

Chimeric Autoantibody Receptor- and/or Peptide-MHC-Based CAR Therapies for Targeted Elimination of Antigen-Specific B or T Cells in Hypersensitivity Disorders Such as Allergies and Autoimmune Diseases

 , , ,
, , ,
Abstract
1. Introduction
2. Methods
2.1. Search Strategy
2.2. Screening and Data Extraction
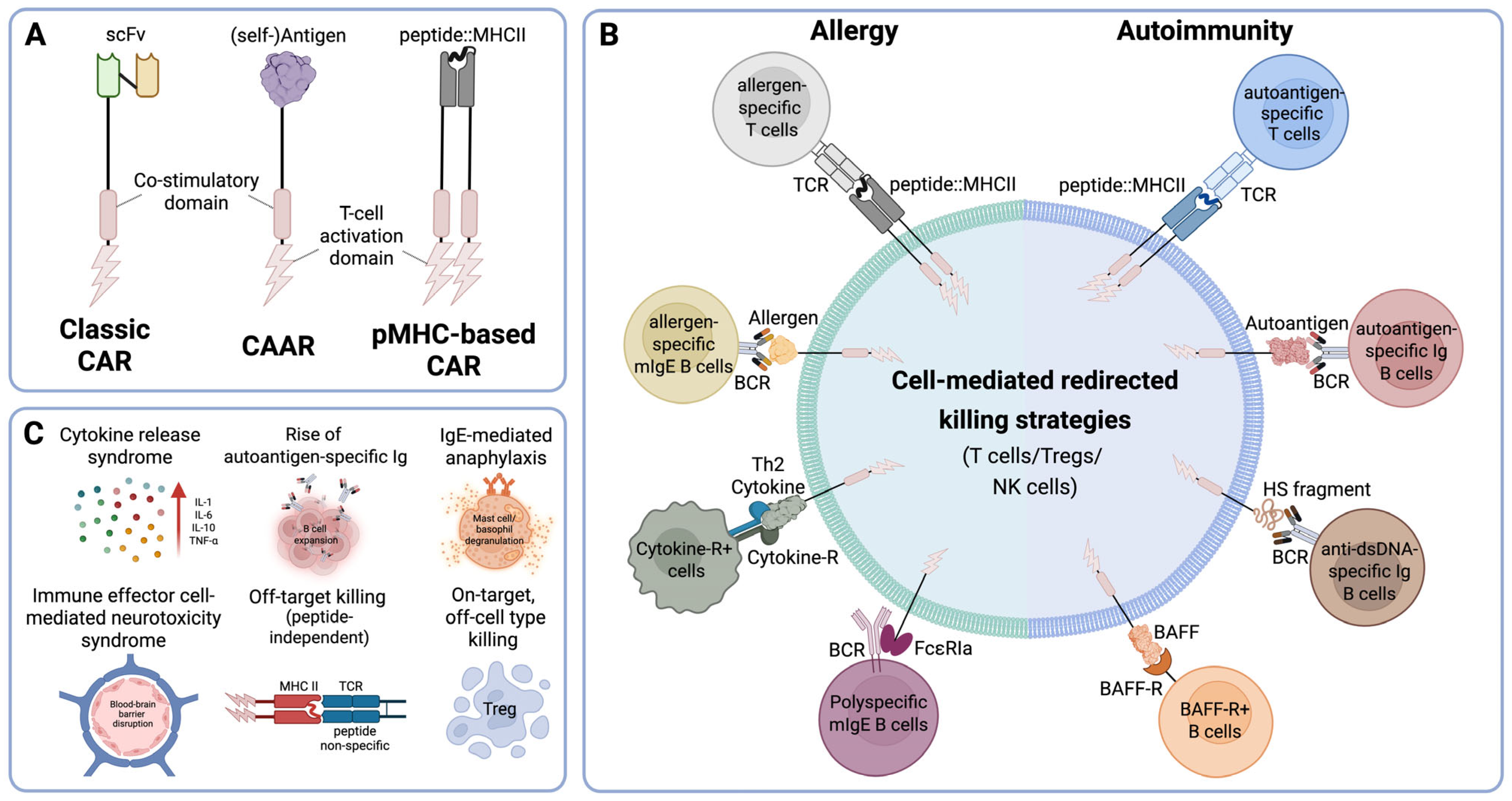
3. Targeting Specificity: CARs vs. CAARs vs. pMHC-Based CAR
4. Hypersensitivity Disorders
4.1. Antigen-Based CARs in Allergy Treatment
4.2. Antigen-Based CAR in the Treatment of Autoimmune Diseases
5. Concluding Remarks: Challenges and Future Strategies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hatzubai, A.; Maloney, D.G.; Levy, R. The use of a monoclonal anti-idiotype antibody to study the biology of a human B cell lymphoma. J. Immunol. 1981, 126, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; GrilloLopez, A.J.; Bodkin, D.J.; White, C.A.; Liles, T.M.; Royston, I.; Varns, C.; Rosenberg, J.; Levy, R. IDEC-C2B8: Results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J. Clin. Oncol. 1997, 15, 3266–3274. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab-The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef]
- Puertas, B.; Mateos, M.V.; Gonzalez-Calle, V. Anti-BCMA CAR T-cell Therapy: Changing the Natural History of Multiple Myeloma. Hemasphere 2022, 6, e691. [Google Scholar] [CrossRef]
- Vicente, A.M.; Ballensiefen, W.; Jonsson, J.I. How personalised medicine will transform healthcare by 2030: The ICPerMed vision. J. Transl. Med. 2020, 18, 180. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- FDA. FDA Approval Brings First Gene Therapy to the United States. Available online: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed on 17 February 2025).
- Zabel, M.; Tauber, P.A.; Pickl, W.F. The making and function of CAR cells. Immunol. Lett. 2019, 212, 53–69. [Google Scholar] [CrossRef]
- Boucher, J.C.; Davila, M.L. Chimeric Antigen Receptor Design Today and Tomorrow. Cancer J. 2021, 27, 92–97. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Taheri, M.; Javan, F.; Poudineh, M.; Athari, S.S. Beyond CAR-T: The rise of CAR-NK cell therapy in asthma immunotherapy. J. Transl. Med. 2024, 22, 736. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Schmitt, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Muller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef]
- Kolanus, W.; Romeo, C.; Seed, B. T cell activation by clustered tyrosine kinases. Cell 1993, 74, 171–183. [Google Scholar] [CrossRef]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgard, G.; Wenthe, J.; Lovgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallbook, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef]
- Bandukwala, H.S.; Wu, Y.; Feuerer, M.; Chen, Y.; Barboza, B.; Ghosh, S.; Stroud, J.C.; Benoist, C.; Mathis, D.; Rao, A.; et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 2011, 34, 479–491. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Dwivedi, A.; Karulkar, A.; Ghosh, S.; Rafiq, A.; Purwar, R. Lymphocytes in Cellular Therapy: Functional Regulation of CAR T Cells. Front. Immunol. 2018, 9, 3180. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Maus, M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019, 37, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.L.; Nguyen, P.; Leitenberg, D.; Flavell, R.A. Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood 2001, 98, 2364–2371. [Google Scholar] [CrossRef]
- Nguyen, P.; Geiger, T.L. Antigen-specific targeting of CD8+ T cells with receptor-modified T lymphocytes. Gene Ther. 2003, 10, 594–604. [Google Scholar] [CrossRef]
- Nguyen, P.; Duthoit, C.T.; Geiger, T.L. Induction of tolerance and immunity by redirected B cell-specific cytolytic T lymphocytes. Gene Ther. 2007, 14, 1739–1749. [Google Scholar] [CrossRef]
- Khan, A.N.; Chowdhury, A.; Karulkar, A.; Jaiswal, A.K.; Banik, A.; Asija, S.; Purwar, R. Immunogenicity of CAR-T Cell Therapeutics: Evidence, Mechanism and Mitigation. Front. Immunol. 2022, 13, 886546. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef]
- Lorenz, F.K.; Ellinger, C.; Kieback, E.; Wilde, S.; Lietz, M.; Schendel, D.J.; Uckert, W. Unbiased Identification of T-Cell Receptors Targeting Immunodominant Peptide–MHC Complexes for T-Cell Receptor mmunotherapy. Hum. Gene Ther. 2017, 28, 1158–1168. [Google Scholar] [CrossRef]
- Spanier, J.A.; Fung, V.; Wardell, C.M.; Alkhatib, M.H.; Chen, Y.; Swanson, L.A.; Dwyer, A.J.; Weno, M.E.; Silva, N.; Mitchell, J.S.; et al. Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J. Clin. Investig. 2023, 133, e168601. [Google Scholar] [CrossRef]
- Hanssens, H.; Meeus, F.; De Veirman, K.; Breckpot, K.; Devoogdt, N. The antigen-binding moiety in the driver’s seat of CARs. Med. Res. Rev. 2022, 42, 306–342. [Google Scholar] [CrossRef]
- Ward, D.E.; Fay, B.L.; Adejuwon, A.; Han, H.; Ma, Z. Chimeric Antigen Receptors Based on Low Affinity Mutants of FcepsilonRI Re-direct T Cell Specificity to Cells Expressing Membrane IgE. Front. Immunol. 2018, 9, 2231. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Liu, Y.; Wang, L.; He, Z.; Zhao, X.; Ma, Y.; Jia, Y.; Li, Z.; Yin, N.; Peng, M. A single infusion of engineered long-lived and multifunctional T cells confers durable remission of asthma in mice. Nat. Immunol. 2024, 25, 1059–1072. [Google Scholar] [CrossRef]
- Abdeladhim, M.; Zhang, A.H.; Kropp, L.E.; Lindrose, A.R.; Venkatesha, S.H.; Mitre, E.; Scott, D.W. Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin. Immunol. 2019, 207, 49–54. [Google Scholar] [CrossRef]
- Jyothi, M.D.; Flavell, R.A.; Geiger, T.L. Targeting autoantigen-specific T cells and suppression of autoimmune encephalomyelitis with receptor-modified T lymphocytes. Nat. Biotechnol. 2002, 20, 1215–1220. [Google Scholar] [CrossRef]
- Mekala, D.J.; Alli, R.S.; Geiger, T.L. IL-10-dependent suppression of experimental allergic encephalomyelitis by Th2-differentiated, anti-TCR redirected T lymphocytes. J. Immunol. 2005, 174, 3789–3797. [Google Scholar] [CrossRef]
- Mekala, D.J.; Geiger, T.L. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood 2005, 105, 2090–2092. [Google Scholar] [CrossRef]
- Mekala, D.J.; Alli, R.S.; Geiger, T.L. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 11817–11822. [Google Scholar] [CrossRef]
- Yi, J.; Miller, A.T.; Archambault, A.S.; Jones, A.J.; Bradstreet, T.R.; Bandla, S.; Hsu, Y.S.; Edelson, B.T.; Zhou, Y.W.; Fremont, D.H.; et al. Antigen-specific depletion of CD4(+) T cells by CAR T cells reveals distinct roles of higher- and lower-affinity TCRs during autoimmunity. Sci. Immunol. 2022, 7, eabo0777. [Google Scholar] [CrossRef]
- Sahlolbei, M.; Azangou-Khyavy, M.; Khanali, J.; Khorsand, B.; Shiralipour, A.; Ahmadbeigi, N.; Madjd, Z.; Ghanbarian, H.; Ardjmand, A.; Hashemi, S.M.; et al. Engineering chimeric autoantibody receptor T cells for targeted B cell depletion in multiple sclerosis model: An in-vitro study. Heliyon 2023, 9, e19763. [Google Scholar] [CrossRef]
- Reincke, S.M.; von Wardenburg, N.; Homeyer, M.A.; Kornau, H.C.; Spagni, G.; Li, L.Y.; Kreye, J.; Sanchez-Sendin, E.; Blumenau, S.; Stappert, D.; et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell 2023, 186, 5084–5097e18. [Google Scholar] [CrossRef]
- Scott, G.S.; Fishman, S.; Khai Siew, L.; Margalit, A.; Chapman, S.; Chervonsky, A.V.; Wen, L.; Gross, G.; Wong, F.S. Immunotargeting of insulin reactive CD8 T cells to prevent diabetes. J. Autoimmun. 2010, 35, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Fishman, S.; Lewis, M.D.; Siew, L.K.; De Leenheer, E.; Kakabadse, D.; Davies, J.; Ziv, D.; Margalit, A.; Karin, N.; Gross, G.; et al. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol. Ther. 2017, 25, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Thelin, M.A.; Parrish, H.L.; Deshpande, N.R.; Lee, M.S.; Karimzadeh, A.; Niewczas, M.A.; Serwold, T.; Kuhns, M.S. A biomimetic five-module chimeric antigen receptor ((5M)CAR) designed to target and eliminate antigen-specific T cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28950–28959. [Google Scholar] [CrossRef] [PubMed]
- Kakabadse, D.; Chen, D.; Fishman, S.; Weinstein-Marom, H.; Davies, J.; Wen, L.; Gross, G.; Wong, F.S. Regulatory CD4+ T cells redirected against pathogenic CD8+ T cells protect NOD mice from development of autoimmune diabetes. Front. Immunol. 2024, 15, 1463971. [Google Scholar] [CrossRef]
- Uppin, V.; Gibbons, H.; Troje, M.; Feinberg, D.; Webber, B.R.; Moriarity, B.S.; Parameswaran, R. CAR-T cell targeting three receptors on autoreactive B cells for systemic lupus erythematosus therapy. J. Autoimmun. 2025, 151, 103369. [Google Scholar] [CrossRef]
- Sole, C.; Royo, M.; Sandoval, S.; Moline, T.; Gabaldon, A.; Cortes-Hernandez, J. Precise Targeting of Autoantigen-Specific B Cells in Lupus Nephritis with Chimeric Autoantibody Receptor T Cells. Int. J. Mol. Sci. 2024, 25, 4226. [Google Scholar] [CrossRef]
- Seifert, L.; Riecken, K.; Zahner, G.; Hambach, J.; Hagenstein, J.; Dubberke, G.; Huber, T.B.; Koch-Nolte, F.; Fehse, B.; Tomas, N.M. An antigen-specific chimeric autoantibody receptor (CAAR) NK cell strategy for the elimination of anti-PLA2R1 and anti-THSD7A antibody-secreting cells. Kidney Int. 2024, 105, 886–889. [Google Scholar] [CrossRef]
- Oh, S.; Mao, X.; Manfredo-Vieira, S.; Lee, J.; Patel, D.; Choi, E.J.; Alvarado, A.; Cottman-Thomas, E.; Maseda, D.; Tsao, P.Y.; et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat. Biotechnol. 2023, 41, 1229–1238. [Google Scholar] [CrossRef]
- Whittington, K.B.; Prislovsky, A.; Beaty, J.; Albritton, L.; Radic, M.; Rosloniec, E.F. CD8+ T Cells Expressing an HLA-DR1 Chimeric Antigen Receptor Target Autoimmune CD4+ T Cells in an Antigen-Specific Manner and Inhibit the Development of Autoimmune Arthritis. J. Immunol. 2022, 208, 16–26. [Google Scholar] [CrossRef]
- Zhu, H.X.; Yang, S.H.; Gao, C.Y.; Bian, Z.H.; Chen, X.M.; Huang, R.R.; Meng, Q.L.; Li, X.; Jin, H.; Tsuneyama, K.; et al. Targeting pathogenic CD8(+) tissue-resident T cells with chimeric antigen receptor therapy in murine autoimmune cholangitis. Nat. Commun. 2024, 15, 2936. [Google Scholar] [CrossRef]
- Zhang, A.H.; Yoon, J.; Kim, Y.C.; Scott, D.W. Targeting Antigen-Specific B Cells Using Antigen-Expressing Transduced Regulatory T Cells. J. Immunol. 2018, 201, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, Y.; Shu, J.; Jiang, H.; Huang, L.; Xu, M.; Liu, J.; Hu, Y.; Mei, H. GPIbalpha CAAR T cells function like a Trojan horse to eliminate autoreactive B cells to treat immune thrombocytopenia. Haematologica 2024, 109, 2256–2270. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Rezaei, N. Hypersensitivity. In Encyclopedia of Infection and Immunity; Elsevier: Amsterdam, The Netherlands, 2022; pp. 243–258. [Google Scholar]
- Hopp, R.J. Hypersensitivity Reactions: An Everyday Occurrence in Pediatric Allergy Clinics. Pediatr. Allergy Immunol. Pulmonol. 2020, 33, 12–18. [Google Scholar] [CrossRef]
- Jutel, M.; Agache, I.; Zemelka-Wiacek, M.; Akdis, M.; Chivato, T.; Del Giacco, S.; Gajdanowicz, P.; Gracia, I.E.; Klimek, L.; Lauerma, A.; et al. Nomenclature of allergic diseases and hypersensitivity reactions: Adapted to modern needs: An EAACI position paper. Allergy 2023, 78, 2851–2874. [Google Scholar] [CrossRef]
- Durham, S.R.; Shamji, M.H. Allergen immunotherapy: Past, present and future. Nat. Rev. Immunol. 2023, 23, 317–328. [Google Scholar] [CrossRef]
- Protic-Rosic, I.; Lopandic, Z.; Popovic, D.; Blagojevic, G.; Gavrovic-Jankulovic, M. rBet v 1a-BanLec(wt) induce upregulation of IL-10 and IFN-gamma gene expression in Caco-2/THP-1 co-culture and secretion of IL-10 and IFN-gamma/IL-4 levels in PBMCs of birch pollen allergic donors. Int. Immunopharmacol. 2024, 129, 111607. [Google Scholar] [CrossRef]
- van Wijk, R.G.; Dahl, R. Pros and cons: Should AIT be considered in all patients with allergic asthma? Authorea 2021. [Google Scholar] [CrossRef]
- Belliveau, P.P. Omalizumab: A monoclonal anti-IgE antibody. MedGenMed 2005, 7, 27. [Google Scholar]
- Schmetterer, K.G.; Haiderer, D.; Leb-Reichl, V.M.; Neunkirchner, A.; Jahn-Schmid, B.; Kung, H.J.; Schuch, K.; Steinberger, P.; Bohle, B.; Pickl, W.F. Bet v 1-specific T-cell receptor/forkhead box protein 3 transgenic T cells suppress Bet v 1-specific T-cell effector function in an activation-dependent manner. J. Allergy Clin. Immunol. 2011, 127, 238–245.e3. [Google Scholar] [CrossRef]
- Limnander, A.; Kaur, N.; Asrat, S.; Tasker, C.; Boyapati, A.; Ben, L.H.; Janczy, J.; Pedraza, P.; Abreu, P.; Chen, W.C.; et al. A therapeutic strategy to target distinct sources of IgE and durably reverse allergy. Sci. Transl. Med. 2023, 15, eadf9561. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Levings, M.K.; Scott, D.W. Engineering therapeutic T cells to suppress alloimmune responses using TCRs, CARs, or BARs. Am. J. Transplant. 2018, 18, 1305–1311. [Google Scholar] [CrossRef]
- Boonpiyathad, T.; Sozener, Z.C.; Akdis, M.; Akdis, C.A. The role of Treg cell subsets in allergic disease. Asian Pac. J. Allergy Immunol. 2020, 38, 139–149. [Google Scholar] [CrossRef]
- Vukovic, J.; Abazovic, D.; Vucetic, D.; Medenica, S. CAR-engineered T cell therapy as an emerging strategy for treating autoimmune diseases. Front. Med. 2024, 11, 1447147. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef]
- Kissler, S. Toward a precision medicine approach for autoimmunity. Proc. Natl. Acad. Sci. USA 2022, 119, e2204841119. [Google Scholar] [CrossRef]
- Waldner, H. The role of innate immune responses in autoimmune disease development. Autoimmun. Rev. 2009, 8, 400–404. [Google Scholar] [CrossRef]
- Sadeqi Nezhad, M.; Seifalian, A.; Bagheri, N.; Yaghoubi, S.; Karimi, M.H.; Adbollahpour-Alitappeh, M. Chimeric Antigen Receptor Based Therapy as a Potential Approach in Autoimmune Diseases: How Close Are We to the Treatment? Front. Immunol. 2020, 11, 603237. [Google Scholar] [CrossRef]
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef]
- Nunez, D.; Patel, D.; Volkov, J.; Wong, S.; Vorndran, Z.; Muller, F.; Aigner, M.; Volkl, S.; Mackensen, A.; Schett, G.; et al. Cytokine and reactivity profiles in SLE patients following anti-CD19 CART therapy. Mol. Ther. Methods Clin. Dev. 2023, 31, 101104. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Sengupta, R.; Gold, R.; Schroers, R.; Haghikia, A.; Lorente, M.; Pendleton, M.; Register, A.; Heesen, C.; Kroger, N.; et al. Successful generation of fully human, second generation, anti-CD19 CAR T cells for clinical use in patients with diverse autoimmune disorders. Cytotherapy 2025, 27, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Granit, V.; Benatar, M.; Kurtoglu, M.; Miljkovic, M.D.; Chahin, N.; Sahagian, G.; Feinberg, M.H.; Slansky, A.; Vu, T.; Jewell, C.M.; et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): A prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. 2023, 22, 578–590. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Hamadani, M.; Miklos, D.B.; Holmes, H.; Hinkle, J.; Kennedy-Wilde, J.; Maller, O.; Weinstein, M.; Galimi, F.; Lai, R.; et al. A phase 1 study of ADI-001- Anti-CD20 CAR-engineered allogeneic gamma delta (γδ) T cells in adults with B-cell malignancies. J. Clin. Oncol. 2022, 40, 7509. [Google Scholar] [CrossRef]
- Doglio, M.; Alexander, T.; Del Papa, N.; Snowden, J.A.; Greco, R.; Autoimmune Diseases Working Party of the European Society. New insights in systemic lupus erythematosus: From regulatory T cells to CAR-T-cell strategies. J. Allergy Clin. Immunol. 2022, 150, 1289–1301. [Google Scholar] [CrossRef]
- Greco, R.; Alexander, T.; Del Papa, N.; Muller, F.; Saccardi, R.; Sanchez-Guijo, F.; Schett, G.; Sharrack, B.; Snowden, J.A.; Tarte, K.; et al. Innovative cellular therapies for autoimmune diseases: Expert-based position statement and clinical practice recommendations from the EBMT practice harmonization and guidelines committee. EClinicalMedicine 2024, 69, 102476. [Google Scholar] [CrossRef]
- Makuch, M.; Wilson, R.; Al-Diwani, A.; Varley, J.; Kienzler, A.K.; Taylor, J.; Berretta, A.; Fowler, D.; Lennox, B.; Leite, M.I.; et al. N-methyl-D-aspartate receptor antibody production from germinal center reactions: Therapeutic implications. Ann. Neurol. 2018, 83, 553–561. [Google Scholar] [CrossRef]
- Frigault, M.J.; Dietrich, J.; Gallagher, K.; Roschewski, M.; Jordan, J.T.; Forst, D.; Plotkin, S.R.; Cook, D.; Casey, K.S.; Lindell, K.A.; et al. Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: A phase 1/2 clinical trial. Blood 2022, 139, 2306–2315. [Google Scholar] [CrossRef]
- WHO. Diabetes—WHO Europe. Available online: https://www.who.int/europe/health-topics/diabetes#tab=tab_2 (accessed on 19 March 2025).
- Purcell, A.W.; Sechi, S.; DiLorenzo, T.P. The Evolving Landscape of Autoantigen Discovery and Characterization in Type 1 Diabetes. Diabetes 2019, 68, 879–886. [Google Scholar] [CrossRef]
- Turner, S.J.; La Gruta, N.L. A subset of immune-system T cells branded as seeds for type 1 diabetes. Nature 2022, 602, 35–36. [Google Scholar] [CrossRef]
- Demirkaya, E.; Sahin, S.; Romano, M.; Zhou, Q.; Aksentijevich, I. New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. J. Clin. Med. 2020, 9, 712. [Google Scholar] [CrossRef]
- Isenberg, D.; Rahman, A. Systemic lupus erythematosus—2005 annus mirabilis? Nat. Clin. Pract. Rheumatol. 2006, 2, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Smulski, C.R.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Pers, J.O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Saraux, A.; Jamin, C.; Youinou, P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef]
- Wong, D.P.; Roy, N.K.; Zhang, K.; Anukanth, A.; Asthana, A.; Shirkey-Son, N.J.; Dunmire, S.; Jones, B.J.; Lahr, W.S.; Webber, B.R.; et al. A BAFF ligand-based CAR-T cell targeting three receptors and multiple B cell cancers. Nat. Commun. 2022, 13, 217. [Google Scholar] [CrossRef]

{kind=link}
| Category | Review Question |
|---|---|
| Targeting Conditions | What hypersensitivity reactions (allergies, autoimmune diseases) are being addressed using pMHC CAR or antigen-based CAAR therapies? |
| CAR Design | What are the key characteristics of pMHC CAR and CAAR constructs designed for these indications? |
| Preclinical and Clinical Development | What stages of development (preclinical models, clinical trials) exist for these therapies? |
| Efficacy and Safety | What are the reported benefits, challenges, and safety concerns of pMHC CAR and CAAR therapies for treating hypersensitivity-related diseases? |
| Category | Criteria |
|---|---|
| Inclusion criteria |
|
| Exclusion criteria |
|
| Databases searched | Medline (via Ovid), Embase, Cochrane CENTRAL |
| Last search date | 3 February 2025 |
| Disease | CAR Type (Extracellular Domain) | Signaling Domains | Targeted Cells | Experiment Setting | Total Doses Administered * | Achieved Result * | Safety Considerations | Citation |
|---|---|---|---|---|---|---|---|---|
| Allergy | CAAR-Jurkat T cells (human FcεRIα, and its 5 mutants) | CD3ζ | Cells expressing membrane-bound IgE (mIgE) | In vitro | - | - | n.a. | [32] |
| CAAR-CD8+ T cells (mouse IL-5 and mouse IL-4 mutein) | CD28-CD3ζ | IL-5Rα+ cells | In vitro In vivo (mouse model) | Single intravenous injection | Long-term remission of asthma | No CRS or toxicity reported | [33] | |
| CAAR-Tregs (ovalbumin) | CD28-CD3ζ | anti-OVA IgE-producing B cells | In vivo (mouse model) | Single intravenous injection | Protection from hypothermia and anaphylaxis | No acute toxicity or anaphylaxis reported | [34] | |
| Experimental allergic encephalomyelitis (mouse model of multiple sclerosis) | pMHC-based CAR- CD8+ T cells (Immunodominant peptide epitope of the myelin basic protein (MBP) autoantigen class II MHC IAs chain) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Single dose | Elimination of autoreactive T cells | No safety data reported | [35] |
| pMHC-based CAR- CD4+ T cells (MBP autoantigen class II MHC IAs chain) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Single intravenous injection | Effective in treating advanced EAE, IL-10 is crucial for the therapeutic activity | No safety data reported | [36] | |
| pMHC-based CAR- CD4+ CD25+ regulatory T cells (MBP autoantigen class II MHC IAs chain) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Single intravenous injection | Suppression of EAE | No safety data reported | [37] | |
| pMHC-based CAR- CD4+ CD25+ regulatory T cells (MBP autoantigen class II MHC IAs chain) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Single intravenous injection | Suppression of the autoreactive T cell response and EAE development in an IL-10-dependent manner | No safety data reported | [38] | |
| pMHC-based CAR CD8+ T cells (MOG97–108 pMHCII (DR4)-CAR) | CD28-CD3ζ | Auto antigen-specific CD4+ T cells | In vitro In vivo (mouse model) | Single intravenous injection | Naïve and activated CD4+ T cells can be efficiently eliminated in vivo using pMHCII-CAR cells in a TCR-specific manner | No safety data reported | [39] | |
| CAAR (myelin basic protein (MBP) | CD137-CD3ζ | Autoreactive B cells | In vitro | - | - | n.a. | [40] | |
| NMDAR encephalitis | CAAR Jurkat T cells/primary human T cells (NMDAR autoantigen) | 4-1BB-CD3ζ | Autoreactive B cells | In vivo in vivo (mouse model) | Single dose | A reduction in serum and brain autoantibodies with no signs of off-target toxicity or adverse events | No off-target toxicity reported | [41] |
| Diabetes type 1 | pMHC-based CAR CD8+ T cells (insulin peptide 15–21 linked to β2-microglobulin) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Single intravenous injection | Reduced insulitis and diabetes after adoptive transfer of insulin-reactive CD8 T cells into NOD.SCID mice | No safety data reported | [42] |
| pMHC-based CAR mouse CD8+ T cells (peptide/β2-microglobulin/peptide: Ins15–23, IGRP206–214) | CD3ζ | T cells expressing insulin-reactive G9C8 or IGRP-reactive NY8.3 TCRs | In vitro In vivo (mouse model) | Single intravenous injection | Protection when InsB15–23/β2m/CD3-ζ mRNA-transfected cells were transferred targeting InsB15–23-reactive T cells | No safety data reported | [43] | |
| pMHCII-based chimeric module receptor CTLs (pMHCII and CD80 extracellular domains; Peptide: InsB9–23; HIP2, HIP6) | TCRα and TCRβ subunits assembled with the CD3γε, δε, and ζζ linked to the pMHCII extracellular domain; p56Lck (Lck) linked to CD80 extracellular domain; | Autoreactive T cells | In vitro In vivo (mouse model) | Single dose | Effective in eliminating CD4+ autoreactive T cells in T1D mouse model with cytotoxicity driven by IFN-γ production, proliferation, and cell killing in a TCR-specific manner | No signs of toxicity or pathology in mice reported | [44] | |
| pMHC-based CAR CD8+ T cells (peptide/β2-microglobulin peptide: Ins15–23, IGRP206–214) | CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Two doses one-week interval | Protection against diabetes and reduced the severity of insulitis | No safety data reported | [45] | |
| Systemic lupus erythematosus ** | CAAR CD8+ T cells (B-cell activating factor (BAFF)) | CD28-OX40-CD3ζ | Autoreactive B cells | In vitro In vivo (mouse model) | Single dose | Complete lysis of SLE patient plasma B cells in mouse model | No safety data reported | [46] |
| Lupus nephritis | DNA CAART (α–actinin, heparan sulphate, histone-1, and C1q) | CD28-CD3ζ | B cells expressing anti-dsDNA autoantibodies | In vitro | - | - | n.a. | [47] |
| Membranous nephropathy | CAAR-NK-92/T cells (Immunodominant regions of the MN antigens PLA2R1 and THSD7A) | membrane-proximal ITAM domains of human CD28, the ITAM domain of human 4-1BB, and the CD3ζ | Autoreactive B cells | In vitro | - | - | n.a. | [48] |
| Pemphigus Vulgaris ** | CAAR T cells (desmoglein (Dsg) 3), or combination Dsg3 with extracellular cadherin (EC) domains1-3, 1-4, and 1-5) | CD137-CD3ζ | Autoreactive B cells | In vitro In vivo (mouse model) | Single dose | Decreased Dsg3 serum autoantibody titers, absence of autoantibody binding and blistering in oral mucosa | No off-target toxicity reported | [28] |
| Muscle-specific tyrosine kinase myasthenia gravis ** | CAAR T cells (Muscle-specific tyrosine kinase (MuSK)) | CD137-CD3ζ | Autoreactive B cells | In vitro In vivo (mouse model) | Single dose | Reduced anti-MuSK IgG without decreasing B cells or total IgG levels, reflecting MuSK-specific B cell depletion | No off-target toxicity reported | [49] |
| Rheumatoid arthritis | pMHC-based CAR CD8+ T cells (peptide from type II collagen (CII) linked to the DRB1 chain, linked to second domains from I-Ed | CD28-CD3ζ | Autoreactive T cells | In vitro In vivo (mouse model) | Two doses | Autoimmune CD4+ T cell response decreased, autoantibody production suppressed, and the incidence and severity of autoimmune arthritis diminished | No safety data reported | [50] |
| Primary biliary cholangitis | CAAR CD8+ T cells (PD-L1) | CD28-CD3ζ | PD-1+ T cells | In vitro In vivo (mouse model) | Two doses | Depleted liver CD8+ T cells and alleviated autoimmune cholangitis | No off-target toxicity; no pathological changes in multiple organs; minimal impact on systemic inflammation reported | [51] |
| Hemophilia A | CAAR Tregs (Human FVIII C2 or human A2) | CD28-CD3ζ | FVIII-specific B cells | In vitro | - | - | n.a. | [52] |
| Primary immune thrombocytopenia | CAAR Jurkat (GPIba) | 4-1BB-CD3ζ | Autoreactive B cells | In vitro In vivo (mouse model) | Single dose | Selectively lysed target cells, led to reduced autoantibody titers, and a lower human platelet clearance rate in a xenograft mouse model. | No off-target toxicity reported | [53] |
| Hypersensitivity Type | Characteristic | Disease |
|---|---|---|
| Type 1 (IgE mediated) | Immediate Orchestrated by Th2 cells Local or systemic |
|
| Type 2 (Antibody-mediated) | IgM and IgG antibodies bind to cell surface molecules, resulting in activation of the complement system or antibody-dependent cell-mediated cytotoxicity |
|
| Type 3 (Immune complex-mediated) | Formation of antibody complexes that bind to basal membranes Systemic |
|
| Type 4 (Cell-mediated) | Delayed Activation of T cells by tissue antigen-presenting cells (APCs) leading to tissue damage |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Protić-Rosić, I.; Sehgal, A.N.A.; Wrighton, S.; Heller, B.; Pickl, W.F. Chimeric Autoantibody Receptor- and/or Peptide-MHC-Based CAR Therapies for Targeted Elimination of Antigen-Specific B or T Cells in Hypersensitivity Disorders Such as Allergies and Autoimmune Diseases. Cells 2025, 14, 753. https://doi.org/10.3390/cells14100753
Protić-Rosić I, Sehgal ANA, Wrighton S, Heller B, Pickl WF. Chimeric Autoantibody Receptor- and/or Peptide-MHC-Based CAR Therapies for Targeted Elimination of Antigen-Specific B or T Cells in Hypersensitivity Disorders Such as Allergies and Autoimmune Diseases. Cells. 2025; 14(10):753. https://doi.org/10.3390/cells14100753
Chicago/Turabian StyleProtić-Rosić, Isidora, Al Nasar Ahmed Sehgal, Sebastian Wrighton, Birgit Heller, and Winfried F. Pickl. 2025. "Chimeric Autoantibody Receptor- and/or Peptide-MHC-Based CAR Therapies for Targeted Elimination of Antigen-Specific B or T Cells in Hypersensitivity Disorders Such as Allergies and Autoimmune Diseases" Cells 14, no. 10: 753. https://doi.org/10.3390/cells14100753
APA StyleProtić-Rosić, I., Sehgal, A. N. A., Wrighton, S., Heller, B., & Pickl, W. F. (2025). Chimeric Autoantibody Receptor- and/or Peptide-MHC-Based CAR Therapies for Targeted Elimination of Antigen-Specific B or T Cells in Hypersensitivity Disorders Such as Allergies and Autoimmune Diseases. Cells, 14(10), 753. https://doi.org/10.3390/cells14100753