
Primary Cell Cultures in Neurobiology: Optimized Protocol for Culture of Mouse Fetal Hindbrain Neurons

 ,
,  ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Cell Culture Media
2.3. Fetal Tissue Dissociation
2.4. Fetal Cell Culture
2.5. Immunofluorescence
2.6. Electrophysiological Analyses
2.7. RT-qPCR
2.8. Cre-Mediated Recombination in Culture
2.9. Experimental Design and Statistical Analyses
3. Results
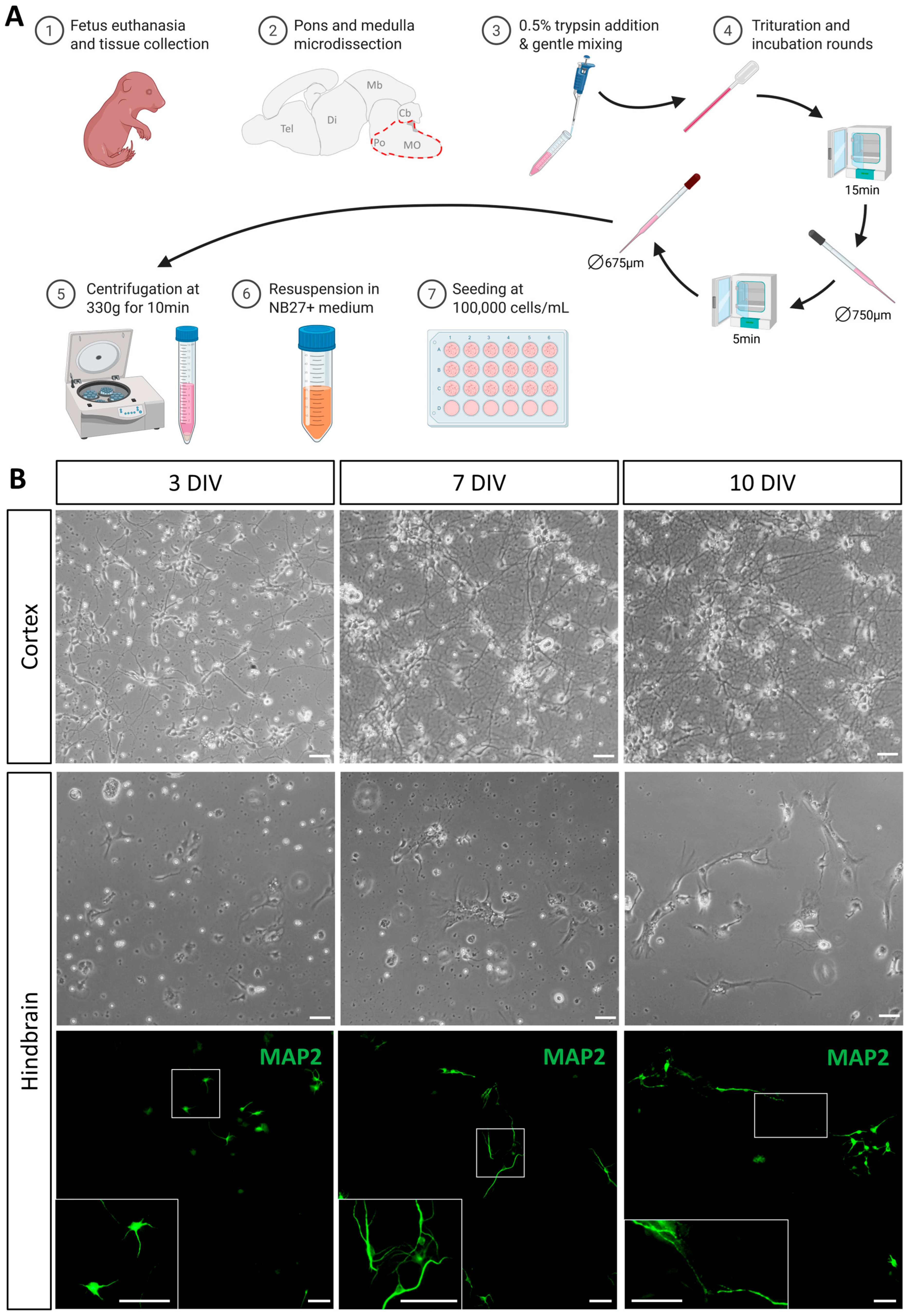
3.1. Isolation of Cells from the Fetal Mouse Hindbrain
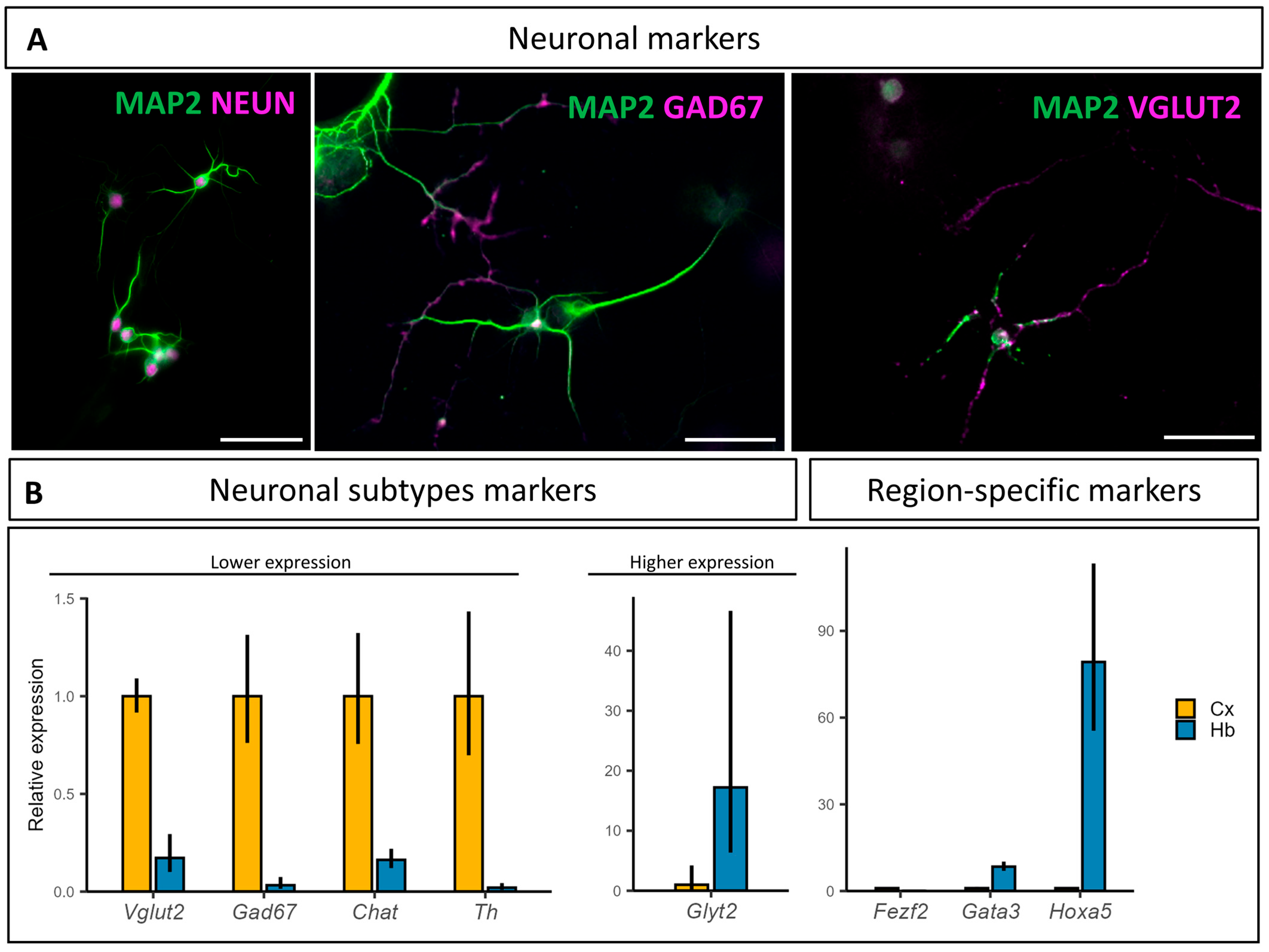
3.2. Characterization of the Hindbrain Neuronal Populations Obtained In Vitro
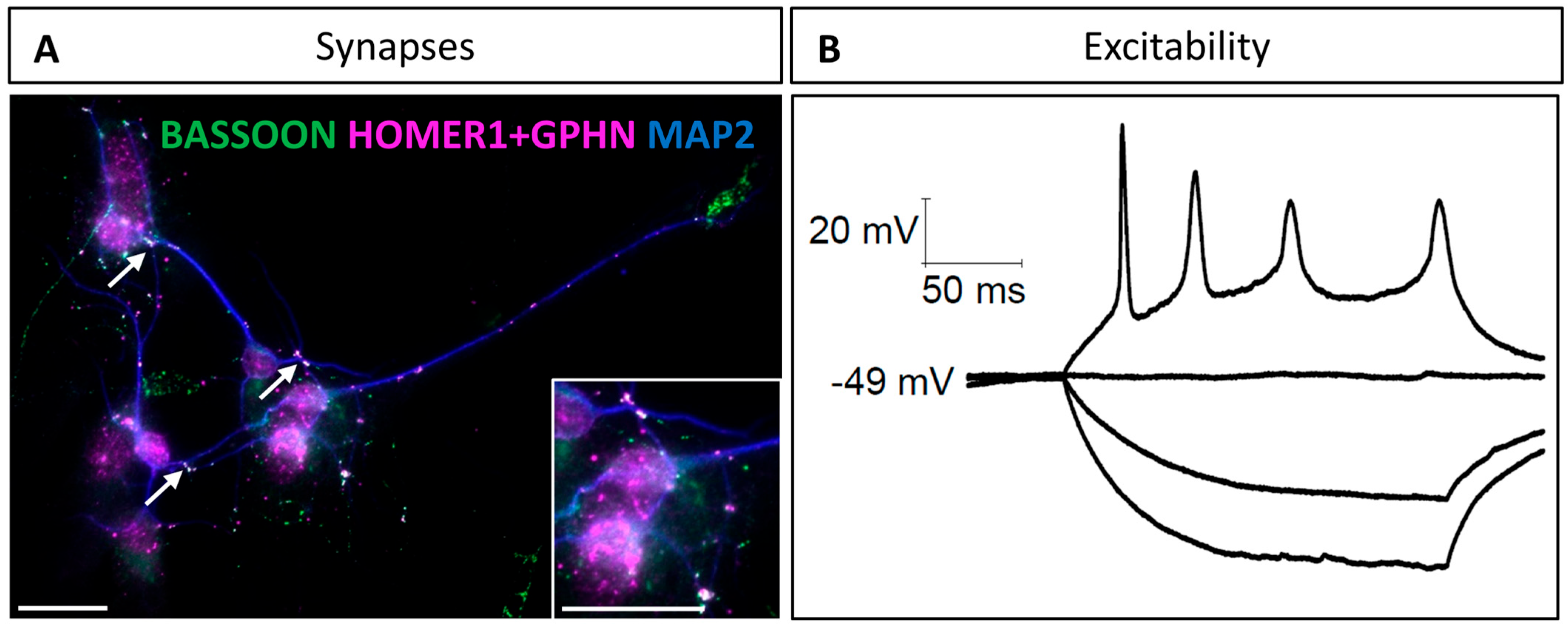
3.3. Synaptic Properties of Hindbrain-Derived Primary Neurons
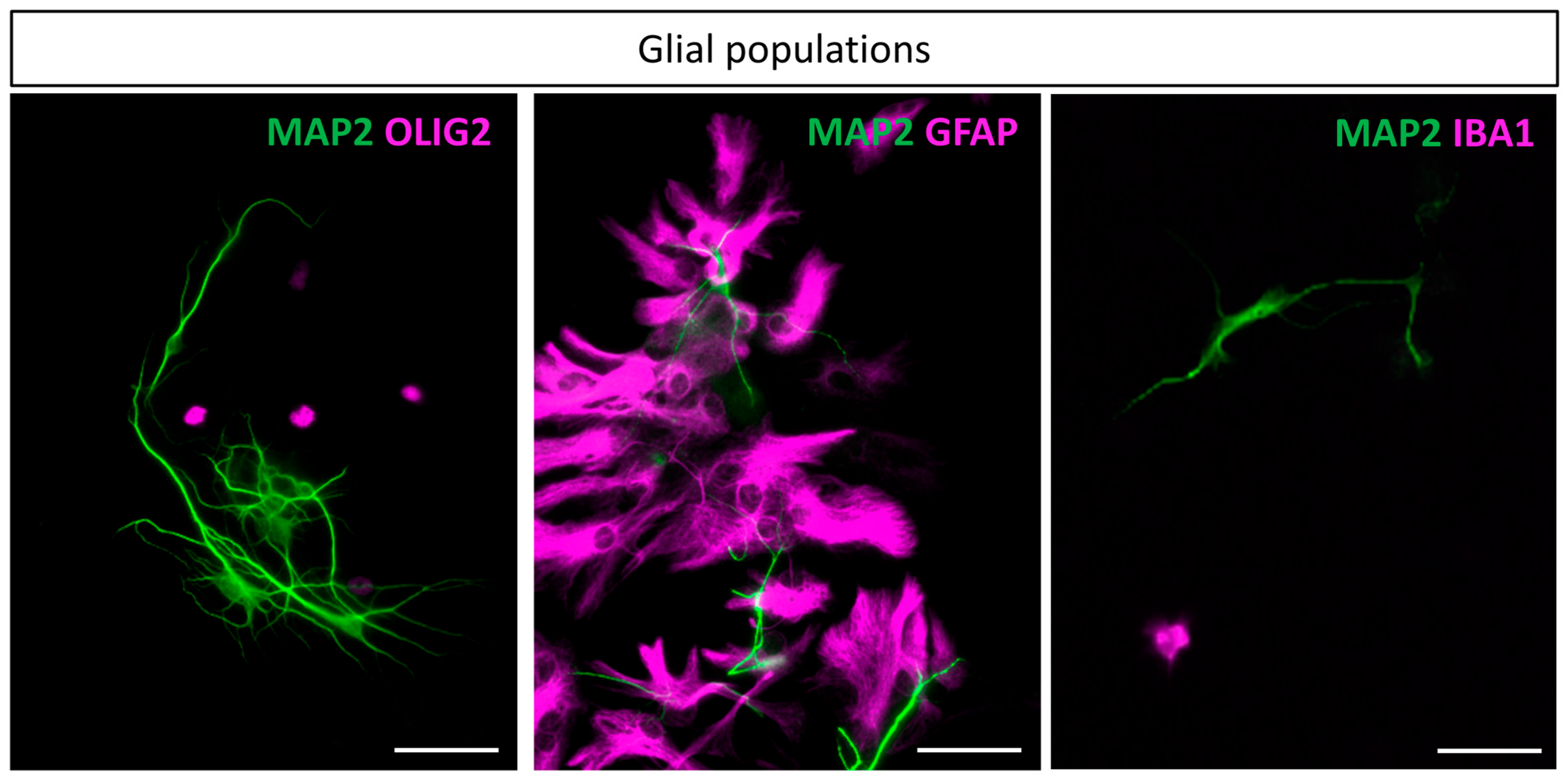
3.4. Glial Populations
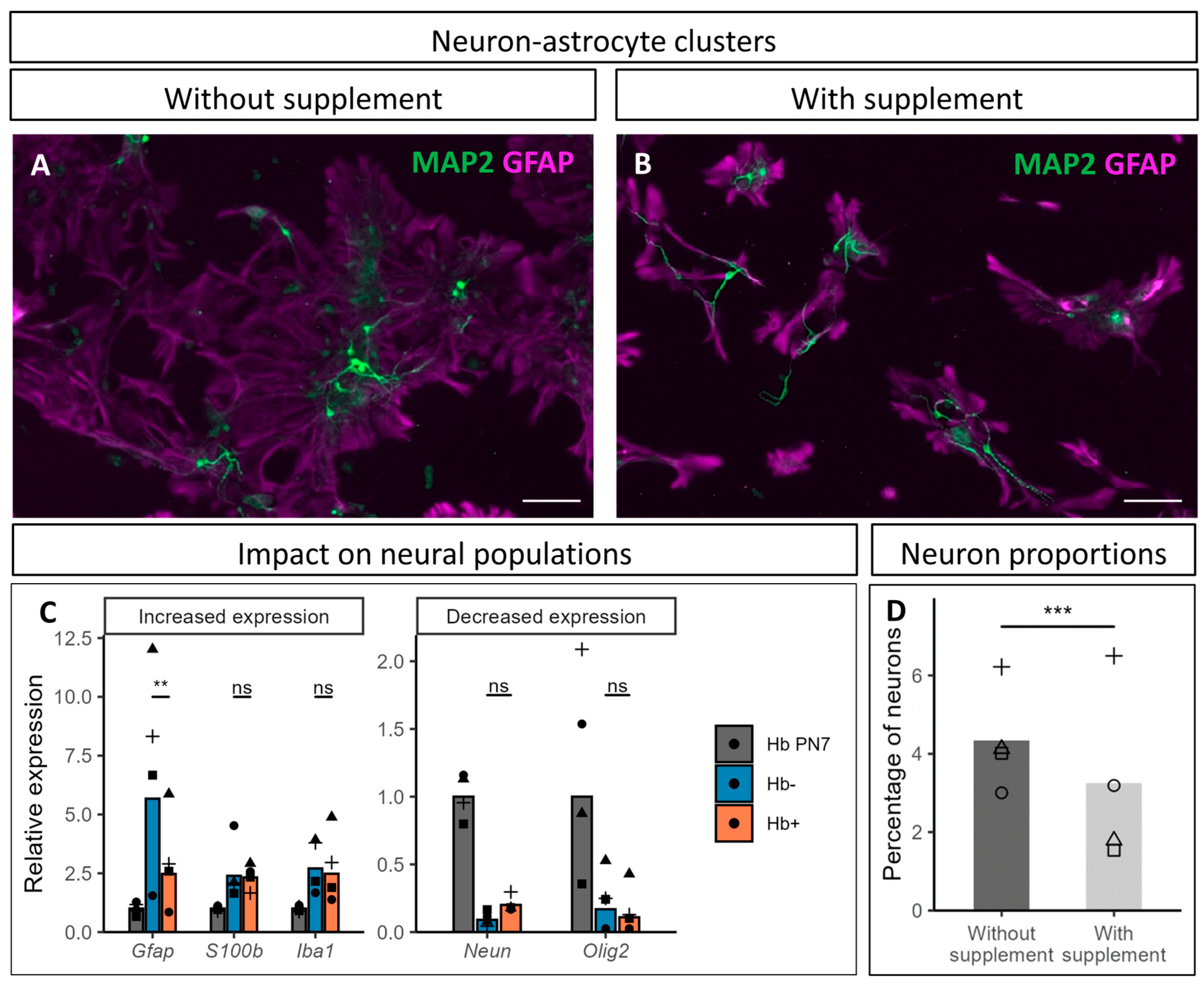
3.5. Impact of CultureOne™ Supplement
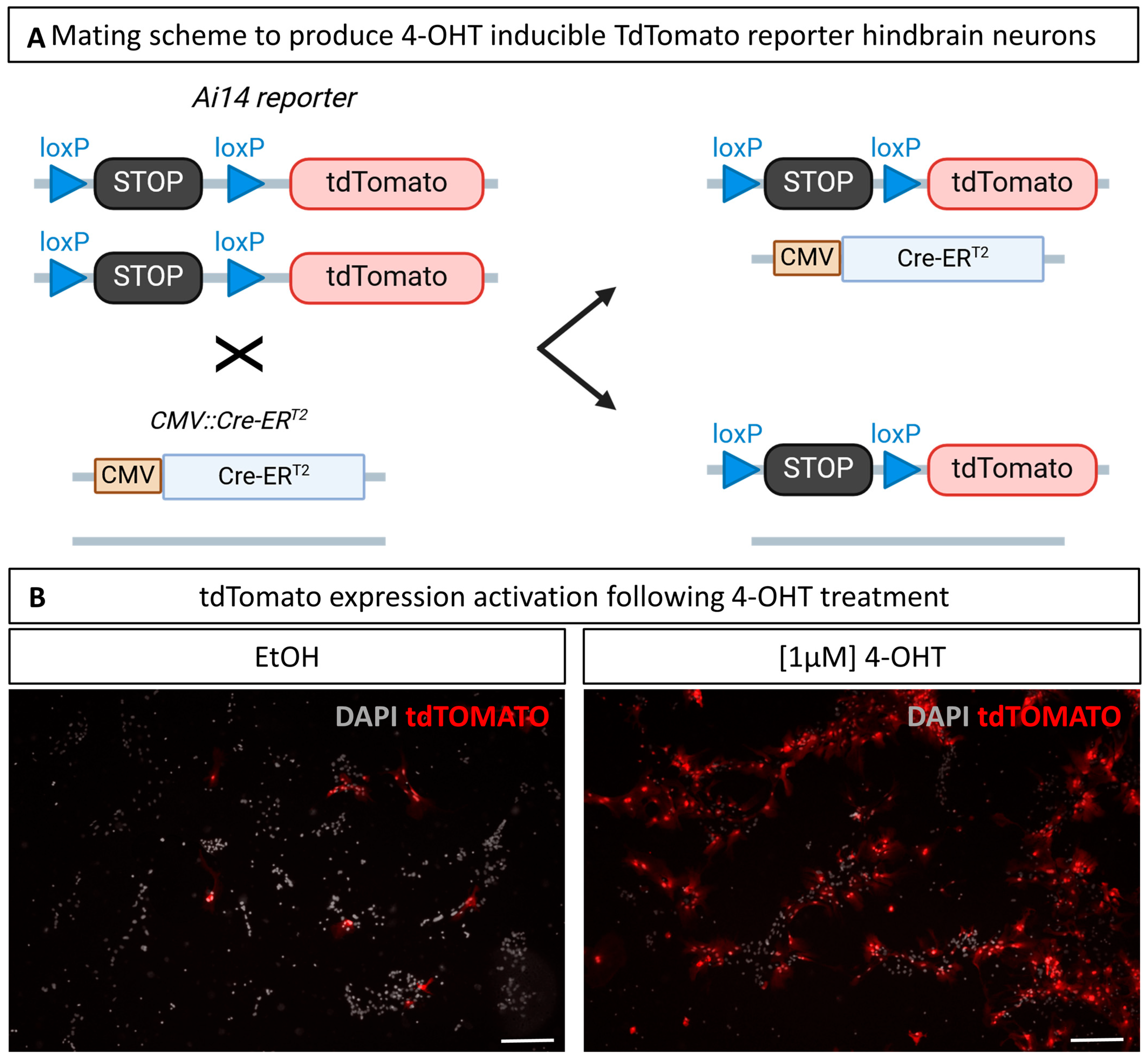
3.6. Culture of Hindbrain Neural Cells from Transgenic Mice and In Vitro Induction of Cre Recombination
4. Discussion
4.1. Neuronal Populations
4.2. Glial Cells Populations
4.3. Applications and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-OHT | 4-hydroxy-tamoxifen |
| Cx | Cortex |
| DIV | days in vitro |
| E | Embryonic day |
| EtOH | Ethanol |
| FBS | Fetal bovine serum |
| GABA | Gamma-aminobutyric acid |
| Hb | Hindbrain |
| HBSS | Hank’s balanced salt solution |
| PBS | Phosphate-buffered saline |
| PN | Postnatal day |
| PCR | Polymerase chain reaction |
| PFA | Paraformaldehyde |
| PLL | Poly-L-Lysine |
| RGC | Retinal ganglion cell |
| RT | Room temperature |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
| TB | Tris-buffered saline |
| TBS-Tx | Tris-buffered saline with Triton X-100 |
References
- Nicholls, J.G.; Paton, J.F.R. Brainstem: Neural Networks Vital for Life. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2447–2451. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Abdala, A.P.L.; Borgmann, A.; Rybak, I.A.; Paton, J.F.R. Brainstem Respiratory Networks: Building Blocks and Microcircuits. Trends Neurosci. 2013, 36, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, P.G. The Sympathetic Control of Blood Pressure. Nat. Rev. Neurosci. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Cullen, K.E. The Vestibular System: Multimodal Integration and Encoding of Self-Motion for Motor Control. Trends Neurosci. 2012, 35, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Luppi, P.-H.; Clement, O.; Sapin, E.; Peyron, C.; Gervasoni, D.; Léger, L.; Fort, P. Brainstem Mechanisms of Paradoxical (REM) Sleep Generation. Pflüg. Arch.-Eur. J. Physiol. 2012, 463, 43–52. [Google Scholar] [CrossRef]
- Ángeles Fernández-Gil, M.; Palacios-Bote, R.; Leo-Barahona, M.; Mora-Encinas, J.P. Anatomy of the Brainstem: A Gaze into the Stem of Life. Semin. Ultrasound CT MRI 2010, 31, 196–219. [Google Scholar] [CrossRef]
- Dubreuil, V.; Barhanin, J.; Goridis, C.; Brunet, J.-F. Breathing with Phox2b. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2477–2483. [Google Scholar] [CrossRef]
- Gillis, R.A.; Dezfuli, G.; Bellusci, L.; Vicini, S.; Sahibzada, N. Brainstem Neuronal Circuitries Controlling Gastric Tonic and Phasic Contractions: A Review. Cell. Mol. Neurobiol. 2022, 42, 333–360. [Google Scholar] [CrossRef]
- Muller, K.J.; Tsechpenakis, G.; Homma, R.; Nicholls, J.G.; Cohen, L.B.; Eugenin, J. Optical Analysis of Circuitry for Respiratory Rhythm in Isolated Brainstem of Foetal Mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2485–2491. [Google Scholar] [CrossRef]
- Cifra, A.; Nani, F.; Sharifullina, E.; Nistri, A. A Repertoire of Rhythmic Bursting Produced by Hypoglossal Motoneurons in Physiological and Pathological Conditions. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2493–2500. [Google Scholar] [CrossRef]
- Homma, R.; Baker, B.J.; Jin, L.; Garaschuk, O.; Konnerth, A.; Cohen, L.B.; Zecevic, D. Wide-Field and Two-Photon Imaging of Brain Activity with Voltage- and Calcium-Sensitive Dyes. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2453–2467. [Google Scholar] [CrossRef] [PubMed]
- Ausborn, J.; Shevtsova, N.A.; Caggiano, V.; Danner, S.M.; Rybak, I.A. Computational Modeling of Brainstem Circuits Controlling Locomotor Frequency and Gait. eLife 2019, 8, e43587. [Google Scholar] [CrossRef]
- Gordon, J.; Amini, S.; White, M.K. General Overview of Neuronal Cell Culture. In Neuronal Cell Culture; Amini, S., White, M.K., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 1–8. ISBN 978-1-62703-639-9. [Google Scholar]
- Kroeger, D.; Ferrari, L.L.; Petit, G.; Mahoney, C.E.; Fuller, P.M.; Arrigoni, E.; Scammell, T.E. Cholinergic, Glutamatergic, and GABAergic Neurons of the Pedunculopontine Tegmental Nucleus Have Distinct Effects on Sleep/Wake Behavior in Mice. J. Neurosci. 2017, 37, 1352–1366. [Google Scholar] [CrossRef]
- Moreira, T.S.; Takakura, A.C.; Falquetto, B.; Ramirez, J.-M.; Oliveira, L.M.; Silva, P.E.; Araujo, E.V. Neuroanatomical and Neurochemical Organization of Brainstem and Forebrain Circuits Involved in Breathing Regulation. J. Neurophysiol. 2025, 133, 1116–1137. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F. Systematic Morphometry of Catecholamine Nuclei in the Brainstem. Front. Neuroanat. 2017, 11, 98. [Google Scholar]
- Westergard, T.; Rothstein, J.D. Astrocyte Diversity: Current Insights and Future Directions. Neurochem. Res. 2020, 45, 1298–1305. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular Basis of Astrocyte Diversity and Morphology across the CNS in Health and Disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef]
- Erö, C.; Gewaltig, M.-O.; Keller, D.; Markram, H. A Cell Atlas for the Mouse Brain. Front. Neuroinform. 2018, 12, 84. [Google Scholar] [CrossRef]
- Kivell, B.M.; McDonald, F.J.; Miller, J.H. Method for Serum-Free Culture of Late Fetal and Early Postnatal Rat Brainstem Neurons. Brain Res. Protoc. 2001, 6, 91–99. [Google Scholar] [CrossRef]
- Gimenez-Cassina, A.; Lim, F.; Cerrato, T.; Palomo, G.M.; Diaz-Nido, J. Mitochondrial Hexokinase II Promotes Neuronal Survival and Acts Downstream of Glycogen Synthase Kinase-3. J. Biol. Chem. 2009, 284, 3001–3011. [Google Scholar] [CrossRef]
- van Niekerk, E.A.; Kawaguchi, R.; Marques de Freria, C.; Groeniger, K.; Marchetto, M.C.; Dupraz, S.; Bradke, F.; Geschwind, D.H.; Gage, F.H.; Tuszynski, M.H. Methods for Culturing Adult CNS Neurons Reveal a CNS Conditioning Effect. Cell Rep. Methods 2022, 2, 100255. [Google Scholar] [CrossRef]
- Santagati, F.; Minoux, M.; Ren, S.-Y.; Rijli, F.M. Temporal Requirement of Hoxa2 in Cranial Neural Crest Skeletal Morphogenesis. Dev. Camb. Engl. 2005, 132, 4927–4936. [Google Scholar] [CrossRef]
- Lizen, B.; Claus, M.; Jeannotte, L.; Rijli, F.M.; Gofflot, F. Perinatal Induction of Cre Recombination with Tamoxifen. Transgenic Res. 2015, 24, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A Robust and High-Throughput Cre Reporting and Characterization System for the Whole Mouse Brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef]
- Gualdani, R.; Gailly, P.; Yuan, J.-H.; Yerna, X.; Di Stefano, G.; Truini, A.; Cruccu, G.; Dib-Hajj, S.D.; Waxman, S.G. A TRPM7 Mutation Linked to Familial Trigeminal Neuralgia: Omega Current and Hyperexcitability of Trigeminal Ganglion Neurons. Proc. Natl. Acad. Sci. USA 2022, 119, e2119630119. [Google Scholar] [CrossRef] [PubMed]
- Gualdani, R.; Barbeau, S.; Yuan, J.-H.; Jacobs, D.S.; Gailly, P.; Dib-Hajj, S.D.; Waxman, S.G. TRPV1 Corneal Neuralgia Mutation: Enhanced pH Response, Bradykinin Sensitization, and Capsaicin Desensitization. Proc. Natl. Acad. Sci. USA 2024, 121, e2406186121. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA Analysis Tool for Deep Sequencing of Plant Small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Lizen, B.; Hutlet, B.; Bissen, D.; Sauvegarde, D.; Hermant, M.; Ahn, M.-T.; Gofflot, F. HOXA5 Localization in Postnatal and Adult Mouse Brain Is Suggestive of Regulatory Roles in Postmitotic Neurons. J. Comp. Neurol. 2017, 525, 1155–1175. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2024. [Google Scholar]
- Posit Team. RStudio: Integrated Development Environment for R. Posit Software, PBC, Boston, MA, USA, 2025. Available online: http://www.posit.co/ (accessed on 14 April 2025).
- Wickham, H. Ggplot2; Use R! Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Verstraelen, P.; Garcia-Diaz Barriga, G.; Verschuuren, M.; Asselbergh, B.; Nuydens, R.; Larsen, P.H.; Timmermans, J.-P.; De Vos, W.H. Systematic Quantification of Synapses in Primary Neuronal Culture. iScience 2020, 23, 101542. [Google Scholar] [CrossRef]
- Richter, K.; Langnaese, K.; Kreutz, M.R.; Olias, G.; Zhai, R.; Scheich, H.; Garner, C.C.; Gundelfinger, E.D. Presynaptic Cytomatrix Protein Bassoon Is Localized at Both Excitatory and Inhibitory Synapses of Rat Brain. J. Comp. Neurol. 1999, 408, 437–448. [Google Scholar] [CrossRef]
- Brakeman, P.R.; Lanahan, A.A.; O’Brien, R.; Roche, K.; Barnes, C.A.; Huganir, R.L.; Worley, P.F. Homer: A Protein That Selectively Binds Metabotropic Glutamate Receptors. Nature 1997, 386, 284–288. [Google Scholar] [CrossRef]
- Betz, H.; Kuhse, J.; Schmieden, V.; Malosio, M.L.; Langosch, D.; Prior, P.; Schmitt, B.; Kirsch, J. How to Build a Glycinergic Postsynaptic Membrane. J. Cell Sci. Suppl. 1991, 15, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, F.L.; Trattnig, C.; Kuhse, J.; Nawrotzki, R.A.; Kirsch, J. Gephyrin: A Key Regulatory Protein of Inhibitory Synapses and Beyond. Histochem. Cell Biol. 2018, 150, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A Novel Gene Iba1 in the Major Histocompatibility Complex Class III Region Encoding an EF Hand Protein Expressed in a Monocytic Lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef]
- ThermoFisher Gibco Application Note: Improve the Purity of Primary Neurons with the CultureOne Supplement. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FBID%2FApplication-Notes%2Fprimary-neurons-cultureone-supplement-app-note.pdf (accessed on 24 July 2024).
- Sturrock, R.R.; Rao, K.A. A Quantitative Histological Study of Neuronal Loss from the Locus Coeruleus of Ageing Mice. Neuropathol. Appl. Neurobiol. 1985, 11, 55–60. [Google Scholar] [CrossRef]
- von Coelln, R.; Thomas, B.; Savitt, J.M.; Lim, K.L.; Sasaki, M.; Hess, E.J.; Dawson, V.L.; Dawson, T.M. Loss of Locus Coeruleus Neurons and Reduced Startle in Parkin Null Mice. Proc. Natl. Acad. Sci. USA 2004, 101, 10744–10749. [Google Scholar] [CrossRef]
- Masuko, S.; Nakajima, Y.; Nakajima, S.; Yamaguchi, K. Noradrenergic Neurons from the Locus Ceruleus in Dissociated Cell Culture: Culture Methods, Morphology, and Electrophysiology. J. Neurosci. Off. J. Soc. Neurosci. 1986, 6, 3229–3241. [Google Scholar] [CrossRef]
- Tan, C.X.; Burrus Lane, C.J.; Eroglu, C. Role of Astrocytes in Synapse Formation and Maturation. Curr. Top. Dev. Biol. 2021, 142, 371–407. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Barres, B.A. Synaptic Efficacy Enhanced by Glial Cells In Vitro. Science 1997, 277, 1684–1687. [Google Scholar] [CrossRef]
- Buosi, A.S.; Matias, I.; Araujo, A.P.B.; Batista, C.; Gomes, F.C.A. Heterogeneity in Synaptogenic Profile of Astrocytes from Different Brain Regions. Mol. Neurobiol. 2018, 55, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte Glypicans 4 and 6 Promote Formation of Excitatory Synapses via GluA1 AMPA Receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Zhao, S.; Umpierre, A.D.; Wu, L.-J. Tuning Neural Circuits and Behaviors by Microglia in the Adult Brain. Trends Neurosci. 2024, 47, 181–194. [Google Scholar] [CrossRef]
- Rodarie, D.; Verasztó, C.; Roussel, Y.; Reimann, M.; Keller, D.; Ramaswamy, S.; Markram, H.; Gewaltig, M.-O. A Method to Estimate the Cellular Composition of the Mouse Brain from Heterogeneous Datasets. PLoS Comput. Biol. 2022, 18, e1010739. [Google Scholar] [CrossRef] [PubMed]
- Roussel, Y.; Verasztó, C.; Rodarie, D.; Damart, T.; Reimann, M.; Ramaswamy, S.; Markram, H.; Keller, D. Mapping of Morpho-Electric Features to Molecular Identity of Cortical Inhibitory Neurons. PLoS Comput. Biol. 2023, 19, e1010058. [Google Scholar] [CrossRef]
- Burry, R.W. Antimitotic Drugs That Enhance Neuronal Survival in Olfactory Bulb Cell Cultures. Brain Res. 1983, 261, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Fracek, S.P.; Guo, L.; Schafer, R. Morphological Characteristics of Cultured Olfactory Bulb Cells. Exp. Brain Res. 1994, 100, 421–436. [Google Scholar] [CrossRef]
- Liu, R.; Lin, G.; Xu, H. An Efficient Method for Dorsal Root Ganglia Neurons Purification with a One-Time Anti-Mitotic Reagent Treatment. PLoS ONE 2013, 8, e60558. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Wallace, T.L.; Johnson, E.M. Cytosine Arabinoside Kills Postmitotic Neurons in a Fashion Resembling Trophic Factor Deprivation: Evidence That a Deoxycytidine-Dependent Process May Be Required for Nerve Growth Factor Signal Transduction. J. Neurosci. Off. J. Soc. Neurosci. 1990, 10, 184–193. [Google Scholar] [CrossRef]
- Wallace, T.L.; Johnson, E.M. Cytosine Arabinoside Kills Postmitotic Neurons: Evidence That Deoxycytidine May Have a Role in Neuronal Survival That Is Independent of DNA Synthesis. J. Neurosci. 1989, 9, 115–124. [Google Scholar] [CrossRef]
- Oorschot, D.E. Effect of Fluorodeoxyuridine on Neurons and Non-Neuronal Cells in Cerebral Explants. Exp. Brain Res. 1989, 78, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Agrawal, S.K. Curcumin Alleviates Oxidative Stress and Mitochondrial Dysfunction in Astrocytes. Neuroscience 2016, 333, 92–103. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers (5′-3′) | PCR Program | |||||
|---|---|---|---|---|---|---|
| Ai14 Rosa26R-tdTomato | 1 | 95 °C | 5 min | |||
| Fw | ggcattaaagcagcgtatcc | 2 | 95 °C | 30 s | 35 cycles | |
| 3 | 50 °C | 1 min | ||||
| Rv | ctgttcctgtacggcatgg | 4 | 72 °C | 40 s | ||
| 5 | 72 °C | 7 min | ||||
| CMV-CreERT2 | 1 | 95 °C | 5 min | |||
| Fw | gtccgggctgccacgaccaa | 2 | 95 °C | 1 min | 35 cycles | |
| 3 | 65 °C | 1 min | ||||
| Rv | acggaaatccatgcgtcgaccagtt | 4 | 72 °C | 30 s | ||
| 5 | 72 °C | 7 min | ||||
| Antigen | (Sub-) Cellular Target | Species | Catalog Number | Batch Number | Dilution | RRID |
|---|---|---|---|---|---|---|
| Bassoon | Presynapse | Mouse | 75-491 | 475-3JS-69 | 1/500 | AB_2716712 |
| CHAT | Cholinergic neurons | Mouse | MAB305 | 2455649 | 1/100 | AB_94647 |
| GAD67 | GABAergic neurons | Mouse | MAB5406 | 2042787 | 1/100 | AB_2278725 |
| Gephyrin | Inhibitory synapse | Rabbit | 147 008 | 1-12 | 1/500 | AB_2619834 |
| GFAP | Astrocytes | Mouse | Sc-58766 | J1308 | 1/500 | AB_783554 |
| Homer1 | Excitatory synapse | Rabbit | 160 003 | 3-72 | 1/500 | AB_887730 |
| MAP2 | Neuronal body and dendrites | Chicken | PA1-16751 | WC3231134A | 1/2000 | AB_2138189 |
| NEUN | Neuronal nucleus | Mouse | MAB377 | 3018822 | 1/500 | AB_2298772 |
| OLIG2 | Oligodendrocyte nucleus | Mouse | MABN50 | 2026416 | 1/100 | AB_10807410 |
| TH | Dopaminergic neurons | Mouse | MAB318 | LV1541610 | 1/100 | AB_2313764 |
| VGLUT2 | Glutamatergic neurons | Mouse | Ab79157 | Gr99208-2 | 1/100 | AB_1603114 |
| IBA1 | Microglia | Rabbit | 019-19741 | SAJ2266 | 1/500 | AB_839504 |
| Serotonin | Serotonergic neurons | Rabbit | 20080 | 542021 | 1/500 | AB_10718516 |
| Target Species | Alexa Fluor | Species | Catalog Number | Batch Number | Dilution | RRID |
|---|---|---|---|---|---|---|
| Anti-chicken | 488 | Goat | A11039 | 1937504 | 1/500 | AB_2534096 |
| Anti-chicken | 647 | Donkey | 703-605-155 | 1/500 | AB_2340379 | |
| Anti-mouse | 488 | Donkey | A21202 | 714258 | 1/500 | AB_141607 |
| Anti-mouse | 555 | Goat | 4409 | 19 | 1/500 | AB_1904022 |
| Anti-rabbit | 555 | Goat | 4413 | 20 | 1/500 | AB_10694110 |
| Target | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| Neun | cttatggagcggtcgtgtatca | taactgtcactgtaggctgct |
| Gfap | catcgagatcgccacctaca | ctggaggttggagaaagtctgt |
| Olig2 | cctggtgtctagtcgcccat | gacacagtccctcctgtgaa |
| Hoxa5 | gcgcaagctgcacattagt | ggcatgagctatttcgatcc |
| Th | ctcctcagttctgtgcgtcg | gtcagagaagcccggatgg |
| S100beta | tggttgccctcattgatgtct | cccatccccatcttcgtcc |
| Iba1 | cagggatttgcagggaggaaa | agtttggacggcagatcctc |
| Vglut2 | tgaaatcagcaaggttggca | cccccgataggcacaatgat |
| Gad67 | gagacaccctgaagtacggg | atgagaacaaacacgggtgc |
| Chat | ccattgtgaagcggtttggg | gccaggcggttgtttagataca |
| Glyt2 | cacgctggagcacaacaatac | cagttccctcgggccttatt |
| Gata3 | ttgataaggggccggttctg | cgggttcggatgtaagtcga |
| Fezf2 | aaattatccatacccaggaaaaacc | ctgtgggtgagcttgtgattc |
| Ppia | aggattcatgtgccagggtg | ccgccagtgccattatgg |
| H2a | gctggtggtggtgtcatcc | tttcttcccgatcagcgatt |
| 36B4 | tgagattcgggatatgctgttg | ttccaatggtgcctctggaga |
| Hprt | gcttgctggtgaaaaggacctctcgaag | ccctgaagtactcattatagtcaagggcat |
| Tbp | acccttcaccaatgactcctatg | atgatgactgcagcaaatcgc |
| Rpl32 | ggcaccagtcagaccgatat | caggatctggcccttgaac |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glibert, H.; Bridoux, L.; Palate, M.; Piget, C.; Ahn, M.-T.; Gualdani, R.; Domínguez-Bajo, A.; Clotman, F.; Rijli, F.M.; Gofflot, F. Primary Cell Cultures in Neurobiology: Optimized Protocol for Culture of Mouse Fetal Hindbrain Neurons. Cells 2025, 14, 758. https://doi.org/10.3390/cells14110758
Glibert H, Bridoux L, Palate M, Piget C, Ahn M-T, Gualdani R, Domínguez-Bajo A, Clotman F, Rijli FM, Gofflot F. Primary Cell Cultures in Neurobiology: Optimized Protocol for Culture of Mouse Fetal Hindbrain Neurons. Cells. 2025; 14(11):758. https://doi.org/10.3390/cells14110758
Chicago/Turabian StyleGlibert, Hadrien, Laure Bridoux, Maëlle Palate, Coralie Piget, Marie-Thérèse Ahn, Roberta Gualdani, Ana Domínguez-Bajo, Frédéric Clotman, Filippo M. Rijli, and Françoise Gofflot. 2025. "Primary Cell Cultures in Neurobiology: Optimized Protocol for Culture of Mouse Fetal Hindbrain Neurons" Cells 14, no. 11: 758. https://doi.org/10.3390/cells14110758
APA StyleGlibert, H., Bridoux, L., Palate, M., Piget, C., Ahn, M.-T., Gualdani, R., Domínguez-Bajo, A., Clotman, F., Rijli, F. M., & Gofflot, F. (2025). Primary Cell Cultures in Neurobiology: Optimized Protocol for Culture of Mouse Fetal Hindbrain Neurons. Cells, 14(11), 758. https://doi.org/10.3390/cells14110758