bai, M.; Wu, M.; Zhang, A. From Physiology to Pathology: The Role of Mitochondria in Acute Kidney Injuries and Chronic Kidney Diseases. Kidney Dis. 2023, 9, 342–357. [Google Scholar] [CrossRef]
- Takasu, M.; Kishi, S.; Nagasu, H.; Kidokoro, K.; Brooks, C.R.; Kashihara, N. The Role of Mitochondria in Diabetic Kidney Disease and Potential Therapeutic Targets. Kidney Int. Rep. 2024, 10, 328–342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, R.; Zhang, C.; Xiang, Z.; Lin, T.; Ling, J.; Hu, H. Role of mitochondria in renal ischemia–reperfusion injury. FEBS J. 2024, 291, 5365–5378. [Google Scholar] [CrossRef] [PubMed]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef]
- Jiang, Y.; Krantz, S.; Qin, X.; Li, S.; Gunasekara, H.; Kim, Y.-M.; Zimnicka, A.; Bae, M.; Ma, K.; Toth, P.T.; et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission—Fusion dynamics and mitophagy. Redox Biol. 2022, 52, 102304. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Nejjar, S.; Jhun, B.S.; Yu, T.; Hsu, W.; Yoon, Y. Transgenic Control of Mitochondrial Fission Induces Mitochondrial Uncoupling and Relieves Diabetic Oxidative Stress. Diabetes 2012, 61, 2093–2104. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Swan, E.J.; Maxwell, A.P.; McKnight, A.J. Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet. Med. 2015, 32, 1110–1115. [Google Scholar] [CrossRef]
- Agil, A.; Chayah, M.; Visiedo, L.; Navarro-Alarcon, M.; Ferrer, J.M.R.; Tassi, M.; Reiter, R.J.; Fernández-Vázquez, G. Melatonin Improves Mitochondrial Dynamics and Function in the Kidney of Zücker Diabetic Fatty Rats. J. Clin. Med. 2020, 9, 2916. [Google Scholar] [CrossRef]
- Orozco-Ibarra, M.; Aparicio-Trejo, O.E.; Jiménez-Uribe, A.P.; Hernández-Cruz, E.Y.; Aranda-Rivera, A.K.; Amador-Martínez, I.; Fernández-Valverde, F.; Pedraza-Chaverri, J. Assessment of Kidney Mitochondrial Function by High-Resolution Respirometry, Transmission Electron Microscopy, and Histological Techniques. Methods Mol. Biol. 2023, 2664, 283–308. [Google Scholar]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.-K.; Garrick, M.D. Iron and Cadmium Entry Into Renal Mitochondria: Physiological and Toxicological Implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Gui, T.; Kullak-Ublick, G.A.; Li, Y.; Visentin, M. The Role of Mitochondria in Drug-Induced Kidney Injury. Front. Physiol. 2020, 11, 1079. [Google Scholar] [CrossRef]
- Boveris, A.; Valdez, L.B.; Zaobornyj, T.; Bustamante, J. Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.; Ciarimboli, G. Editorial: Mitochondria in Renal Health and Disease. Front. Physiol. 2021, 12, 707175. [Google Scholar] [CrossRef]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin–Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef]
- Gujarati, N.A.; Vasquez, J.M.; Bogenhagen, D.F.; Mallipattu, S.K. The complicated role of mitochondria in the podocyte. Am. J. Physiol. Renal. Physiol. 2020, 319, F955–F965. [Google Scholar] [CrossRef]
- Liu, S.; Yuan, Y.; Xue, Y.; Xing, C.; Zhang, B. Podocyte Injury in Diabetic Kidney Disease: A Focus on Mitochondrial Dysfunction. Front. Cell Dev. Biol. 2022, 10, 832887. [Google Scholar] [CrossRef]
- Hejazian, S.M.; Ardalan, M.; Hosseiniyan Khatibi, S.M.; Rahbar Saadat, Y.; Barzegari, A.; Gueguen, V.; Meddahi-Pellé, A.; Anagnostou, F.; Zununi Vahed, S.; Pavon-Djavid, G. Biofactors regulating mitochondrial function and dynamics in podocytes and podocytopathies. J. Cell. Physiol. 2023, 238, 2206–2227. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, J.; Zhu, W.; Niu, Y.; Wu, M.; Zhang, A. Therapeutic Potential Targeting Podocyte Mitochondrial Dysfunction in Focal Segmental Glomerulosclerosis. Kidney Dis. 2023, 9, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xin, W.; Xiong, J.; Yao, M.; Zhang, B.; Zhao, J. The Intestinal Microbiota and Metabolites in the Gut-Kidney-Heart Axis of Chronic Kidney Disease. Front. Pharmacol. 2022, 13, 837500. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Dong, J.; Wang, R.; Bi, G.; Xu, D.; Zhang, Y.; Deng, Y.; Lin, W.; Yang, Z.; et al. HDAC3 aberration-incurred GPX4 suppression drives renal ferroptosis and AKI-CKD progression. Redox Biol. 2023, 68, 102939. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Guo, Y.; Che, R.; Wang, P.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Physiol. 2024, 326, F768–F779. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Hildebrandt, F. Disease mechanisms of monogenic congenital anomalies of the kidney and urinary tract. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 325–343. [Google Scholar] [CrossRef]
- Westland, R.; Sanna-Cherchi, S. Recessive mutations in CAKUT and VACTERL association. Kidney Int. 2014, 85, 1253–1255. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pract. 2019, 38, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraten, C.A.; Hoenderop, J.G.; de Baaij, J.H.F. Mitochondrial Dysfunction in Kidney Tubulopathies. Annu. Rev. Physiol. 2024, 86, 379–403. [Google Scholar] [CrossRef]
- Noble, R.A.; Lucas, B.J.; Selby, N.M. Long-Term Outcomes in Patients with Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2020, 15, 423–429. [Google Scholar] [CrossRef]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Yao, C.; Li, Z.; Sun, K.; Zhang, Y.; Shou, S.; Jin, H. Mitochondrial dysfunction in acute kidney injury. Ren. Fail. 2024, 46, 2393262. [Google Scholar] [CrossRef]
- Clare, C.S. Identifying and managing acute kidney injury. Nurs. Stand. 2022, 37, 59–66. [Google Scholar] [CrossRef]
- Rahman, M.; Shad, F.; Smith, M.C. Acute kidney injury: A guide to diagnosis and management. Am. Fam. Physician 2012, 86, 631–639. [Google Scholar] [PubMed]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Tomar, B.; Sharma, D.; Rath, S.K. Mitochondrial dysfunction and oxidative stress: Role in chronic kidney disease. Life Sci. 2023, 319, 121432. [Google Scholar] [CrossRef]
- Gaitonde, D.Y.; Cook, D.L.; Rivera, I.M. Chronic Kidney Disease: Detection and Evaluation. Am. Fam. Physician 2017, 96, 776–783. [Google Scholar]
- Pettitt, R.M.; Brumbaugh, A.P.; Gartman, M.F.; Jackson, A.M. Chronic kidney disease: Detection and evaluation. Am. Fam. Physician 2020, 12, 14–19. [Google Scholar]
- Chouhan, A.S.; Hingway, S.; Kaple, M.N. A Brief Review of Diagnostic Techniques and Clinical Management in Chronic Kidney Disease. Cureus 2023, 15, e49030. [Google Scholar] [CrossRef] [PubMed]
- Saisawat, P.; Kohl, S.; Hilger, A.C.; Hwang, D.-Y.; Gee, H.Y.; Dworschak, G.C.; Tasic, V.; Pennimpede, T.; Natarajan, S.; Sperry, E.; et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 2014, 85, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.Y.X.; Winyard, P.; Marlais, M. Congenital anomalies of the kidney and urinary tract: Antenatal diagnosis, management and counselling of families. Pediatr. Nephrol. 2023, 39, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Soares dos Santos, A.C., Jr.; Marques de Miranda, D.; Simões e Silva, A.C. Congenital anomalies of the kidney and urinary tract: An embryogenetic review. Birth Defects Res. C Embryo Today 2014, 102, 374–381. [Google Scholar] [CrossRef]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef]
- Lv, T.; Zhang, Y.; Ji, X.; Sun, S.; Xu, L.; Ma, W.; Liu, Y.; Wan, Q. GCN5L1-mediated TFAM acetylation at K76 participates in mitochondrial biogenesis in acute kidney injury. J. Transl. Med. 2022, 20, 571. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29. [Google Scholar] [CrossRef]
- Yu, C.; Li, T.; Li, J.; Cui, B.; Liu, N.; Bayliss, G.; Zhuang, S. Inhibition of polycomb repressive complex 2 by targeting EED protects against cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2022, 26, 4061–4075. [Google Scholar] [CrossRef]
- de Oliveira Cruz, J.; Silva, A.O.; Ribeiro, J.M.; Luizon, M.R.; Ceron, C.S. Epigenetic Regulation of the N-Terminal Truncated Isoform of Matrix Metalloproteinase-2 (NTT-MMP-2) and Its Presence in Renal and Cardiac Diseases. Front. Genet. 2021, 12, 637148. [Google Scholar] [CrossRef]
- Ge, Q.-M.; Huang, C.-M.; Zhu, X.-Y.; Bian, F.; Pan, S.-M. Differentially expressed miRNAs in sepsis-induced acute kidney injury target oxidative stress and mitochondrial dysfunction pathways. PLoS ONE 2017, 12, e0173292. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.K.; Kota, S.B. Noncoding RNA and epigenetic gene regulation in renal diseases. Drug Discov. Today 2017, 22, 1112–1122. [Google Scholar] [CrossRef]
- Hajarnis, S.; Lakhia, R.; Yheskel, M.; Williams, D.; Sorourian, M.; Liu, X.; Aboudehen, K.; Zhang, S.; Kersjes, K.; Galasso, R.; et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 2017, 8, 14395. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.-W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Jin, H.; Shao, X.; Zhu, X.; Wu, J.; Zhang, M.; Zhang, Z.; Shen, J.; et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 2020, 17, 2975–2990. [Google Scholar] [CrossRef]
- Yang, K.; Li, T.; Geng, Y.; Zou, X.; Peng, F.; Gao, W. The role of mitophagy in the development of chronic kidney disease. PeerJ 2024, 12, e17260. [Google Scholar] [CrossRef]
- Bhatia, D.; Choi, M.E. Autophagy and mitophagy: Physiological implications in kidney inflammation and diseases. Am. J. Physiol. Physiol. 2023, 325, F1–F21. [Google Scholar] [CrossRef]
- Cui, X.; Zhou, Z.; Tu, H.; Wu, J.; Zhou, J.; Yi, Q.; Liu, O.; Dai, X. Mitophagy in fibrotic diseases: Molecular mechanisms and therapeutic applications. Front. Physiol. 2024, 15, 1430230. [Google Scholar] [CrossRef]
- Maglica, M.; Kelam, N.; Perutina, I.; Racetin, A.; Rizikalo, A.; Filipović, N.; Prusac, I.K.; Mišković, J.; Vukojević, K. Immunoexpression Pattern of Autophagy-Related Proteins in Human Congenital Anomalies of the Kidney and Urinary Tract. Int. J. Mol. Sci. 2024, 25, 6829. [Google Scholar] [CrossRef] [PubMed]
- Bayjanov, J.R.; Doornbos, C.; Ozisik, O.; Shin, W.; Queralt-Rosinach, N.; Wijnbergen, D.; Saulnier-Blache, J.-S.; Schanstra, J.P.; Buffin-Meyer, B.; Klein, J.; et al. Integrative analysis of multi-omics data reveals importance of collagen and the PI3K AKT signalling pathway in CAKUT. Sci. Rep. 2024, 14, 20731. [Google Scholar] [CrossRef]
- Ruggiero, C.; Ehrenshaft, M.; Cleland, E.; Stadler, K. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am. J. Physiol. Metab. 2011, 300, E1047–E1058. [Google Scholar] [CrossRef]
- Chen, J.; Muntner, P.; Hamm, L.L.; Jones, D.W.; Batuman, V.; Fonseca, V.; Whelton, P.K.; He, J. The Metabolic Syndrome and Chronic Kidney Disease in U.S. Adults. Ann. Intern. Med. 2004, 140, 167–174. [Google Scholar] [CrossRef]
- Abrass, C.K. Overview: Obesity: What does it have to do with kidney disease? J. Am. Soc. Nephrol. 2004, 15, 2768–2772. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Franczyk, B.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Effect of Diet on the Survival of Patients with Chronic Kidney Disease. Nutrients 2017, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Declèves, A.-E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK Mediates the Initiation of Kidney Disease Induced by a High-Fat Diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-Carboxamide Ribonucleoside: A Specific Method for Activating AMP-Activated Protein Kinase in Intact Cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Prem, P.N.; Kurian, G.A. High-Fat Diet Increased Oxidative Stress and Mitochondrial Dysfunction Induced by Renal Ischemia-Reperfusion Injury in Rat. Front. Physiol. 2021, 12, 715693. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, J.; Li, S.; Guo, F.; Li, A.; Wu, H.; Chen, J.; Pan, Q.; Liao, S.; Liu, H.-F.; et al. High-Fat Diet-Induced Renal Proximal Tubular Inflammatory Injury: Emerging Risk Factor of Chronic Kidney Disease. Front. Physiol. 2021, 12, 786599. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Kalantar-Zadeh, K. Back to the future: Restricted protein intake for conservative management of CKD, triple goals of renoprotection, uremia mitigation, and nutritional health. Int. Urol. Nephrol. 2016, 48, 725–729. [Google Scholar] [CrossRef]
- Bellizzi, V. Low-protein diet or nutritional therapy in chronic kidney disease? Blood Purif. 2013, 36, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Fouque, D.; Aparicio, M. Eleven reasons to control the protein intake of patients with chronic kidney disease. Nat. Clin. Pract. Nephrol. 2007, 3, 383–392. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, Y.-S.; Kim, Y.H.; Chung, W.; Park, S.K.; Choi, K.H.; Ahn, C.; Oh, K.-H. Dietary Protein Intake, Protein Energy Wasting, and the Progression of Chronic Kidney Disease: Analysis from the KNOW-CKD Study. Nutrients 2019, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Riccio, E.; Di Nuzzi, A.; Pisani, A. Nutritional treatment in chronic kidney disease: The concept of nephroprotection. Clin. Exp. Nephrol. 2014, 19, 161–167. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Moore, L.W.; Tortorici, A.R.; Chou, J.A.; St-Jules, D.E.; Aoun, A.; Rojas-Bautista, V.; Tschida, A.K.; Rhee, C.M.; Shah, A.A.; et al. North American experience with Low protein diet for Non-dialysis-dependent chronic kidney disease. BMC Nephrol. 2016, 17, 90. [Google Scholar] [CrossRef]
- Iseki, K. Nutrition and quality of life in chronic kidney disease patients: A practical approach for salt restriction. Kidney Res. Clin. Pract. 2022, 41, 657–669. [Google Scholar] [CrossRef]
- Garofalo, C.; Borrelli, S.; Provenzano, M.; De Stefano, T.; Vita, C.; Chiodini, P.; Minutolo, R.; De Nicola, L.; Conte, G. Dietary Salt Restriction in Chronic Kidney Disease: A Meta-Analysis of Randomized Clinical Trials. Nutrients 2018, 10, 732. [Google Scholar] [CrossRef]
- Crawford-Faucher, A.; Finer, L.B.; Zolna, M.R. Declines in unintended pregnancy in the United States. N. Engl. J. Med. 2017, 374, 843–852. Available online: www.aafp.org/afp (accessed on 1 May 2025).
- McMahon, E.J.; Campbell, K.L.; Bauer, J.D.; Mudge, D.W.; Kelly, J.T. Altered dietary salt intake for people with chronic kidney disease. Cochrane Database Syst. Rev. 2021, 6, CD010070. [Google Scholar] [CrossRef]
- Abe, Y.; Nishiwaki, H.; Suzuki, T.; Noma, H.; Watanabe, Y.; Ota, E.; Hasegawa, T. Renoprotective effects of coenzyme Q10 supplementation in patients with chronic kidney disease: A protocol for a systematic review. BMJ Open 2024, 14, e084088. [Google Scholar] [CrossRef] [PubMed]
- Tkaczenko, H.; Kurhaluk, N. Antioxidant-Rich Functional Foods and Exercise: Unlocking Metabolic Health Through Nrf2 and Related Pathways. Int. J. Mol. Sci. 2025, 26, 1098. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-Y.; Chen, S.; Du, Y. Estrogen and estrogen receptors in kidney diseases. Ren. Fail. 2021, 43, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Kafami, M.; Hosseini, M.; Niazmand, S.; Farrokhi, E.; Hajzadeh, M.A.-R.; Nazemi, S. The effects of estradiol and testosterone on renal tissues oxidative after central injection of angiotensin II in female doca—Salt treated rats. Horm. Mol. Biol. Clin. Investig. 2019, 37, 3. [Google Scholar] [CrossRef]
- Rzewuska-Lech, E.; Jayachandran, M.; Fitzpatrick, L.A.; Miller, V.M. Differential effects of 17β-estradiol and raloxifene on VSMC phenotype and expression of osteoblast-associated proteins. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E105–E112. [Google Scholar] [CrossRef]
- Chang, Y.; Han, Z.; Zhang, Y.; Zhou, Y.; Feng, Z.; Chen, L.; Li, X.; Li, L.; Si, J.-Q. G protein-coupled estrogen receptor activation improves contractile and diastolic functions in rat renal interlobular artery to protect against renal ischemia reperfusion injury. Biomed. Pharmacother. 2019, 112, 108666. [Google Scholar] [CrossRef]
- Guajardo-Correa, E.; Silva-Agüero, J.F.; Calle, X.; Chiong, M.; Henríquez, M.; García-Rivas, G.; Latorre, M.; Parra, V. Estrogen signaling as a bridge between the nucleus and mitochondria in cardiovascular diseases. Front. Cell Dev. Biol. 2022, 10, 968373. [Google Scholar] [CrossRef]
- Tian, H.; Gao, Z.; Wang, G.; Li, H.; Zheng, J. Estrogen potentiates reactive oxygen species (ROS) tolerance to initiate carcinogenesis and promote cancer malignant transformation. Tumor Biol. 2015, 37, 141–150. [Google Scholar] [CrossRef]
- Razmara, A.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007, 1176, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Sowers, J.R. Estrogen and mitochondria function in cardiorenal metabolic syndrome. Prog. Mol. Biol. Transl. Sci. 2014, 127, 229–249. [Google Scholar] [PubMed]
- Wang, X.X.; Myakala, K.; Libby, A.E.; Krawczyk, E.; Panov, J.; Jones, B.A.; Bhasin, K.; Shults, N.; Qi, Y.; Krausz, K.W.; et al. Estrogen-Related Receptor Agonism Reverses Mitochondrial Dysfunction and Inflammation in the Aging Kidney. Am. J. Pathol. 2023, 193, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Liu, H.; Kim, J.K. Estrogen Protects the Female Heart from Ischemia/Reperfusion Injury through Manganese Superoxide Dismutase Phosphorylation by Mitochondrial p38β at Threonine 79 and Serine 106. PLoS ONE 2016, 11, e0167761. [Google Scholar] [CrossRef]
- Lee, W.-L.; Cheng, M.-H.; Tarng, D.-C.; Yang, W.-C.; Lee, F.-K.; Wang, P.-H. The benefits of estrogen or selective estrogen receptor modulator on kidney and its related disease—Chronic kidney disease—Mineral and bone disorder: Osteoporosis. J. Chin. Med. Assoc. 2013, 76, 365–371. [Google Scholar] [CrossRef]
- Rhee, C.M. The interaction between thyroid and kidney disease: An overview of the evidence. Curr. Opin. Endocrinol. Diabetes 2016, 23, 407–415. [Google Scholar] [CrossRef]
- Basu, G.; Mohapatra, A. Interactions between thyroid disorders and kidney disease. Indian J. Endocrinol. Metab. 2012, 16, 204–213. [Google Scholar] [CrossRef]
- Sivertsson, E.; Friederich-Persson, M.; Persson, P.; Nangaku, M.; Hansell, P.; Palm, F. Thyroid hormone increases oxygen metabolism causing intrarenal tissue hypoxia; A pathway to kidney disease. PLoS ONE 2022, 17, e0264524. [Google Scholar] [CrossRef]
- Amorim Amato, A.; Martins Santos, G.; de Assis Rocha Neves, F. Thyroid hormone action in chronic kidney disease. Curr. Opin. Endocrinol. Diabetes 2008, 15, 459–465. [Google Scholar] [CrossRef]
- Harper, M.-E.; Seifert, E.L. Thyroid Hormone Effects on Mitochondrial Energetics. Thyroid 2008, 18, 145–156. [Google Scholar] [CrossRef]
- Rajeev, G.; Rayappa, W.D.S.C.; Vijayalakshmi, R.; Swathi, M.; Kumar, S. Evaluation of thyroid hormone levels in chronic kidney disease patients. Saudi J. Kidney Dis. Transplant. 2015, 26, 90–93. [Google Scholar] [CrossRef]
- Alsagheer, M.M.M.; Rahman, A.A.A.; Abdelmeguid, M.M.; Abdelhafez, A.A. Evaluation of Thyroid Hormones Level in Chronic Kidney Disease Patients. Al-Azhar Int. Med. J. 2023, 4, 9. [Google Scholar] [CrossRef]
- Qin, S.; Liu, C.; Chen, Y.; Yao, M.; Liao, S.; Xin, W.; Gong, S.; Guan, X.; Li, Y.; Xiong, J.; et al. Cobaltosic oxide-polyethylene glycol-triphenylphosphine nanoparticles ameliorate the acute-to-chronic kidney disease transition by inducing BNIP3-mediated mitophagy. Kidney Int. 2023, 103, 903–916. [Google Scholar] [CrossRef]
- Cao, Y.; Xiong, J.; Guan, X.; Yin, S.; Chen, J.; Yuan, S.; Liu, H.; Lin, S.; Zhou, Y.; Qiu, J.; et al. Paeoniflorin suppresses kidney inflammation by regulating macrophage polarization via KLF4-mediated mitophagy. Phytomedicine 2023, 116, 154901. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, W.; Li, B.; Qiao, X.; Wang, X.; Yang, G.; Li, S. The potential role of hydrogen sulfide in regulating macrophage phenotypic changes via PINK1/parkin-mediated mitophagy in sepsis-related cardiorenal syndrome. Immunopharmacol. Immunotoxicol. 2024, 46, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Suga, N.; Ikeda, Y.; Yoshikawa, S.; Matsuda, S. Inspiring Tactics with the Improvement of Mitophagy and Redox Balance for the Development of Innovative Treatment against Polycystic Kidney Disease. Biomolecules 2024, 14, 207. [Google Scholar] [CrossRef]
- Fan, X.; Wu, L.; Wang, F.; Liu, D.; Cen, X.; Xia, H. Mitophagy Regulates Kidney Diseases. Kidney Dis. 2024, 10, 73–587. [Google Scholar] [CrossRef]
- Tang, C.-Y.; Lai, C.-C.; Yang, A.-H.; Chiang, S.-C.; Huang, P.-C.; Tseng, K.-W.; Huang, C.-H. Magnolol reduces myocardial injury induced by renal ischemia and reperfusion. J. Chin. Med. Assoc. 2022, 85, 584–596. [Google Scholar] [CrossRef]
- Zhu, Z.; Luan, G.; Peng, S.; Fang, Y.; Fang, Q.; Shen, S.; Wu, K.; Qian, S.; Jia, W.; Ye, J.; et al. Huangkui capsule attenuates diabetic kidney disease through the induction of mitophagy mediated by STING1/PINK1 signaling in tubular cells. Phytomedicine 2023, 119, 154975. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Feng, X.; Xu, Y.; Zhou, L.; Wang, C.; Wang, M. Astragaloside IV attenuates fatty acid-induced renal tubular injury in diabetic kidney disease by inhibiting fatty acid transport protein-2. Phytomedicine 2024, 134, 155991. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-κB via PP2A activation. Life Sci. 2020, 246, 117382. [Google Scholar] [CrossRef]
- Hurtado, K.A.; Schnellmann, R.G. Mitophagy regulates mitochondrial number following pharmacological induction of mitochondrial biogenesis in renal proximal tubule cells. Front. Pharmacol. 2024, 15, 1344075. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, M.; Bai, X.; Li, J.; Nie, P.; Li, B.; Luo, P. SS-31, a Mitochondria-Targeting Peptide, Ameliorates Kidney Disease. Oxid. Med. Cell. Longev. 2022, 2022, 1295509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Song, Y.; Chen, L.; Chen, P.; Yuan, M.; Meng, Y.; Wang, Q.; Zheng, G.; Qiu, Z. Urolithin A Attenuates Hyperuricemic Nephropathy in Fructose-Fed Mice by Impairing STING-NLRP3 Axis-Mediated Inflammatory Response via Restoration of Parkin-Dependent Mitophagy. Front. Pharmacol. 2022, 13, 907209. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhang, R.; Zhou, J.; Guo, P.; Li, P.; Ye, M.; Liu, Y.; Shi, S. Pharmacokinetics and safety of pirfenidone in individuals with chronic kidney disease stage G2 and G3a: A single-dose, Phase I, bridging study. J. Pharm. Sci. 2025, 114, 1087–1094. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perkovic, V.; Tuttle, K.R.; Pergola, P.E.; Mahaffey, K.W.; Patel, U.D.; Ishida, J.H.; Kuo, A.; Chen, F.; Kustra, F.; et al. Selonsertib in Patients with Diabetic Kidney Disease: A Phase 2b Randomized Active Run-In Clinical Trial. J. Am. Soc. Nephrol. 2024, 35, 1726–1736. [Google Scholar] [CrossRef]
- Dagar, N.; Habshi, T.; Shelke, V.; Jadhav, H.R.; Gaikwad, A.B. Renoprotective effect of esculetin against ischemic acute kidney injury-diabetic comorbidity. Free. Radic. Res. 2024, 58, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Gross, O.; Wang, F.; de la Rosa, R.J.E.; Hall, M.; Sayer, J.A.; Appel, G.; Hariri, A.; Liu, S.; Maski, M.; et al. A Randomized Controlled Clinical Trial Testing Effects of Lademirsen on Kidney Function Decline in Adults with Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2024, 19, 995–1004. [Google Scholar] [CrossRef]
- Landa-Moreno, C.I.; Trejo-Hurtado, C.M.; la Cruz, J.L.-D.; Peña-Montes, D.J.; Murillo-Villicaña, M.; Huerta-Cervantes, M.; Montoya-Pérez, R.; Salgado-Garciglia, R.; Manzo-Avalos, S.; Cortés-Rojo, C.; et al. Antioxidant Effect of the Ethyl Acetate Extract of Potentilla indica on Kidney Mitochondria of Streptozotocin-Induced Diabetic Rats. Plants 2023, 12, 3196. [Google Scholar] [CrossRef]
- Ito, S.; Nakashima, M.; Ishikiriyama, T.; Nakashima, H.; Yamagata, A.; Imakiire, T.; Kinoshita, M.; Seki, S.; Kumagai, H.; Oshima, N. Effects of L-Carnitine Treatment on Kidney Mitochondria and Macrophages in Mice with Diabetic Nephropathy. Kidney Blood Press. Res. 2022, 47, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Li, H.; Saitta, B.; Sepehr, S.; Huang, P.; Pham, J.; Wang, J.; Mancino, V.; Chung, E.J.; Pinkosky, S.L.; et al. Beneficial effects of bempedoic acid treatment in polycystic kidney disease cells and mice. Front. Mol. Biosci. 2022, 9, 1001941. [Google Scholar] [CrossRef]
- Xie, W.; He, Q.; Zhang, Y.; Xu, X.; Wen, P.; Cao, H.; Zhou, Y.; Luo, J.; Yang, J.; Jiang, L. Pyruvate kinase M2 regulates mitochondrial homeostasis in cisplatin-induced acute kidney injury. Cell Death Dis. 2023, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Cui, W.; Ma, W.; Li, J.; Liu, Z.; Lin, Y. Typhaneoside-Tetrahedral Framework Nucleic Acids System: Mitochondrial Recovery and Antioxidation for Acute Kidney Injury treatment. ACS Nano 2023, 17, 8767–8781. [Google Scholar] [CrossRef]
- Lan, T.; Guo, H.; Lu, X.; Geng, K.; Wu, L.; Luo, Y.; Zhu, J.; Shen, X.; Guo, Q.; Wu, S. Dual-Responsive Curcumin-Loaded Nanoparticles for the Treatment of Cisplatin-Induced Acute Kidney Injury. Biomacromolecules 2022, 23, 5253–5266. [Google Scholar] [CrossRef]
- Burnstock, G.; Evans, L.C.; Bailey, M.A. Purinergic signalling in the kidney in health and disease. Purinergic Signal. 2013, 10, 71–101. [Google Scholar] [CrossRef] [PubMed]
- Menzies, R.I.; Tam, F.W.; Unwin, R.J.; Bailey, M.A. Purinergic signaling in kidney disease. Kidney Int. 2017, 91, 315–323. [Google Scholar] [CrossRef]
- Jabbari, H.; Roushandeh, A.M.; Rostami, M.K.; Razavi-Toosi, M.T.; Shokrgozar, M.A.; Jahanian-Najafabadi, A.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165809. [Google Scholar] [CrossRef]
- Cho, J.; Doo, S.W.; Song, N.; Lee, M.; Lee, H.; Kim, H.; Jeon, J.S.; Noh, H.; Kwon, S.H. Dapagliflozin Reduces Urinary Kidney Injury Biomarkers in Chronic Kidney Disease Irrespective of Albuminuria Level. Clin. Pharmacol. Ther. 2024, 115, 1441–1449. [Google Scholar] [CrossRef]
- Yadav, B.; Prasad, N.; Kushwaha, R.S.; Patel, M.R.; Bhadauria, D.S.; Kaul, A. Urinary Mitochondrial Deoxyribonucleic Acid: A Novel Biomarker of Coronavirus Disease 2019-associated Acute Kidney Injury in Renal Transplant Recipients. Indian J. Transplant. 2023, 17, 287–293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Treatment | Type | Stage | Target Pathway | Key Indication | Reference |
|---|---|---|---|---|---|
| TJ0113 | Small molecule | Phase II | PINK1/Parkin activation | AS | [137] |
| Magnolol | Natural metabolite | Experimental | FUNDC1/BNIP3 activation | CKD fibrosis | [138] |
| Huangkui Capsule | Herbal formulation | Phase IV | STING1/PINK1 axis | DCKD | [139] |
| Astragaloside IV | Botanical extract | Experimental | PINK1/Parkin modulation | DCKD-TI | [140] |
| Metformin | AMPK agonist | Off-label use | AMPK/PINK1/Parkin | DCKD | [141] |
| LY344864 | 5-HT1F receptor agonist | Preclinical | Mitochondrial biogenesis | AKI | [142] |
| MitoQ | Synthetic antioxidant | Phase II | mtROS | DCKD/HCKD | [143] |
| Elamipretide | Mitochondrial peptide | Phase II | Mitochondrial cristae | DCKD | [144] |
| Urolithin A | Natural metabolite | Experimental | Mitophagy in tubular cells | HN | [145] |
| Pirfenidone | Anti-fibrotic | Phase II | Reduces kidney fibrosis | DCKD | [146] |
| Selonsertib | ASK1 inhibitor | Phase II | JNK pathway | DCKD | [147] |
| Esculetin | Coumarin derivative | Experimental | PINK1/Parkin mitophagy | DOX-induced KI | [148] |
| Lademirsen | miR-21 antagonist | Phase II | Inflammatory pathways | AS | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlović, N.; Križanac, M.; Kumrić, M.; Vukojević, K.; Božić, J. Mitochondrial Dysfunction: The Silent Catalyst of Kidney Disease Progression. Cells 2025, 14, 794. https://doi.org/10.3390/cells14110794
Pavlović N, Križanac M, Kumrić M, Vukojević K, Božić J. Mitochondrial Dysfunction: The Silent Catalyst of Kidney Disease Progression. Cells. 2025; 14(11):794. https://doi.org/10.3390/cells14110794
Chicago/Turabian StylePavlović, Nikola, Marinela Križanac, Marko Kumrić, Katarina Vukojević, and Joško Božić. 2025. "Mitochondrial Dysfunction: The Silent Catalyst of Kidney Disease Progression" Cells 14, no. 11: 794. https://doi.org/10.3390/cells14110794
APA StylePavlović, N., Križanac, M., Kumrić, M., Vukojević, K., & Božić, J. (2025). Mitochondrial Dysfunction: The Silent Catalyst of Kidney Disease Progression. Cells, 14(11), 794. https://doi.org/10.3390/cells14110794