
Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy


Abstract

1. Introduction
Search Strategy
2. Macrophage Origin, Polarization, and Characterization
2.1. Macrophage Differentiation in the TME
2.2. Recent Technological Advances in TAM Characterization
2.2.1. Integrated Multi-Omics Strategies
2.2.2. AI-Driven Image Analysis
2.2.3. Functional Enrichment and Survival Analysis
2.2.4. Prognostic Biomarkers in Hematologic Malignancies
2.2.5. Pan-Cancer Biomarkers
2.2.6. Clinical Implications of Technological Advances
3. Macrophages Inside of the TME Accelerate Tumor Development
3.1. Implications of M1-Type TAM for the Regression of Tumors
3.2. M2-Type TAM’s Role in Driving Tumor Growth
3.3. Dual Roles of TAMs in Pro-/Anti-Tumor Immunity
3.4. TAM Regulation in Solid and Liquid Tumors
3.4.1. TAMs in Solid Tumors
3.4.2. TAMs in Liquid Tumors
3.5. Regulation of TAM-Associated Metabolic Reprogramming in TME
4. Crosstalk Between TAMs and Tumor Cells
5. Macrophages in Immunoregulation
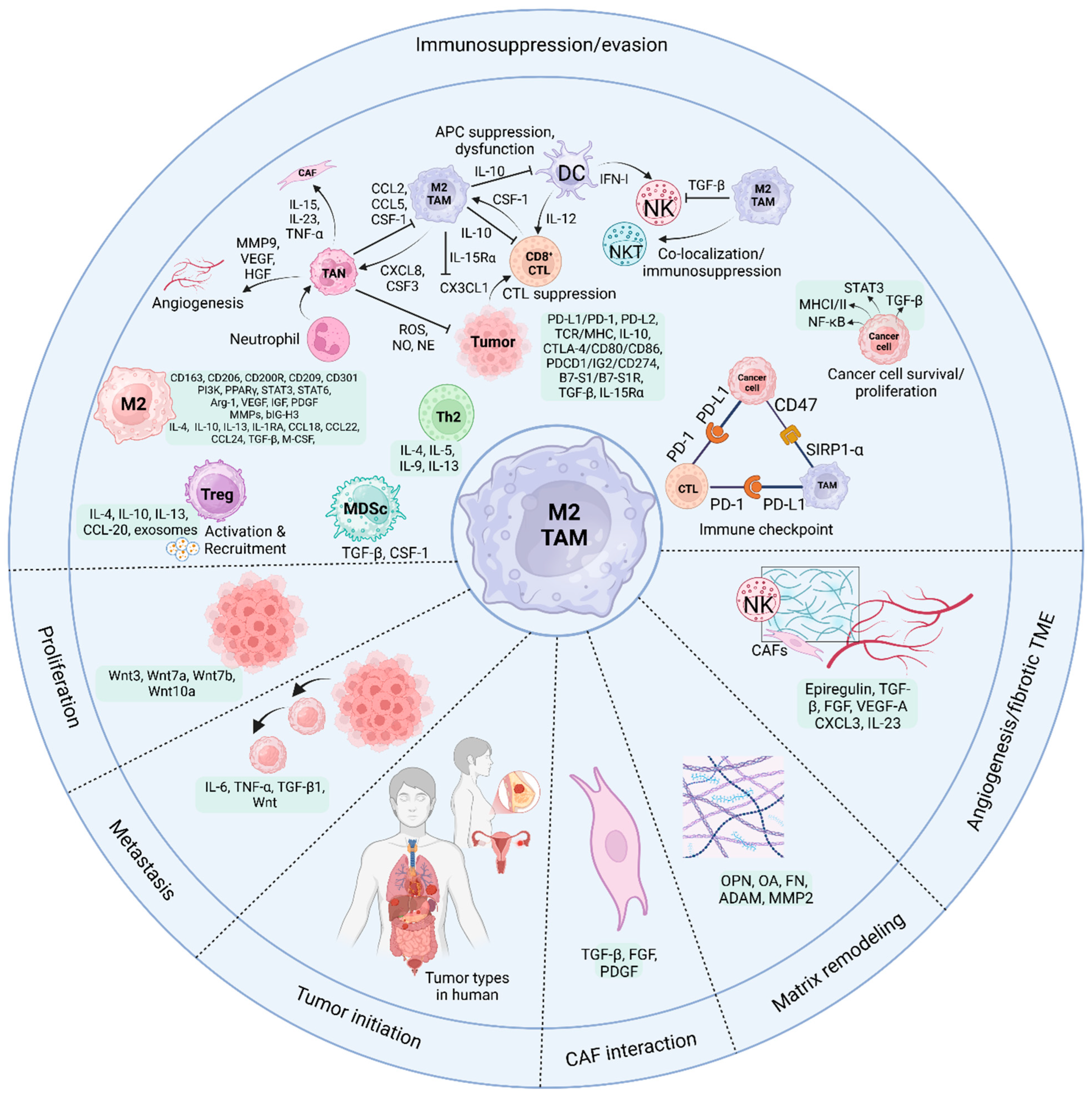
5.1. TAMs and CD8+ CTLs
5.2. TAMs and NK Cells
5.3. TAMs and NKT Cells
5.4. TAMs and CD4 T Cells
5.5. TAMs and DCs
5.6. TAMs and Neutrophils
5.7. TAMs and MDSCs
5.8. TAMs and γδ T Cells
6. Immunotherapy Employing Macrophages and Anti-PD-1/PD-L1
6.1. Effects of TAMs on PD-1/PD-L1 Expression
6.2. TAMs and Resistance to Anti-PD-1
6.3. Macrophage Immune Responses to Anti-PD-1/PD-L1 Therapy
6.4. Clinical Efficacy of Anti-PD-1/PD-L1 Therapy
7. Targeting TAM Immunotherapy
7.1. TAMs in Clinical Therapy
7.1.1. Chemokine Inhibitor
7.1.2. CSF1R Inhibitor
7.1.3. Antibody-Targeting CSF1R
7.1.4. Anti-CD40 Agonist
7.2. Clinical Trial Simulation and TAM Physiology Integration for Cancer Therapy
7.3. Reduced TAM Levels
TAM Elimination with CAR-T Cells
7.4. TAM Reprogramming
7.5. Therapy Using CAR–Macrophages
7.6. Integrating Anti-PD-1 Treatment with Macrophages Targeting Cancer
8. Contradictory Results Regarding TAM Depletion Versus Reprogramming Strategies
Future Therapeutic Perspectives
9. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Gao, L.; Wan, X.C.; Li, J.; Tian, T.; Hu, J.; Zhang, Q.L.; Su, Y.F.; Zeng, Y.P.; Hu, Z.J.; et al. Clinicopathologic features and abnormal signaling pathways in plasmablastic lymphoma: A multicenter study in China. BMC Med. 2022, 20, 483. [Google Scholar] [CrossRef]
- Zheng, H.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Zhang, S.; He, G.; Liu, J.; Fan, Q.; et al. Targeting tumor-associated macrophages in hepatocellular carcinoma: Biology, strategy, and immunotherapy. Cell Death Discov. 2023, 9, 65. [Google Scholar] [CrossRef]
- Guan, F.; Wang, R.; Yi, Z.; Luo, P.; Liu, W.; Xie, Y.; Liu, Z.; Xia, Z.; Zhang, H.; Cheng, Q. Tissue macrophages: Origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct. Target. Ther. 2025, 10, 93. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, Y.; Zhu, Z.; Lu, C.; Zhang, C.; Zeng, L.; Xie, F.; Zhang, L.; Zhou, F. Mucosal immune response in biology, disease prevention and treatment. Signal Transduct. Target. Ther. 2025, 10, 7. [Google Scholar] [CrossRef]
- Feng, X.; Yang, C.; Wang, T.; Zhang, J.; Zhou, H.; Ma, B.; Xu, M.; Deng, G. IFN-τ Maintains Immune Tolerance by Promoting M2 Macrophage Polarization via Modulation of Bta-miR-30b-5p in Early Uterine Pregnancy in Dairy Cows. Cells 2025, 14, 87. [Google Scholar] [CrossRef]
- He, Y.; Hong, Q.; Chen, S.; Zhou, J.; Qiu, S. Reprogramming tumor-associated macrophages in gastric cancer: A pathway to enhanced immunotherapy. Front. Immunol. 2025, 16, 1558091. [Google Scholar] [CrossRef]
- Wu, Y.; Park, J.; Xu, E.; Kim, D.; Lee, J.; Oh, Y.K. MicroRNA-induced reprogramming of tumor-associated macrophages for modulation of tumor immune microenvironment. J. Control. Release 2025, 381, 113593. [Google Scholar] [CrossRef]
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): Results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–2308. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Deng, C.; Zhou, X.Y.; Deng, R. The biology and treatment of Epstein-Barr virus-positive diffuse large B cell lymphoma, NOS. Heliyon 2024, 10, e23921. [Google Scholar] [CrossRef]
- Goc, J.; Lv, M.; Bessman, N.J.; Flamar, A.L.; Sahota, S.; Suzuki, H.; Teng, F.; Putzel, G.G.; Eberl, G.; Withers, D.R.; et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 2021, 184, 5015–5030.e16. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X.; Guo, C.; Wang, W. Tumour-associated macrophages heterogeneity drives resistance to clinical therapy. Expert Rev. Mol. Med. 2022, 24, e17. [Google Scholar] [CrossRef]
- Han, J.; Dong, L.; Wu, M.; Ma, F. Dynamic polarization of tumor-associated macrophages and their interaction with intratumoral T cells in an inflamed tumor microenvironment: From mechanistic insights to therapeutic opportunities. Front. Immunol. 2023, 14, 1160340. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, M.U.; Azhar Ud Din, M.; Khan, I.M.; Khan, M.I.; Bungau, S.; Hassan, S.S.U. Reprogramming tumor-associated macrophages as a unique approach to target tumor immunotherapy. Front. Immunol. 2023, 14, 1166487. [Google Scholar] [CrossRef]
- Toledo, B.; Zhu Chen, L.; Paniagua-Sancho, M.; Marchal, J.A.; Perán, M.; Giovannetti, E. Deciphering the performance of macrophages in tumour microenvironment: A call for precision immunotherapy. J. Hematol. Oncol. 2024, 17, 44. [Google Scholar] [CrossRef]
- Shao, Y.; Han, S.; Hou, Z.; Yang, C.; Zhao, Y. Tumor-associated macrophages within the immunological milieu: An emerging focal point for therapeutic intervention. Heliyon 2024, 10, e36839. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021, 39, 1479–1496.e1418. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kim, Y.S.; Song, H. Macrophages: A double-edged sword in female reproduction and disorders. Exp. Mol. Med. 2025, 57, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, D.J.; Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 2023, 186, 1627–1651. [Google Scholar] [CrossRef] [PubMed]
- Lösslein, A.K.; Henneke, P. Macrophage Differentiation and Metabolic Adaptation in Mycobacterial Infections. Annu. Rev. Immunol. 2025, 43, 423–450. [Google Scholar] [CrossRef] [PubMed]
- Verona, F.; Di Bella, S.; Schirano, R.; Manfredi, C.; Angeloro, F.; Bozzari, G.; Todaro, M.; Giannini, G.; Stassi, G.; Veschi, V. Cancer stem cells and tumor-associated macrophages as mates in tumor progression: Mechanisms of crosstalk and advanced bioinformatic tools to dissect their phenotypes and interaction. Front. Immunol. 2025, 16, 1529847. [Google Scholar] [CrossRef]
- López-Collazo, E.; Hurtado-Navarro, L. Cell fusion as a driver of metastasis: Re-evaluating an old hypothesis in the age of cancer heterogeneity. Front. Immunol. 2025, 16, 1524781. [Google Scholar] [CrossRef]
- Cao, Y.; Yi, Y.; Han, C.; Shi, B. NF-κB signaling pathway in tumor microenvironment. Front. Immunol. 2024, 15, 1476030. [Google Scholar] [CrossRef]
- Daniel, B.; Nagy, G.; Horvath, A.; Czimmerer, Z.; Cuaranta-Monroy, I.; Poliska, S.; Hays, T.T.; Sauer, S.; Francois-Deleuze, J.; Nagy, L. The IL-4/STAT6/PPARγ signaling axis is driving the expansion of the RXR heterodimer cistrome, providing complex ligand responsiveness in macrophages. Nucleic Acids Res. 2018, 46, 4425–4439. [Google Scholar] [CrossRef]
- Liu, H.; Amakye, W.K.; Ren, J. Codonopsis pilosula polysaccharide in synergy with dacarbazine inhibits mouse melanoma by repolarizing M2-like tumor-associated macrophages into M1-like tumor-associated macrophages. Biomed. Pharmacother. 2021, 142, 112016. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, Z.; Ma, R.; Zhang, Y.; Zhao, L.; Yan, Y.; Lv, X.; Zhang, L.; Su, P.; Bi, J.; et al. lncRNA-Xist/miR-101-3p/KLF6/C/EBPα axis promotes TAM polarization to regulate cancer cell proliferation and migration. Mol. Ther. Nucleic Acids 2021, 23, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Zhang, Z.; Dayyani, F.; Zhang, Z.; Yaghmai, V.; Choi, A.; Valerin, J.; Imagawa, D.; Abi-Jaoudeh, N. Modulation of Tumor-Associated Macrophages to Overcome Immune Suppression in the Hepatocellular Carcinoma Microenvironment. Cancers 2024, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Snijckers, R.P.M.; Foks, A.C. Adaptive immunity and atherosclerosis: Aging at its crossroads. Front. Immunol. 2024, 15, 1350471. [Google Scholar] [CrossRef]
- Dong, X.; Dong, C.; Li, B. Effects of macrophages in OSCC progression. Front. Immunol. 2024, 15, 1517886. [Google Scholar] [CrossRef]
- Fatfat, Z.; Hussein, M.; Fatfat, M.; Gali-Muhtasib, H. Omics technologies as powerful approaches to unravel colorectal cancer complexity and improve its management. Mol. Cells 2025, 48, 100200. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Dai, Y.; Zhao, Z.; Zhu, J.; Guo, H.; Yang, R. Single-cell sequencing to multi-omics: Technologies and applications. Biomark. Res. 2024, 12, 110. [Google Scholar] [CrossRef]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Gonzalez Castro, L.N.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M.; et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 2024, 187, 2485–2501.e2426. [Google Scholar] [CrossRef]
- Xin, Y.; Ma, Q.; Deng, Q.; Wang, T.; Wang, D.; Wang, G. Analysis of single-cell and spatial transcriptomics in TNBC cell-cell interactions. Front. Immunol. 2025, 16, 1521388. [Google Scholar] [CrossRef]
- Lei, K.; Lei, Y.; Wang, Z.; Ye, Z.; Liu, J.; Chen, W.; Zhou, C.; Tan, J.; Chen, S.; Zhang, Y.; et al. Integrative multi-omics and Mendelian randomization analysis reveal SPP1+ tumor-associated macrophage-driven prognostic signature for hepatocellular carcinoma. Front. Mol. Biosci. 2025, 12, 1594610. [Google Scholar] [CrossRef]
- Shi, L.; Mao, H.; Ma, J. Integrated analysis of tumor-associated macrophages and M2 macrophages in CRC: Unraveling molecular heterogeneity and developing a novel risk signature. BMC Med. Genom. 2024, 17, 145. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Ma, Y.; Wang, M.; Wang, S.; Yu, W.; Dong, S.; Deng, W.; Bie, L.; Zhang, C.; Shen, W.; et al. Tumor-associated macrophage clusters linked to immunotherapy in a pan-cancer census. NPJ Precis. Oncol. 2024, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.Y.; Ng, C.W.; Rajapakse, M.P.; Ang, N.; Yeong, J.P.S.; Lau, M.C. The promise and challenge of spatial omics in dissecting tumour microenvironment and the role of AI. Front. Oncol. 2023, 13, 1172314. [Google Scholar] [CrossRef]
- Tian, H.; Tian, Y.; Li, D.; Zhao, M.; Luo, Q.; Kong, L.; Qin, T. Artificial intelligence model predicts M2 macrophage levels and HCC prognosis with only globally labeled pathological images. Front. Oncol. 2024, 14, 1474155. [Google Scholar] [CrossRef]
- Rigamonti, A.; Viatore, M.; Polidori, R.; Rahal, D.; Erreni, M.; Fumagalli, M.R.; Zanini, D.; Doni, A.; Putignano, A.R.; Bossi, P.; et al. Integrating AI-Powered Digital Pathology and Imaging Mass Cytometry Identifies Key Classifiers of Tumor Cells, Stroma, and Immune Cells in Non-Small Cell Lung Cancer. Cancer Res. 2024, 84, 1165–1177. [Google Scholar] [CrossRef]
- Lauwers, Y.; De Groof, T.W.M.; Vincke, C.; Van Craenenbroeck, J.; Jumapili, N.A.; Barthelmess, R.M.; Courtoy, G.; Waelput, W.; De Pauw, T.; Raes, G.; et al. Imaging of tumor-associated macrophage dynamics during immunotherapy using a CD163-specific nanobody-based immunotracer. Proc. Natl. Acad. Sci. USA 2024, 121, e2409668121. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, Y.; Ruan, L.; Huang, M.; Chen, W.; Sun, X.; Liu, J.; Jiang, Z. Integrated single-cell and bulk transcriptomic analysis identifies a novel macrophage subtype associated with poor prognosis in breast cancer. Cancer Cell Int. 2025, 25, 119. [Google Scholar] [CrossRef]
- Xu, L.; Chen, Y.; Liu, L.; Hu, X.; He, C.; Zhou, Y.; Ding, X.; Luo, M.; Yan, J.; Liu, Q.; et al. Tumor-associated macrophage subtypes on cancer immunity along with prognostic analysis and SPP1-mediated interactions between tumor cells and macrophages. PLoS Genet. 2024, 20, e1011235. [Google Scholar] [CrossRef]
- Jiang, H.; Pang, J.; Li, T.; Akofala, A.; Zhou, X.; Yi, C.; Ning, S.; Gao, X.; Qiao, Y.; Kou, J. PD-1 regulates the anti-tumor immune function of macrophages through JAK2-STAT3 signaling pathway in colorectal cancer tumor microenvironment. J. Transl. Med. 2025, 23, 502. [Google Scholar] [CrossRef]
- Hofman, P.; Badoual, C.; Henderson, F.; Berland, L.; Hamila, M.; Long-Mira, E.; Lassalle, S.; Roussel, H.; Hofman, V.; Tartour, E.; et al. Multiplexed Immunohistochemistry for Molecular and Immune Profiling in Lung Cancer-Just About Ready for Prime-Time? Cancers 2019, 11, 283. [Google Scholar] [CrossRef]
- Zhu, Y.; Song, Y.; Jiang, W.; Zhang, J.; Yin, L.; Lin, X.; Lu, Y.; Tao, D.; Ma, Y. A novel tumor-associated macrophage risk signature predicts prognosis and immunotherapy response in lung adenocarcinoma. Am. J. Cancer Res. 2025, 15, 876–893. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, J.H.; Hollmén, M. Clinical landscape of macrophage-reprogramming cancer immunotherapies. Br. J. Cancer 2024, 131, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Gkiokas, A.; Papadatou-Gigante, M.; Gkioka, A.I.; Koudouna, A.; Tryfou, T.M.; Alexandropoulos, A.; Bartzi, V.; Kafasi, N.; Kyrtsonis, M.C. Tumor-Associated Macrophage (TAM)-Related Cytokines, sCD163, CCL2, and CCL4, as Novel Biomarkers for Overall Survival and Time to Treatment in Waldenstrom’s Macroglobulinemia: Emphasis on Asymptomatic WM. Cells 2025, 14, 275. [Google Scholar] [CrossRef]
- Ren, F.; Pang, X.; Jin, F.; Luan, N.; Guo, H.; Zhu, L. Integration of scRNA-seq and bulk RNA-seq to reveal the association and potential molecular mechanisms of metabolic reprogramming regulated by lactylation and chemotherapy resistance in ovarian cancer. Front. Immunol. 2025, 16, 1513806. [Google Scholar] [CrossRef]
- Lin, D.; Zheng, T.; Huang, S.; Liu, R.; Guan, S.; Zhang, Z. Identification of a novel macrophage-related prognostic signature in colorectal cancer. Sci. Rep. 2024, 14, 2767. [Google Scholar] [CrossRef]
- Wu, L.; Liang, F.; Chen, C.; Zhang, Y.; Huang, H.; Pan, Y. Identification of prognostic and therapeutic biomarkers associated with macrophage and lipid metabolism in pancreatic cancer. Sci. Rep. 2025, 15, 14584. [Google Scholar] [CrossRef]
- Yan, H.; Ju, X.; Huang, A.; Yuan, J. Advancements in technology for characterizing the tumor immune microenvironment. Int. J. Biol. Sci. 2024, 20, 2151–2167. [Google Scholar] [CrossRef]
- Matsutani, T.; Akbay, E.; Elkord, E. Editorial: Novel biomarkers in tumor immunity and immunotherapy. Front. Immunol. 2024, 15, 1405082. [Google Scholar] [CrossRef]
- Petralia, F.; Ma, W.; Yaron, T.M.; Caruso, F.P.; Tignor, N.; Wang, J.M.; Charytonowicz, D.; Johnson, J.L.; Huntsman, E.M.; Marino, G.B.; et al. Pan-cancer proteogenomics characterization of tumor immunity. Cell 2024, 187, 1255–1277.e1227. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Layeghi, S.M.; Falakian, Z.; Golestani, S.; Kobravi, S.; Talebi, S.; Yousefi, M. An update to experimental and clinical aspects of tumor-associated macrophages in cancer development: Hopes and pitfalls. Clin. Exp. Med. 2024, 24, 156. [Google Scholar] [CrossRef]
- Shao, S.; Miao, H.; Ma, W. Unraveling the enigma of tumor-associated macrophages: Challenges, innovations, and the path to therapeutic breakthroughs. Front. Immunol. 2023, 14, 1295684. [Google Scholar] [CrossRef] [PubMed]
- Motevasseli, M.; Darvishi, M.; Khoshnevisan, A.; Zeinalizadeh, M.; Saffar, H.; Bayat, S.; Najafi, A.; Abbaspour, M.J.; Mamivand, A.; Olson, S.B.; et al. Distinct tumor-TAM interactions in IDH-stratified glioma microenvironments unveiled by single-cell and spatial transcriptomics. Acta Neuropathol. Commun. 2024, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Bied, M.; Ho, W.W.; Ginhoux, F.; Blériot, C. Roles of macrophages in tumor development: A spatiotemporal perspective. Cell. Mol. Immunol. 2023, 20, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Davidson, D.; Li, R.; Zhong, M.C.; Qian, J.; Chen, J.; Veillette, A. Inflammatory macrophages exploit unconventional pro-phagocytic integrins for phagocytosis and anti-tumor immunity. Cell Rep. 2021, 37, 110111. [Google Scholar] [CrossRef]
- Zheng, X.; Jiang, Q.; Han, M.; Ye, F.; Wang, M.; Qiu, Y.; Wang, J.; Gao, M.; Hou, F.; Wang, H. FBXO38 regulates macrophage polarization to control the development of cancer and colitis. Cell. Mol. Immunol. 2023, 20, 1367–1378. [Google Scholar] [CrossRef]
- Zhang, G.; Gao, Z.; Guo, X.; Ma, R.; Wang, X.; Zhou, P.; Li, C.; Tang, Z.; Zhao, R.; Gao, P. CAP2 promotes gastric cancer metastasis by mediating the interaction between tumor cells and tumor-associated macrophages. J. Clin. Investig. 2023, 133, e166224. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Park, M.D.; Reyes-Torres, I.; LeBerichel, J.; Hamon, P.; LaMarche, N.M.; Hegde, S.; Belabed, M.; Troncoso, L.; Grout, J.A.; Magen, A.; et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat. Immunol. 2023, 24, 792–801. [Google Scholar] [CrossRef]
- Yu, Y.; Dai, K.; Gao, Z.; Tang, W.; Shen, T.; Yuan, Y.; Wang, J.; Liu, C. Sulfated polysaccharide directs therapeutic angiogenesis via endogenous VEGF secretion of macrophages. Sci. Adv. 2021, 7, eabd8217. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e1512. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of lung macrophages in health and disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Masmoudi, D.; Villalba, M.; Alix-Panabières, C. Natural killer cells: The immune frontline against circulating tumor cells. J. Exp. Clin. Cancer Res. 2025, 44, 118. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Nakahata, S.; Iha, H. Complex Role of Regulatory T Cells (Tregs) in the Tumor Microenvironment: Their Molecular Mechanisms and Bidirectional Effects on Cancer Progression. Int. J. Mol. Sci. 2024, 25, 7346. [Google Scholar] [CrossRef]
- Kazakova, A.; Sudarskikh, T.; Kovalev, O.; Kzhyshkowska, J.; Larionova, I. Interaction of tumor-associated macrophages with stromal and immune components in solid tumors: Research progress. Int. J. Oncol. 2023, 62, 32. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Liu, X.; Ren, B.; Ren, J.; Gu, M.; You, L.; Zhao, Y. The significant role of amino acid metabolic reprogramming in cancer. Cell Commun. Signal. 2024, 22, 380. [Google Scholar] [CrossRef]
- Biray Avci, C.; Goker Bagca, B.; Nikanfar, M.; Takanlou, L.S.; Takanlou, M.S.; Nourazarian, A. Tumor microenvironment and cancer metastasis: Molecular mechanisms and therapeutic implications. Front. Pharmacol. 2024, 15, 1442888. [Google Scholar] [CrossRef] [PubMed]
- Kzhyshkowska, J.; Shen, J.; Larionova, I. Targeting of TAMs: Can we be more clever than cancer cells? Cell. Mol. Immunol. 2024, 21, 1376–1409. [Google Scholar] [CrossRef] [PubMed]
- Piwocka, O.; Piotrowski, I.; Suchorska, W.M.; Kulcenty, K. Dynamic interactions in the tumor niche: How the cross-talk between CAFs and the tumor microenvironment impacts resistance to therapy. Front. Mol. Biosci. 2024, 11, 1343523. [Google Scholar] [CrossRef]
- Cesano, A.; Augustin, R.; Barrea, L.; Bedognetti, D.; Bruno, T.C.; Carturan, A.; Hammer, C.; Ho, W.S.; Kather, J.N.; Kirchhoff, T.; et al. Advances in the understanding and therapeutic manipulation of cancer immune responsiveness: A Society for Immunotherapy of Cancer (SITC) review. J. Immunother. Cancer 2025, 13, e008876. [Google Scholar] [CrossRef]
- Jahandideh, A.; Yarizadeh, M.; Noei-Khesht Masjedi, M.; Fatehnejad, M.; Jahandideh, R.; Soheili, R.; Eslami, Y.; Zokaei, M.; Ahmadvand, A.; Ghalamkarpour, N.; et al. Macrophage’s role in solid tumors: Two edges of a sword. Cancer Cell Int. 2023, 23, 150. [Google Scholar] [CrossRef]
- Li, S.; Sheng, J.; Zhang, D.; Qin, H. Targeting tumor-associated macrophages to reverse antitumor drug resistance. Aging 2024, 16, 10165–10196. [Google Scholar] [CrossRef]
- Karimova, A.F.; Khalitova, A.R.; Suezov, R.; Markov, N.; Mukhamedshina, Y.; Rizvanov, A.A.; Huber, M.; Simon, H.U.; Brichkina, A. Immunometabolism of tumor-associated macrophages: A therapeutic perspective. Eur. J. Cancer 2025, 220, 115332. [Google Scholar] [CrossRef]
- Mattioda, C.; Voena, C.; Ciardelli, G.; Mattu, C. In Vitro 3D Models of Haematological Malignancies: Current Trends and the Road Ahead? Cells 2025, 14, 38. [Google Scholar] [CrossRef]
- Huang, R.; Kang, T.; Chen, S. The role of tumor-associated macrophages in tumor immune evasion. J. Cancer Res. Clin. Oncol. 2024, 150, 238. [Google Scholar] [CrossRef]
- Li, M.Y.; Ye, W.; Luo, K.W. Immunotherapies Targeting Tumor-Associated Macrophages (TAMs) in Cancer. Pharmaceutics 2024, 16, 865. [Google Scholar] [CrossRef]
- Liu, H.; Lv, Z.; Zhang, G.; Yan, Z.; Bai, S.; Dong, D.; Wang, K. Molecular understanding and clinical aspects of tumor-associated macrophages in the immunotherapy of renal cell carcinoma. J. Exp. Clin. Cancer Res. 2024, 43, 242. [Google Scholar] [CrossRef] [PubMed]
- Crezee, T.; Rabold, K.; de Jong, L.; Jaeger, M.; Netea-Maier, R.T. Metabolic programming of tumor associated macrophages in the context of cancer treatment. Ann. Transl. Med. 2020, 8, 1028. [Google Scholar] [CrossRef] [PubMed]
- Mojsilovic, S.S.; Mojsilovic, S.; Villar, V.H.; Santibanez, J.F. The Metabolic Features of Tumor-Associated Macrophages: Opportunities for Immunotherapy? Anal. Cell. Pathol. 2021, 2021, 5523055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ding, X.; Zhang, X.; Li, Y.; Xu, R.; Li, H.J.; Zuo, D.; Chen, G. Unveiling the contribution of tumor-associated macrophages in driving epithelial-mesenchymal transition: A review of mechanisms and therapeutic Strategies. Front. Pharmacol. 2024, 15, 1404687. [Google Scholar] [CrossRef]
- Nan, D.; Yao, W.; Huang, L.; Liu, R.; Chen, X.; Xia, W.; Sheng, H.; Zhang, H.; Liang, X.; Lu, Y. Glutamine and cancer: Metabolism, immune microenvironment, and therapeutic targets. Cell Commun. Signal. 2025, 23, 45. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, Q.; Peng, H. Tumor-associated macrophages: New insights on their metabolic regulation and their influence in cancer immunotherapy. Front. Immunol. 2023, 14, 1157291. [Google Scholar] [CrossRef]
- De Simone, G.; Soldani, C.; Morabito, A.; Franceschini, B.; Ferlan, F.; Costa, G.; Pastorelli, R.; Donadon, M.; Brunelli, L. Implication of metabolism in the polarization of tumor-associated-macrophages: The mass spectrometry-based point of view. Front. Immunol. 2023, 14, 1193235. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Z.; Chen, Y.; Tian, H.; Chai, P.; Shen, Y.; Yao, Y.; Xu, S.; Ge, S.; Jia, R. Lactate and lactylation in cancer. Signal Transduct. Target. Ther. 2025, 10, 38. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, T.; Li, S.; Wang, X.; Hou, R.; Guan, Z.; Liu, D.; Zheng, J.; Shi, M. A new perspective on the therapeutic potential of tumor metastasis: Targeting the metabolic interactions between TAMs and tumor cells. Int. J. Biol. Sci. 2024, 20, 5109–5126. [Google Scholar] [CrossRef]
- Aizaz, M.; Khan, A.; Khan, F.; Khan, M.; Musad Saleh, E.A.; Nisar, M.; Baran, N. The cross-talk between macrophages and tumor cells as a target for cancer treatment. Front. Oncol. 2023, 13, 1259034. [Google Scholar] [CrossRef]
- Gao, J.; Xiong, A.; Liu, J.; Li, X.; Wang, J.; Zhang, L.; Liu, Y.; Xiong, Y.; Li, G.; He, X. PANoptosis: Bridging apoptosis, pyroptosis, and necroptosis in cancer progression and treatment. Cancer Gene Ther. 2024, 31, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: An effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, C.; Insinga, G.; Gimigliano, F.; Morrione, A.; Giordano, A.; Giurisato, E. Insights into CSF-1R Expression in the Tumor Microenvironment. Biomedicines 2024, 12, 2381. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, C.; Zeng, C.; Sun, C.; Xia, L.; Wang, G.; Chen, X.; Zhang, B.; Liu, J.; Ding, Z.Y. Cancer stem cells and niches: Challenges in immunotherapy resistance. Mol. Cancer 2025, 24, 52. [Google Scholar] [CrossRef]
- Yao, J.; Ji, L.; Wang, G.; Ding, J. Effect of neutrophils on tumor immunity and immunotherapy resistance with underlying mechanisms. Cancer Commun. 2025, 45, 15–42. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, J.; Ruan, S.; Zhang, B.; Zhu, H.; Que, Y.; Ying, S.; Li, X.; Hu, Y.; Song, Z. Overcoming immunotherapy resistance in gastric cancer: Insights into mechanisms and emerging strategies. Cell Death Dis. 2025, 16, 75. [Google Scholar] [CrossRef]
- Wu, C.; Dong, S.; Huang, R.; Chen, X. Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers 2023, 15, 726. [Google Scholar] [CrossRef]
- Liu, Y.; Weng, L.; Wang, Y.; Zhang, J.; Wu, Q.; Zhao, P.; Shi, Y.; Wang, P.; Fang, L. Deciphering the role of CD47 in cancer immunotherapy. J. Adv. Res. 2024, 63, 129–158. [Google Scholar] [CrossRef]
- Brady, R.V.; Thamm, D.H. Tumor-associated macrophages: Prognostic and therapeutic targets for cancer in humans and dogs. Front. Immunol. 2023, 14, 1176807. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Xu, H.; Fu, X.; Wang, S.; Ge, Y.; Zhang, L.; Li, J.; Zhang, F.; Yang, Y.; He, Y.; Sun, Y.; et al. Immunoglobulin-like transcript 5 polarizes M2-like tumor-associated macrophages for immunosuppression in non-small cell lung cancer. Int. J. Cancer 2025, 156, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, B.; Chen, J.; Li, T. Role of exosomal miRNAs and macrophage polarization in gastric cancer: A novel therapeutic strategy. Eur. J. Pharmacol. 2025, 990, 177268. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, J.P.; Haruki, K.; Lau, M.C.; Väyrynen, S.A.; Zhong, R.; Dias Costa, A.; Borowsky, J.; Zhao, M.; Fujiyoshi, K.; Arima, K.; et al. The Prognostic Role of Macrophage Polarization in the Colorectal Cancer Microenvironment. Cancer Immunol. Res. 2021, 9, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Filippopoulou, C.; Besta, S.; Samuels, M.; Betrán, A.L.; Abu Ajamieh, M.; Vella, V.; Jones, W.; Giamas, G. Spatial biology—Unravelling complexity within the glioblastoma microenvironment. Trends Mol. Med. 2025, in press. [CrossRef]
- Gangadaran, P.; Onkar, A.; Rajendran, R.L.; Goenka, A.; Oh, J.M.; Khan, F.; Nagarajan, A.K.; Muthu, S.; Krishnan, A.; Hong, C.M.; et al. Noninvasive in vivo imaging of macrophages: Understanding tumor microenvironments and delivery of therapeutics. Biomark. Res. 2025, 13, 20. [Google Scholar] [CrossRef]
- Cai, D.; Tian, F.; Zhang, D.; Tu, J.; Wang, Y. CRABP2 (Cellular Retinoic Acid Binding Protein 2D): A novel biomarker for the diagnosis and prognosis involved in immune infiltration of lung adenocarcinoma. J. Cancer 2025, 16, 1631–1646. [Google Scholar] [CrossRef]
- Qi, Y.Q.; Xiong, F.; Chen, Y.J. The correlation between tumor-associated macrophages and the prognosis of east Asian hepatocellular carcinoma patients: A systematic review and meta-analysis. Pathol. Res. Pract. 2023, 252, 154919. [Google Scholar] [CrossRef]
- Nedeljković, M.; Vuletić, A.; Mirjačić Martinović, K. Divide and Conquer-Targeted Therapy for Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2025, 26, 1396. [Google Scholar] [CrossRef]
- Jeong, H.; Hwang, I.; Kang, S.H.; Shin, H.C.; Kwon, S.Y. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J. Breast. Cancer 2019, 22, 38–51. [Google Scholar] [CrossRef]
- Baghel, K.S.; Tewari, B.N.; Shrivastava, R.; Malik, S.A.; Lone, M.U.; Jain, N.K.; Tripathi, C.; Kanchan, R.K.; Dixit, S.; Singh, K.; et al. Macrophages promote matrix protrusive and invasive function of breast cancer cells via MIP-1β dependent upregulation of MYO3A gene in breast cancer cells. Oncoimmunology 2016, 5, e1196299. [Google Scholar] [CrossRef]
- Mei, J.; Xiao, Z.; Guo, C.; Pu, Q.; Ma, L.; Liu, C.; Lin, F.; Liao, H.; You, Z.; Liu, L. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget 2016, 7, 34217–34228. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Neufeld, L.; McGaha, T.L. Linking macrophage metabolism to function in the tumor microenvironment. Nat. Cancer 2025, 6, 239–252. [Google Scholar] [CrossRef]
- Ma, M.; Zhang, Y.; Pu, K.; Tang, W. Nanomaterial-enabled metabolic reprogramming strategies for boosting antitumor immunity. Chem. Soc. Rev. 2025, 54, 653–714. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, X.; Li, A.; Qiao, X.; Xu, Y. The mechanism of action and therapeutic potential of tumor-associated macrophages in tumor immune evasion. Front. Immunol. 2025, 16, 1545928. [Google Scholar] [CrossRef]
- Bao, C.; Ma, Q.; Ying, X.; Wang, F.; Hou, Y.; Wang, D.; Zhu, L.; Huang, J.; He, C. Histone lactylation in macrophage biology and disease: From plasticity regulation to therapeutic implications. EBioMedicine 2025, 111, 105502. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Li, Y.; Gong, Q.; Luo, K. Metformin-based nanomedicines for reprogramming tumor immune microenvironment. Theranostics 2025, 15, 993–1016. [Google Scholar] [CrossRef]
- He, S.; Zheng, L.; Qi, C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol. Cancer 2025, 24, 5. [Google Scholar] [CrossRef]
- Ahn, J.; Kim, B.; Bello, A.B.; Moon, J.J.; Arai, Y.; Lee, S.H. Regenerative Functions of Regulatory T Cells and Current Strategies Utilizing Mesenchymal Stem Cells in Immunomodulatory Tissue Regeneration. Tissue Eng. Regen. Med. 2025, 22, 167–180. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.S.; Kim, W.; Lee, E.H.; Kim, S.; Kim, T.; Shin, E.A.; Pyo, K.H.; Lee, H.; Jin, S.H.; et al. Isoxazole-based molecules restore NK cell immune surveillance in hepatocarcinogenesis by targeting TM4SF5 and SLAMF7 linkage. Signal Transduct. Target. Ther. 2025, 10, 15. [Google Scholar] [CrossRef]
- Fanijavadi, S.; Hansen, T.F.; Zedan, A.H. NK Cell-Microbiota Interaction Biomarker Strategy: Advancing Prostate Cancer Management. Biomolecules 2025, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Jabbarzadeh Kaboli, P.; Babaeizad, A.; Maghsoudloo, M. Neoantigen mRNA vaccines and A(2)A receptor antagonism: A strategy to enhance T cell immunity. Hum. Vaccin. Immunother. 2025, 21, 2458936. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.I.; Cosgrove, P.A.; Medina, E.F.; Nath, A.; Chen, J.; Adler, F.R.; Chang, J.T.; Khan, Q.J.; Bild, A.H. Cellular interactions within the immune microenvironment underpins resistance to cell cycle inhibition in breast cancers. Nat. Commun. 2025, 16, 2132. [Google Scholar] [CrossRef]
- Li, Z.; Duan, D.; Li, L.; Peng, D.; Ming, Y.; Ni, R.; Liu, Y. Tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for hepatocellular carcinoma: Recent research progress. Front. Pharmacol. 2024, 15, 1382256. [Google Scholar] [CrossRef]
- Toghraie, F.S.; Bayat, M.; Hosseini, M.S.; Ramezani, A. Tumor-infiltrating myeloid cells; mechanisms, functional significance, and targeting in cancer therapy. Cell. Oncol. 2025, 1–32. [Google Scholar] [CrossRef]
- Hensler, M.; Kasikova, L.; Fiser, K.; Rakova, J.; Skapa, P.; Laco, J.; Lanickova, T.; Pecen, L.; Truxova, I.; Vosahlikova, S.; et al. M2-like macrophages dictate clinically relevant immunosuppression in metastatic ovarian cancer. J. Immunother. Cancer 2020, 8, e000979. [Google Scholar] [CrossRef]
- Trebska-McGowan, K.; Chaib, M.; Alvarez, M.A.; Kansal, R.; Pingili, A.K.; Shibata, D.; Makowski, L.; Glazer, E.S. TGF-β Alters the Proportion of Infiltrating Immune Cells in a Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Surg. 2022, 26, 113–121. [Google Scholar] [CrossRef]
- Mirlekar, B. Tumor promoting roles of IL-10, TGF-β, IL-4, and IL-35: Its implications in cancer immunotherapy. SAGE Open Med. 2022, 10, 20503121211069012. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, S.; Xia, L.; Sun, Z.; Chan, K.M.; Bernards, R.; Qin, W.; Chen, J.; Xia, Q.; Jin, H. Hepatocellular carcinoma: Signaling pathways and therapeutic advances. Signal Transduct. Target. Ther. 2025, 10, 35. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, J.B.; Sun, R.; Wang, N.; Weng, X.Q.; Xu, T.Y.; Fu, D.; Feng, Y.; Xu, P.P.; Cheng, S.; et al. Dual targeting PD-L1 and 4-1BB to overcome dendritic cell-mediated lenalidomide resistance in follicular lymphoma. Signal Transduct. Target. Ther. 2025, 10, 29. [Google Scholar] [CrossRef]
- Xiong, Z.; Huang, Y.; Cao, S.; Huang, X.; Zhang, H. A new strategy for the treatment of advanced ovarian cancer: Utilizing nanotechnology to regulate the tumor microenvironment. Front. Immunol. 2025, 16, 1542326. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E. Tumor-induced metabolic immunosuppression: Mechanisms and therapeutic targets. Cell Rep. 2025, 44, 115206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, Y.; Liu, J.; Guo, L.; Guo, Q.; Liu, W. Bone marrow immune cells and drug resistance in acute myeloid leukemia. Exp. Biol. Med. 2025, 250, 10235. [Google Scholar] [CrossRef]
- Menjivar, R.E.; Nwosu, Z.C.; Du, W.; Donahue, K.L.; Hong, H.S.; Espinoza, C.; Brown, K.; Velez-Delgado, A.; Yan, W.; Lima, F.; et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. Elife 2023, 12, e80721. [Google Scholar] [CrossRef]
- Yang, L.; Chu, Z.; Liu, M.; Zou, Q.; Li, J.; Liu, Q.; Wang, Y.; Wang, T.; Xiang, J.; Wang, B. Amino acid metabolism in immune cells: Essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 59. [Google Scholar] [CrossRef]
- Chi, H.; Pepper, M.; Thomas, P.G. Principles and therapeutic applications of adaptive immunity. Cell 2024, 187, 2052–2078. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, H.; Zhao, P. Compliant immune response of silk-based biomaterials broadens application in wound treatment. Front. Pharmacol. 2025, 16, 1548837. [Google Scholar] [CrossRef]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 2022, 40, 624–638.e629. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Soriano-Cruz, M.; Vázquez-González, W.G.; Molina-Vargas, P.; Faustino-Trejo, A.; Chávez-Rueda, A.K.; Legorreta-Haquet, M.V.; Aguilar-Ruíz, S.R.; Chávez-Sánchez, L. Exosomes as Regulators of Macrophages in Cardiovascular Diseases. Biomedicines 2024, 12, 2683. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Li, Y.; Zhao, H. Fibroblast growth factor signaling in macrophage polarization: Impact on health and diseases. Front. Immunol. 2024, 15, 1390453. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Tharp, K.M.; Kersten, K.; Maller, O.; Timblin, G.A.; Stashko, C.; Canale, F.P.; Menjivar, R.E.; Hayward, M.K.; Berestjuk, I.; Ten Hoeve, J.; et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer 2024, 5, 1045–1062. [Google Scholar] [CrossRef]
- Sato, T.; Sugiyama, D.; Koseki, J.; Kojima, Y.; Hattori, S.; Sone, K.; Nishinakamura, H.; Ishikawa, T.; Ishikawa, Y.; Kato, T.; et al. Sustained inhibition of CSF1R signaling augments antitumor immunity through inhibiting tumor-associated macrophages. JCI Insight 2025, 10, e178146. [Google Scholar] [CrossRef]
- Luan, X.; Lei, T.; Fang, J.; Liu, X.; Fu, H.; Li, Y.; Chu, W.; Jiang, P.; Tong, C.; Qi, H.; et al. Blockade of C5a receptor unleashes tumor-associated macrophage antitumor response and enhances CXCL9-dependent CD8+ T cell activity. Mol. Ther. 2024, 32, 469–489. [Google Scholar] [CrossRef]
- Xie, M.; Lin, Z.; Ji, X.; Luo, X.; Zhang, Z.; Sun, M.; Chen, X.; Zhang, B.; Liang, H.; Liu, D.; et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J. Hepatol. 2023, 79, 109–125. [Google Scholar] [CrossRef]
- Bader, J.E.; Wolf, M.M.; Lupica-Tondo, G.L.; Madden, M.Z.; Reinfeld, B.I.; Arner, E.N.; Hathaway, E.S.; Steiner, K.K.; Needle, G.A.; Hatem, Z.; et al. Obesity induces PD-1 on macrophages to suppress anti-tumour immunity. Nature 2024, 630, 968–975. [Google Scholar] [CrossRef]
- Kerzel, T.; Giacca, G.; Beretta, S.; Bresesti, C.; Notaro, M.; Scotti, G.M.; Balestrieri, C.; Canu, T.; Redegalli, M.; Pedica, F.; et al. In vivo macrophage engineering reshapes the tumor microenvironment leading to eradication of liver metastases. Cancer Cell 2023, 41, 1892–1910.e10. [Google Scholar] [CrossRef]
- Coënon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell Death Dis. 2024, 15, 614. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Khafaga, D.S.R.; El-Morsy, M.T.; Farhan, M.; Aatif, M.; Hosney, M. Targeting tumor-associated macrophages with nanocarrier-based treatment for breast cancer: A step toward developing innovative anti-cancer therapeutics. Heliyon 2024, 10, e37217. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, H.; Jounaidi, Y. Comprehensive snapshots of natural killer cells functions, signaling, molecular mechanisms and clinical utilization. Signal Transduct. Target. Ther. 2024, 9, 302. [Google Scholar] [CrossRef]
- Vidal-Manrique, M.; Nieuwenstein, T.; Hooijmaijers, L.; de Jonge, P.; Djojoatmo, M.; Jansen, J.; van der Waart, A.B.; Brock, R.; Dolstra, H. IL-15 transpresentation by ovarian cancer cells improves CD34+ progenitor-derived NK cell’s anti-tumor functionality. Oncoimmunology 2025, 14, 2465010. [Google Scholar] [CrossRef]
- Jin, P.; Bai, M.; Li, J.; Jia, W.; Yu, J.; Meng, X. Synergistic enhancement of radio-immunotherapy efficacy by IL-15 via macrophage activation and memory T cell response. Cancer Lett. 2025, 613, 217511. [Google Scholar] [CrossRef]
- Horta, A.L.; Gigley, J.; Boutet, M.; Lavau, G.; Weiss, L.M.; Huang, H. Memory-like NK Cells Are a Critical Component of Vaccine-Induced Immunity to Trypanosoma cruzi Infection. J. Immunol. 2024, 212, 617–631. [Google Scholar] [CrossRef]
- Guo, R.; Wang, R.; Zhang, W.; Li, Y.; Wang, Y.; Wang, H.; Li, X.; Song, J. Macrophage Polarisation in the Tumour Microenvironment: Recent Research Advances and Therapeutic Potential of Different Macrophage Reprogramming. Cancer Control 2025, 32, 10732748251316604. [Google Scholar] [CrossRef]
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife 2014, 3, e04177. [Google Scholar] [CrossRef]
- Aftabi, S.; Barzegar Behrooz, A.; Cordani, M.; Rahiman, N.; Sadeghdoust, M.; Aligolighasemabadi, F.; Pistorius, S.; Alavizadeh, S.H.; Taefehshokr, N.; Ghavami, S. Therapeutic targeting of TGF-β in lung cancer. FEBS J. 2025, 292, 1520–1557. [Google Scholar] [CrossRef]
- Mathews, J.A.; Borovsky, D.T.; Reid, K.T.; Murphy, J.M.; Colpitts, S.J.; Carreira, A.S.; Moya, T.A.; Chung, D.C.; Novitzky-Basso, I.; Mattsson, J.; et al. Single cell profiling of hematopoietic stem cell transplant recipients reveals TGF-β1 and IL-2 confer immunoregulatory functions to NK cells. iScience 2024, 27, 111416. [Google Scholar] [CrossRef]
- Shen, K.Y.; Zhu, Y.; Xie, S.Z.; Qin, L.X. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: Current status and prospectives. J. Hematol. Oncol. 2024, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.; Krzywinska, E.; Castells, M.; Gotthardt, D.; Putz, E.M.; Kantari-Mimoun, C.; Chikdene, N.; Meinecke, A.K.; Schrödter, K.; Helfrich, I.; et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat. Commun. 2016, 7, 12528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Jing, S.; Sheng, G.; Jia, F. The basic biology of NK cells and its application in tumor immunotherapy. Front. Immunol. 2024, 15, 1420205. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef]
- Tredicine, M.; Mucci, M.; Recchiuti, A.; Mattoscio, D. Immunoregulatory mechanisms of the arachidonic acid pathway in cancer. FEBS Lett. 2025, 599, 927–951. [Google Scholar] [CrossRef]
- Tognarelli, E.I.; Gutiérrez-Vera, C.; Palacios, P.A.; Pasten-Ferrada, I.A.; Aguirre-Muñoz, F.; Cornejo, D.A.; González, P.A.; Carreño, L.J. Natural Killer T Cell Diversity and Immunotherapy. Cancers 2023, 15, 5737. [Google Scholar] [CrossRef]
- Ji, S.; Shi, Y.; Yin, B. Macrophage barrier in the tumor microenvironment and potential clinical applications. Cell Commun. Signal. 2024, 22, 74. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Chai, D.; Dang, Y.; Zheng, J.; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl. Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef]
- Díaz-Basabe, A.; Strati, F.; Facciotti, F. License to Kill: When iNKT Cells Are Granted the Use of Lethal Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 3909. [Google Scholar] [CrossRef]
- Cruz, S.M.; Sholevar, C.J.; Judge, S.J.; Darrow, M.A.; Iranpur, K.R.; Farley, L.E.; Lammers, M.; Razmara, A.M.; Dunai, C.; Gingrich, A.A.; et al. Intratumoral NKp46+ natural killer cells are spatially distanced from T and MHC-I+ cells with prognostic implications in soft tissue sarcoma. Front. Immunol. 2023, 14, 1230534. [Google Scholar] [CrossRef]
- Lundgren, S.; Micke, P.; Elebro, J.; Heby, M.; Hrynchyk, I.; Nodin, B.; Leandersson, K.; Mezheyeuski, A.; Jirström, K. Topographical Distribution and Spatial Interactions of Innate and Semi-Innate Immune Cells in Pancreatic and Other Periampullary Adenocarcinoma. Front. Immunol. 2020, 11, 558169. [Google Scholar] [CrossRef] [PubMed]
- Delfanti, G.; Dellabona, P.; Casorati, G.; Fedeli, M. Adoptive Immunotherapy With Engineered iNKT Cells to Target Cancer Cells and the Suppressive Microenvironment. Front. Med. 2022, 9, 897750. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Mohammed, A.; Bryant, T.; Ritchie, R.; Stratton, N.; Jackson, L.; Lightfoot, S.; Benbrook, D.M.; Asch, A.S.; Lang, M.L.; et al. Loss of natural killer T cells promotes pancreatic cancer in LSL-Kras(G12D/+) mice. Immunology 2017, 152, 36–51. [Google Scholar] [CrossRef]
- Cruz, M.S.; Loureiro, J.P.; Oliveira, M.J.; Macedo, M.F. The iNKT Cell-Macrophage Axis in Homeostasis and Disease. Int. J. Mol. Sci. 2022, 23, 1640. [Google Scholar] [CrossRef]
- Li, Y.R.; Zhou, Y.; Yu, J.; Zhu, Y.; Lee, D.; Zhu, E.; Li, Z.; Kim, Y.J.; Zhou, K.; Fang, Y.; et al. Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy. Mol. Ther. 2024, 32, 1849–1874. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Dou, Z.; Delfanti, G.; Tsahouridis, O.; Pellegry, C.M.; Zingarelli, M.; Atassi, G.; Woodcock, M.G.; Casorati, G.; et al. CAR-redirected natural killer T cells demonstrate superior antitumor activity to CAR-T cells through multimodal CD1d-dependent mechanisms. Nat. Cancer 2024, 5, 1607–1621. [Google Scholar] [CrossRef]
- Chatzileontiadou, D.S.M.; Sloane, H.; Nguyen, A.T.; Gras, S.; Grant, E.J. The Many Faces of CD4+ T Cells: Immunological and Structural Characteristics. Int. J. Mol. Sci. 2020, 22, 73. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Basic, J.; Stojanovic, I. Dissecting the immune response of CD4+ T cells in Alzheimer’s disease. Rev. Neurosci. 2025, 36, 139–168. [Google Scholar] [CrossRef]
- Khalaf, K.; Chamieh, M.; Welc, N.; Singh, C.; Kaouk, J.L.; Kaouk, A.; Mackiewicz, A.; Kaczmarek, M.; Perek, B. Cellular aspects of immunity involved in the development of atherosclerosis. Front. Immunol. 2025, 16, 1461535. [Google Scholar] [CrossRef]
- Martinenaite, E.; Lecoq, I.; Aaboe-Jørgensen, M.; Ahmad, S.M.; Perez-Penco, M.; Glöckner, H.J.; Chapellier, M.; Lara de la Torre, L.; Olsen, L.R.; Rømer, A.M.A.; et al. Arginase-1-specific T cells target and modulate tumor-associated macrophages. J. Immunother. Cancer 2025, 13, e009930. [Google Scholar] [CrossRef]
- Bertrand, L.; Nelde, A.; Ramirez, B.C.; Hatin, I.; Arbes, H.; François, P.; Demais, S.; Labaronne, E.; Decimo, D.; Guiguettaz, L.; et al. Unveiling conserved HIV-1 open reading frames encoding T cell antigens using ribosome profiling. Nat. Commun. 2025, 16, 1707. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.J.; Alber, B.; Saultz, J.N. The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers 2024, 16, 2615. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Sun, L.; Lin, H.; Liao, Y.; Yang, H.; Mao, Y. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct. Target. Ther. 2024, 9, 68. [Google Scholar] [CrossRef]
- Tsomidis, I.; Voumvouraki, A.; Kouroumalis, E. Immune Checkpoints and the Immunology of Liver Fibrosis. Livers 2025, 5, 5. [Google Scholar] [CrossRef]
- Ge, Y.; Zhou, Q.; Pan, F.; Wang, R. Utilizing Nanoparticles to Overcome Anti-PD-1/PD-L1 Immunotherapy Resistance in Non-Small Cell Lung cancer: A Potential Strategy. Int. J. Nanomed. 2025, 20, 2371–2394. [Google Scholar] [CrossRef]
- Xia, Y.; Huang, C.; Zhong, M.; Zhong, H.; Ruan, R.; Xiong, J.; Yao, Y.; Zhou, J.; Deng, J. Targeting HGF/c-MET signaling to regulate the tumor microenvironment: Implications for counteracting tumor immune evasion. Cell Commun. Signal. 2025, 23, 46. [Google Scholar] [CrossRef]
- Bos, J.; Groen-van Schooten, T.S.; Brugman, C.P.; Jamaludin, F.S.; van Laarhoven, H.W.M.; Derks, S. The tumor immune composition of mismatch repair deficient and Epstein-Barr virus-positive gastric cancer: A systematic review. Cancer Treat. Rev. 2024, 127, 102737. [Google Scholar] [CrossRef]
- Jumaniyazova, E.; Lokhonina, A.; Dzhalilova, D.; Miroshnichenko, E.; Kosyreva, A.; Fatkhudinov, T. The Role of Macrophages in Various Types of Tumors and the Possibility of Their Use as Targets for Antitumor Therapy. Cancers 2025, 17, 342. [Google Scholar] [CrossRef]
- Vilbois, S.; Xu, Y.; Ho, P.C. Metabolic interplay: Tumor macrophages and regulatory T cells. Trends Cancer 2024, 10, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e429. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.T.; Jo, Y.; Chiale, C.; Macal, M.; Fang, Z.; Khatri, F.S.; Codrington, A.L.; Kazane, K.R.; Akbulut, E.; Swaminathan, S.; et al. Metabolic deficiencies underlie reduced plasmacytoid dendritic cell IFN-I production following viral infection. Nat. Commun. 2025, 16, 1460. [Google Scholar] [CrossRef]
- Niemetz, L.; Bodmer, B.S.; Olal, C.; Escudero-Pérez, B.; Hoehn, K.; Bencsik, A.; Vickers, M.A.; Rodríguez, E.; Oestereich, L.; Hoenen, T.; et al. Ebola Virus Infection of Flt3-Dependent, Conventional Dendritic Cells and Antigen Cross-presentation Leads to High Levels of T-Cell Activation. J. Infect. Dis. 2024, 231, 501–511. [Google Scholar] [CrossRef]
- Vafaeian, A.; Rajabi, F.; Rezaei, N. Toll-like receptors in atopic dermatitis: Pathogenesis and therapeutic implications. Heliyon 2025, 11, e42226. [Google Scholar] [CrossRef]
- Chen, M.Y.; Zhang, F.; Goedegebuure, S.P.; Gillanders, W.E. Dendritic cell subsets and implications for cancer immunotherapy. Front. Immunol. 2024, 15, 1393451. [Google Scholar] [CrossRef]
- Niveau, C.; Cettour-Cave, M.; Mouret, S.; Sosa Cuevas, E.; Pezet, M.; Roubinet, B.; Gil, H.; De Fraipont, F.; Landemarre, L.; Charles, J.; et al. MCT1 lactate transporter blockade re-invigorates anti-tumor immunity through metabolic rewiring of dendritic cells in melanoma. Nat. Commun. 2025, 16, 1083. [Google Scholar] [CrossRef]
- Guo, F.; Kong, W.; Li, D.; Zhao, G.; Anwar, M.; Xia, F.; Zhang, Y.; Ma, C.; Ma, X. M2-type tumor-associated macrophages upregulated PD-L1 expression in cervical cancer via the PI3K/AKT pathway. Eur. J. Med. Res. 2024, 29, 357. [Google Scholar] [CrossRef]
- Ma, X.; Guo, Z.; Wei, X.; Zhao, G.; Han, D.; Zhang, T.; Chen, X.; Cao, F.; Dong, J.; Zhao, L.; et al. Spatial Distribution and Predictive Significance of Dendritic Cells and Macrophages in Esophageal Cancer Treated With Combined Chemoradiotherapy and PD-1 Blockade. Front. Immunol. 2021, 12, 786429. [Google Scholar] [CrossRef]
- Xie, D.; Lu, G.; Mai, G.; Guo, Q.; Xu, G. Tissue-resident memory T cells in diseases and therapeutic strategies. MedComm 2025, 6, e70053. [Google Scholar] [CrossRef]
- Li, Y.; Guo, C.; Zhang, F.; Cheng, S.; Li, Y.; Luo, S.; Zeng, Y.; Zhao, Y.; Wu, K. DNMT1 inhibition improves the activity of memory-like natural killer cells by enhancing the level of autophagy. Mol. Biol. Rep. 2024, 52, 68. [Google Scholar] [CrossRef]
- Li, L.; Xu, T.; Qi, X. Balanced regulation of ROS production and inflammasome activation in preventing early development of colorectal cancer. Immunol. Rev. 2025, 329, e13417. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Tu, C.; Xiong, H.; Xu, Y.; Shi, X.; Zhang, X.; Yang, R.; Zhang, N.; Lin, B.; Liu, M.; et al. GITRL enhances cytotoxicity and persistence of CAR-T cells in cancer therapy. Mol. Ther. 2025. [Google Scholar] [CrossRef] [PubMed]
- Luyang, H.; Zeng, F.; Lei, Y.; He, Q.; Zhou, Y.; Xu, J. Bidirectional role of neutrophils in tumor development. Mol. Cancer 2025, 24, 22. [Google Scholar] [CrossRef]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar] [CrossRef]
- Wilczak, M.; Surman, M.; PrzybyłO, M. Melanoma-derived extracellular vesicles transfer proangiogenic factors. Oncol. Res. 2025, 33, 245–262. [Google Scholar] [CrossRef]
- Mehdikhani, F.; Hajimehdipoor, H.; Tansaz, M.; Maresca, M.; Rajabi, S. Sesquiterpene Lactones as Promising Phytochemicals to Cease Metastatic Propagation of Cancer. Biomolecules 2025, 15, 268. [Google Scholar] [CrossRef]
- García-Navas, R.; Gajate, C.; Mollinedo, F. Neutrophils drive endoplasmic reticulum stress-mediated apoptosis in cancer cells through arginase-1 release. Sci. Rep. 2021, 11, 12574. [Google Scholar] [CrossRef]
- Yang, Q.; Cui, M.; Wang, J.; Zhao, Y.; Yin, W.; Liao, Z.; Liang, Y.; Jiang, Z.; Li, Y.; Guo, J.; et al. Circulating mitochondrial DNA promotes M2 polarization of tumor associated macrophages and HCC resistance to sorafenib. Cell Death Dis. 2025, 16, 153. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Rao, A.S.; Stadanlick, J.; Bruns, K.; Sullivan, N.T.; Bermudez, A.; Honig-Frand, A.; Krouse, R.; Arambepola, S.; Guo, E.; et al. Human Tumor-Associated Macrophages and Neutrophils Regulate Antitumor Antibody Efficacy through Lethal and Sublethal Trogocytosis. Cancer Res. 2024, 84, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Zhen, S.; Zhang, L.; Zhao, Q.; Yang, L.; Zhang, Y. A2AR-mediated CXCL5 upregulation on macrophages promotes NSCLC progression via NETosis. Cancer Immunol. Immunother. 2024, 73, 108. [Google Scholar] [CrossRef]
- Schmidt, E.; Distel, L.; Erber, R.; Büttner-Herold, M.; Rosahl, M.C.; Ott, O.J.; Strnad, V.; Hack, C.C.; Hartmann, A.; Hecht, M.; et al. Tumor-Associated Neutrophils Are a Negative Prognostic Factor in Early Luminal Breast Cancers Lacking Immunosuppressive Macrophage Recruitment. Cancers 2024, 16, 3160. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, J.; Xu, T.; Cai, A.; Han, B.; Li, Y.; Fang, Z.; Yu, D.; Wang, S.; Zhou, J.; et al. VSIG4+ tumor-associated macrophages mediate neutrophil infiltration and impair antigen-specific immunity in aggressive cancers through epigenetic regulation of SPP1. J. Exp. Clin. Cancer Res. 2025, 44, 45. [Google Scholar] [CrossRef]
- Fanijavadi, S.; Thomassen, M.; Jensen, L.H. Targeting Triple NK Cell Suppression Mechanisms: A Comprehensive Review of Biomarkers in Pancreatic Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 515. [Google Scholar] [CrossRef]
- Chu, X.; Tian, Y.; Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 2024, 23, 150. [Google Scholar] [CrossRef]
- Werner, W.; Kuzminskaya, M.; Lurje, I.; Tacke, F.; Hammerich, L. Overcoming Resistance to Immune Checkpoint Blockade in Liver Cancer with Combination Therapy: Stronger Together? Semin. Liver Dis. 2024, 44, 159–179. [Google Scholar] [CrossRef]
- Yin, Y.; Feng, W.; Chen, J.; Chen, X.; Wang, G.; Wang, S.; Xu, X.; Nie, Y.; Fan, D.; Wu, K.; et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: From bench to bedside. Exp. Hematol. Oncol. 2024, 13, 72. [Google Scholar] [CrossRef]
- Xiao, G.; Wang, X.; Sheng, J.; Lu, S.; Yu, X.; Wu, J.D. Soluble NKG2D ligand promotes MDSC expansion and skews macrophage to the alternatively activated phenotype. J. Hematol. Oncol. 2015, 8, 13. [Google Scholar] [CrossRef]
- Mitra, A.; Kumar, A.; Amdare, N.P.; Pathak, R. Current Landscape of Cancer Immunotherapy: Harnessing the Immune Arsenal to Overcome Immune Evasion. Biology 2024, 13, 307. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gao, L.; Jiang, X.; Hou, Z.; Wang, Y.; Hou, S.; Qu, H. Single-cell profiling reveals altered immune landscape and impaired NK cell function in gastric cancer liver metastasis. Oncogene 2024, 43, 2635–2646. [Google Scholar] [CrossRef]
- Tabachnick-Cherny, S.; Pulliam, T.; Rodriguez, H.J.; Fan, X.; Hippe, D.S.; Jones, D.C.; Moshiri, A.S.; Smythe, K.S.; Kulikauskas, R.M.; Zaba, L.C.; et al. Characterization of Immunosuppressive Myeloid Cells in Merkel Cell Carcinoma: Correlation with Resistance to PD-1 Pathway Blockade. Clin. Cancer Res. 2024, 30, 1189–1199. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Gu, Y.; Liu, T.; Zhao, X.; Cheng, S.; Duan, L.; Huang, C.; Wu, S.; Gao, S. Complement C3 of tumor-derived extracellular vesicles promotes metastasis of RCC via recruitment of immunosuppressive myeloid cells. Proc. Natl. Acad. Sci. USA 2025, 122, e2420005122. [Google Scholar] [CrossRef]
- Guo, F.; Song, Y.; Dong, S.; Wei, J.; Li, B.; Xu, T.; Wang, H. Characterization and anti-tuberculosis effects of γδ T cells expanded and activated by Mycobacterium tuberculosis heat-resistant antigen. Virulence 2025, 16, 2462092. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, Y.; Niu, S.; Lyu, Z.; Tian, Y.; Shen, X.; Li, Y.R.; Yang, L. Biological functions and therapeutic applications of human mucosal-associated invariant T cells. J. Biomed. Sci. 2025, 32, 32. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, Q.; Li, Y.; Lu, L.; Xiang, Z.; Yin, Z.; Kabelitz, D.; Wu, Y. γδ T cells: Origin and fate, subsets, diseases and immunotherapy. Signal Transduct. Target. Ther. 2023, 8, 434. [Google Scholar] [CrossRef]
- Revesz, I.A.; Joyce, P.; Ebert, L.M.; Prestidge, C.A. Effective γδ T-cell clinical therapies: Current limitations and future perspectives for cancer immunotherapy. Clin. Transl. Immunol. 2024, 13, e1492. [Google Scholar] [CrossRef]
- Petruk, N.; Sousa, S.; Croset, M.; Polari, L.; Zlatev, H.; Selander, K.; Mönkkönen, J.; Clézardin, P.; Määttä, J. Liposome-encapsulated zoledronate increases inflammatory macrophage population in TNBC tumours. Eur. J. Pharm. Sci. 2023, 190, 106571. [Google Scholar] [CrossRef]
- Wu, S.; Lin, X.; Cui, X. Effect of Liposome-Encapsulated Zoledronic Acid on Microenvironment of Hepatocellular Carcinoma May Depend on the Ratio Between M1 and M2 Polarized Macrophages. Bull. Exp. Biol. Med. 2020, 170, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wendong, Y.; Hengwu, X.; Yanhong, C.; Yingying, X.; Feng, Z.; Zeng, W.; Xinjun, C. Mannose modified co-loaded zoledronic liposomes deplete M2-tumor-associated macrophages to enhance anti-tumor effect of doxorubicin on TNBC. J. Drug Deliv. Sci. Technol. 2022, 74, 103551. [Google Scholar] [CrossRef]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In Vivo PET Tracking of (89)Zr-Labeled Vγ9Vδ2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2019, 27, 219–229. [Google Scholar] [CrossRef]
- Parente-Pereira, A.C.; Shmeeda, H.; Whilding, L.M.; Zambirinis, C.P.; Foster, J.; van der Stegen, S.J.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.K.; et al. Adoptive immunotherapy of epithelial ovarian cancer with Vγ9Vδ2 T cells, potentiated by liposomal alendronic acid. J. Immunol. 2014, 193, 5557–5566. [Google Scholar] [CrossRef]
- Gao, Z.; Bai, Y.; Lin, A.; Jiang, A.; Zhou, C.; Cheng, Q.; Liu, Z.; Chen, X.; Zhang, J.; Luo, P. Gamma delta T-cell-based immune checkpoint therapy: Attractive candidate for antitumor treatment. Mol. Cancer 2023, 22, 31. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, H.; Dai, J.; Lin, J.; Zhao, K.; Hu, H.; Zhong, C. Targeting macrophages in cancer immunotherapy: Frontiers and challenges. J. Adv. Res. 2025, in press. [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: Interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet 2023, 402, 2197–2208. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J. Clin. Oncol. 2023, 41, 715–723. [Google Scholar] [CrossRef]
- Li, J.W.; Shi, D.; Wan, X.C.; Hu, J.; Su, Y.F.; Zeng, Y.P.; Hu, Z.J.; Yu, B.H.; Zhang, Q.L.; Wei, P.; et al. Universal extracellular vesicles and PD-L1+ extracellular vesicles detected by single molecule array technology as circulating biomarkers for diffuse large B cell lymphoma. Oncoimmunology 2021, 10, 1995166. [Google Scholar] [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Liu, J.; Dang, P.; Hu, S.; Yuan, W.; Sun, Z.; Liu, Y.; Wang, C. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol. Cancer 2023, 22, 58. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Ning, J.; Hou, X.; Hao, J.; Zhang, W.; Shi, Y.; Huang, Y.; Ruan, X.; Zheng, X.; Gao, M. METTL3 inhibition induced by M2 macrophage-derived extracellular vesicles drives anti-PD-1 therapy resistance via M6A-CD70-mediated immune suppression in thyroid cancer. Cell Death Differ. 2023, 30, 2265–2279. [Google Scholar] [CrossRef]
- You, Q.; Wang, F.; Du, R.; Pi, J.; Wang, H.; Huo, Y.; Liu, J.; Wang, C.; Yu, J.; Yang, Y.; et al. m(6) A Reader YTHDF1-Targeting Engineered Small Extracellular Vesicles for Gastric Cancer Therapy via Epigenetic and Immune Regulation. Adv. Mater. 2023, 35, e2204910. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Hu, H.; Qin, G.; Wu, X.; Bai, F.; Zhang, J.; Cai, Y.; Huang, Y.; Wang, C.; et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell 2023, 41, 1152–1169.e1157. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Xia, H.; Yan, Y.; Zhu, X.; Sun, F.; Sun, L.; Li, S.; Li, D.; Wang, J.; et al. Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing. Genome Med. 2023, 15, 14. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Nam, R.H.; Choi, S.I.; Jang, J.Y.; Kim, J.W.; Na, H.Y.; Lee, H.N. Combination treatment with 17β-estradiol and anti-PD-L1 suppresses MC38 tumor growth by reducing PD-L1 expression and enhancing M1 macrophage population in MC38 colon tumor model. Cancer Lett. 2022, 543, 215780. [Google Scholar] [CrossRef]
- Low, J.T.; Ho, P.C.; Matsushita, M. TAM-tastic: From resistance to resilience in cancer. Trends Pharmacol. Sci. 2024, 45, 953–954. [Google Scholar] [CrossRef]
- Jiang, L.R.; Zhang, N.; Chen, S.T.; He, J.; Liu, Y.H.; Han, Y.Q.; Shi, X.Q.; Yang, J.J.; Mu, D.Y.; Fu, G.H.; et al. PD-1-Positive Tumor-Associated Macrophages Define Poor Clinical Outcomes in Patients With Muscle Invasive Bladder Cancer Through Potential CD68/PD-1 Complex Interactions. Front. Oncol. 2021, 11, 679928. [Google Scholar] [CrossRef]
- Metropulos, A.E.; Munshi, H.G.; Principe, D.R. The difficulty in translating the preclinical success of combined TGFβ and immune checkpoint inhibition to clinical trial. EBioMedicine 2022, 86, 104380. [Google Scholar] [CrossRef]
- Wang, L.; Guo, W.; Guo, Z.; Yu, J.; Tan, J.; Simons, D.L.; Hu, K.; Liu, X.; Zhou, Q.; Zheng, Y.; et al. PD-L1-expressing tumor-associated macrophages are immunostimulatory and associate with good clinical outcome in human breast cancer. Cell Rep. Med. 2024, 5, 101420. [Google Scholar] [CrossRef]
- Xu, S.; Wang, C.; Yang, L.; Wu, J.; Li, M.; Xiao, P.; Xu, Z.; Xu, Y.; Wang, K. Targeting immune checkpoints on tumor-associated macrophages in tumor immunotherapy. Front. Immunol. 2023, 14, 1199631. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Kluger, H.; George, S.; Tykodi, S.S.; Kuzel, T.M.; Perets, R.; Nair, S.; Procopio, G.; Carducci, M.A.; Castonguay, V.; et al. FRACTION-RCC: Nivolumab plus ipilimumab for advanced renal cell carcinoma after progression on immuno-oncology therapy. J. Immunother. Cancer 2022, 10, e005780. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Haag, G.M.; Springfeld, C.; Grün, B.; Apostolidis, L.; Zschäbitz, S.; Dietrich, M.; Berger, A.K.; Weber, T.F.; Zoernig, I.; Schaaf, M.; et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer—The PICCASSO phase I trial. Eur. J. Cancer 2022, 167, 112–122. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.; Le Tourneau, C.; Toulmonde, M.; D’Angelo, S.P.; Weber, K.; Loirat, D.; Jacob, W.; Jegg, A.M.; et al. Long-term clinical activity, safety and patient-reported quality of life for emactuzumab-treated patients with diffuse-type tenosynovial giant-cell tumour. Eur. J. Cancer 2020, 141, 162–170. [Google Scholar] [CrossRef]
- Gomez-Roca, C.; Cassier, P.; Zamarin, D.; Machiels, J.P.; Perez Gracia, J.L.; Stephen Hodi, F.; Taus, A.; Martinez Garcia, M.; Boni, V.; Eder, J.P.; et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004076. [Google Scholar] [CrossRef]
- Machiels, J.P.; Gomez-Roca, C.; Michot, J.M.; Zamarin, D.; Mitchell, T.; Catala, G.; Eberst, L.; Jacob, W.; Jegg, A.M.; Cannarile, M.A.; et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother. Cancer 2020, 8, e001153. [Google Scholar] [CrossRef]
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Calvo Aller, E.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; Shah, M.A.; et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001006. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Dudek, A.Z.; Sukari, A.; Call, J.; Kunk, P.R.; Lewis, K.; Gainor, J.F.; Sarantopoulos, J.; Lee, P.; Golden, A.; et al. ARRY-382 in Combination with Pembrolizumab in Patients with Advanced Solid Tumors: Results from a Phase 1b/2 Study. Clin. Cancer Res. 2022, 28, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef]
- Bauer, C.; Kühnemuth, B.; Duewell, P.; Ormanns, S.; Gress, T.; Schnurr, M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett. 2016, 381, 259–268. [Google Scholar] [CrossRef]
- Chen, K.; Li, X.; Dong, S.; Guo, Y.; Luo, Z.; Zhuang, S.M.; Liu, J.; Liu, T.; Liao, J.; Wen, W. Modulating tumor-associated macrophages through CSF1R inhibition: A potential therapeutic strategy for HNSCC. J. Transl. Med. 2025, 23, 27. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Cao, J.; Chow, L.; Dow, S. Strategies to overcome myeloid cell induced immune suppression in the tumor microenvironment. Front. Oncol. 2023, 13, 1116016. [Google Scholar] [CrossRef]
- Cherney, R.J.; Anjanappa, P.; Selvakumar, K.; Batt, D.G.; Brown, G.D.; Rose, A.V.; Vuppugalla, R.; Chen, J.; Pang, J.; Xu, S.; et al. BMS-813160: A Potent CCR2 and CCR5 Dual Antagonist Selected as a Clinical Candidate. ACS Med. Chem. Lett. 2021, 12, 1753–1758. [Google Scholar] [CrossRef]
- Manji, G.A.; Stanton, L.J.; Hirbe, A.C.; Ge, L.; Sta Ana, S.; Titus, S.; Labadie, B.W.; May, M.S.; Lyu, Y.; Chrisinger, J.S.A.; et al. Phase II Study of Pexidartinib Plus Sirolimus in Unresectable Malignant Peripheral Nerve Sheath Tumors Identifies M2 Macrophage Activation. JCO Oncol. Adv. 2025, 2, e2400083. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Liaw, K.; Reddy, R.; Sharma, A.; Li, J.; Chang, M.; Sharma, R.; Salazar, S.; Kannan, S.; Kannan, R.M. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioeng. Transl. Med. 2021, 6, e10205. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.A.; Klebanoff, C.A.; Schaer, D.; Kauh, J.S.W.; Slovin, S.F.; Adamow, M.; Blinder, V.S.; Brahmachary, M.; Carlsen, M.; Comen, E.; et al. Immunomodulatory Activity of a Colony-stimulating Factor-1 Receptor Inhibitor in Patients with Advanced Refractory Breast or Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2020, 26, 5609–5620. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Weiss, S.A.; Djureinovic, D.; Jessel, S.; Krykbaeva, I.; Zhang, L.; Jilaveanu, L.; Ralabate, A.; Johnson, B.; Levit, N.S.; Anderson, G.; et al. A Phase I Study of APX005M and Cabiralizumab with or without Nivolumab in Patients with Melanoma, Kidney Cancer, or Non-Small Cell Lung Cancer Resistant to Anti-PD-1/PD-L1. Clin. Cancer Res. 2021, 27, 4757–4767. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic Opportunities and Clinical Challenges. Cancers 2021, 13, 2860. [Google Scholar] [CrossRef]
- Zhao, Y.; Ni, Q.; Zhang, W.; Yu, S. Progress in reeducating tumor-associated macrophages in tumor microenvironment. Discov. Oncol. 2024, 15, 312. [Google Scholar] [CrossRef]
- Bradshaw, E.L.; Spilker, M.E.; Zang, R.; Bansal, L.; He, H.; Jones, R.D.O.; Le, K.; Penney, M.; Schuck, E.; Topp, B.; et al. Applications of Quantitative Systems Pharmacology in Model-Informed Drug Discovery: Perspective on Impact and Opportunities. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 777–791. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, C.; Santa-Maria, C.A.; Emens, L.A.; Popel, A.S. Dynamics of tumor-associated macrophages in a quantitative systems pharmacology model of immunotherapy in triple-negative breast cancer. iScience 2022, 25, 104702. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, T.; Zhang, R.; Chen, C.; Li, J. New insights into the role of macrophages in cancer immunotherapy. Front. Immunol. 2024, 15, 1381225. [Google Scholar] [CrossRef]
- Wang, H.; Arulraj, T.; Anbari, S.; Popel, A.S. Quantitative systems pharmacology modeling of macrophage-targeted therapy combined with PD-L1 inhibition in advanced NSCLC. Clin. Transl. Sci. 2024, 17, e13811. [Google Scholar] [CrossRef] [PubMed]
- Shojaee, P.; Mornata, F.; Deutsch, A.; Locati, M.; Hatzikirou, H. The impact of tumor associated macrophages on tumor biology under the lens of mathematical modelling: A review. Front. Immunol. 2022, 13, 1050067. [Google Scholar] [CrossRef]
- Siddiqui, B.A.; Chapin, B.F.; Jindal, S.; Duan, F.; Basu, S.; Yadav, S.S.; Gu, A.D.; Espejo, A.B.; Kinder, M.; Pettaway, C.A.; et al. Immune and pathologic responses in patients with localized prostate cancer who received daratumumab (anti-CD38) or edicotinib (CSF-1R inhibitor). J. Immunother. Cancer 2023, 11, e006262. [Google Scholar] [CrossRef]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef]
- Omstead, A.N.; Paskewicz, M.; Gorbunova, A.; Zheng, P.; Salvitti, M.S.; Mansoor, R.; Reed, P.; Ballengee, S.; Wagner, P.L.; Jobe, B.A.; et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinogenesis 2022, 43, 842–850. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef]
- Kim, D.; An, L.; Moon, J.; Maymi, V.I.; McGurk, A.I.; Rudd, B.D.; Fowell, D.J.; White, A.C. Ccr2+ Monocyte-Derived Macrophages Influence Trajectories of Acquired Therapy Resistance in Braf-Mutant Melanoma. Cancer Res. 2023, 83, 2328–2344. [Google Scholar] [CrossRef]
- Miyamoto, T.; Murakami, R.; Hamanishi, J.; Tanigaki, K.; Hosoe, Y.; Mise, N.; Takamatsu, S.; Mise, Y.; Ukita, M.; Taki, M.; et al. B7-H3 Suppresses Antitumor Immunity via the CCL2-CCR2-M2 Macrophage Axis and Contributes to Ovarian Cancer Progression. Cancer Immunol. Res. 2022, 10, 56–69. [Google Scholar] [CrossRef]
- Trac, N.; Chen, L.Y.; Zhang, A.; Liao, C.P.; Poon, C.; Wang, J.; Ando, Y.; Joo, J.; Garri, C.; Shen, K.; et al. CCR2-targeted micelles for anti-cancer peptide delivery and immune stimulation. J. Control. Release 2021, 329, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, X.; Si, M.; Yang, J.; Sun, S.; Wu, H.; Cui, S.; Qu, X.; Yu, X. Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett. 2020, 474, 36–52. [Google Scholar] [CrossRef]
- Kocher, F.; Puccini, A.; Untergasser, G.; Martowicz, A.; Zimmer, K.; Pircher, A.; Baca, Y.; Xiu, J.; Haybaeck, J.; Tymoszuk, P.; et al. Multi-omic Characterization of Pancreatic Ductal Adenocarcinoma Relates CXCR4 mRNA Expression Levels to Potential Clinical Targets. Clin. Cancer Res. 2022, 28, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Dong, C.; Jia, W.; Ma, B. Exosomal miR-655-3p inhibits growth, and invasion and macrophage M2 polarization through targeting CXCR4 in papillary thyroid carcinoma. Acta Biochim. Pol. 2022, 69, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.; Khan, H.; Kaur, G.; Kumar, P.; Singh, T.G. Therapeutic targeting of angiopoietins in tumor angiogenesis and cancer development. Biochem. Biophys. Res. Commun. 2023, 687, 149130. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Yang, Z.; Lu, Q.; Liu, Z.; Wang, L.; Du, J.; Li, Y.; Yang, D.H.; Wu, S. Reshaping the tumor immune microenvironment to improve CAR-T cell-based cancer immunotherapy. Mol. Cancer 2024, 23, 175. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef]
- Truxova, I.; Cibula, D.; Spisek, R.; Fucikova, J. Targeting tumor-associated macrophages for successful immunotherapy of ovarian carcinoma. J. Immunother. Cancer 2023, 11, e005968. [Google Scholar] [CrossRef]
- Li, Y.R.; Wilson, M.; Yang, L. Target tumor microenvironment by innate T cells. Front. Immunol. 2022, 13, 999549. [Google Scholar] [CrossRef]
- Xu, C.; Chen, J.; Tan, M.; Tan, Q. The role of macrophage polarization in ovarian cancer: From molecular mechanism to therapeutic potentials. Front. Immunol. 2025, 16, 1543096. [Google Scholar] [CrossRef]
- Park, S.Y.; Pylaeva, E.; Bhuria, V.; Gambardella, A.R.; Schiavoni, G.; Mougiakakos, D.; Kim, S.H.; Jablonska, J. Harnessing myeloid cells in cancer. Mol. Cancer 2025, 24, 69. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Wang, T.; Wang, X.F.; Zhang, Y.Q.; Song, S.X.; Ma, C.Q. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol. Res. 2022, 175, 106036. [Google Scholar] [CrossRef]
- Ahmed, E.N.; Cutmore, L.C.; Marshall, J.F. Syngeneic Mouse Models for Pre-Clinical Evaluation of CAR T Cells. Cancers 2024, 16, 3186. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Gadd, M.E.; Qie, Y.; Otamendi-Lopez, A.; Sanchez-Garavito, J.E.; Brooks, M.M.; Ulloa Navas, M.J.; Hundal, T.; Li, S.; Jones, V.K.; et al. Solid cancer-directed CAR T cell therapy that attacks both tumor and immunosuppressive cells via targeting PD-L1. Mol. Ther. Oncol. 2024, 32, 200891. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, P.; Zeng, X.; Guo, J.; Zhang, C.; Fan, Z.; Wang, Z.; Zhu, P.; Chen, Z. CAR-T therapy dilemma and innovative design strategies for next generation. Cell Death Dis. 2025, 16, 211. [Google Scholar] [CrossRef]
- Satapathy, B.P.; Sheoran, P.; Yadav, R.; Chettri, D.; Sonowal, D.; Dash, C.P.; Dhaka, P.; Uttam, V.; Yadav, R.; Jain, M.; et al. The synergistic immunotherapeutic impact of engineered CAR-T cells with PD-1 blockade in lymphomas and solid tumors: A systematic review. Front. Immunol. 2024, 15, 1389971. [Google Scholar] [CrossRef]
- Yang, X.; Liu, X.; Li, J.; Zhang, P.; Li, H.; Chen, G.; Zhang, W.; Wang, T.; Frazer, I.; Ni, G. Caerin 1.1/1.9 Enhances Antitumour Immunity by Activating the IFN-α Response Signalling Pathway of Tumour Macrophages. Cancers 2022, 14, 5785. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Repolarizing macrophages improves breast cancer therapy. Cell Res. 2017, 27, 963–964. [Google Scholar] [CrossRef]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F.; Ren, X.; Sun, R.; Guo, X.; Liu, W.; Wang, B.; Yang, Y.; Zhang, X.; Xu, Y.; et al. Phosphoinositide-Binding Protein TIPE1 Promotes Alternative Activation of Macrophages and Tumor Progression via PIP3/Akt/TGFβ Axis. Cancer Res. 2022, 82, 1603–1616. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Jogalekar, M.P.; Rajendran, R.L.; Khan, F.; Dmello, C.; Gangadaran, P.; Ahn, B.C. CAR T-Cell-Based gene therapy for cancers: New perspectives, challenges, and clinical developments. Front. Immunol. 2022, 13, 925985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Tichet, M.; Wullschleger, S.; Chryplewicz, A.; Fournier, N.; Marcone, R.; Kauzlaric, A.; Homicsko, K.; Deak, L.C.; Umaña, P.; Klein, C.; et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor-reactive CD8+ T cells and reprogramming macrophages. Immunity 2023, 56, 162–179.e166. [Google Scholar] [CrossRef]
- Wang, Y.N.; Wang, Y.Y.; Wang, J.; Bai, W.J.; Miao, N.J.; Wang, J. Vinblastine resets tumor-associated macrophages toward M1 phenotype and promotes antitumor immune response. J. Immunother. Cancer 2023, 11, e007253. [Google Scholar] [CrossRef]
- Zhou, C.; Weng, J.; Liu, C.; Liu, S.; Hu, Z.; Xie, X.; Gao, D.; Zhou, Q.; Sun, J.; Xu, R.; et al. Disruption of SLFN11 Deficiency-Induced CCL2 Signaling and Macrophage M2 Polarization Potentiates Anti-PD-1 Therapy Efficacy in Hepatocellular Carcinoma. Gastroenterology 2023, 164, 1261–1278. [Google Scholar] [CrossRef]
- Zhu, R.; Huang, J.; Qian, F. The role of tumor-associated macrophages in lung cancer. Front. Immunol. 2025, 16, 1556209. [Google Scholar] [CrossRef]
- Kulakova, K.; Lawal, T.R.; McCarthy, E.; Floudas, A. The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets. Cells 2024, 13, 1586. [Google Scholar] [CrossRef]
- Li, Z.; Ding, Y.; Liu, J.; Wang, J.; Mo, F.; Wang, Y.; Chen-Mayfield, T.J.; Sondel, P.M.; Hong, S.; Hu, Q. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat. Commun. 2022, 13, 1845. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Ji, C.; Liu, X.; Gu, B.; Dong, T. Macrophage polarization in the tumor microenvironment: Emerging roles and therapeutic potentials. Biomed. Pharmacother. 2024, 177, 116930. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Gunassekaran, G.R.; Yoo, J.D.; Kwon, T.H.; Hur, K.; Chae, S.; Lee, B. Epigenetic therapy reprograms M2-type tumor-associated macrophages into an M1-like phenotype by upregulating miR-7083-5p. Front. Immunol. 2022, 13, 976196. [Google Scholar] [CrossRef]
- Rodriguez-Perdigon, M.; Haeni, L.; Rothen-Rutishauser, B.; Rüegg, C. Dual CSF1R inhibition and CD40 activation demonstrates anti-tumor activity in a 3D macrophage- HER2+ breast cancer spheroid model. Front. Bioeng. Biotechnol. 2023, 11, 1159819. [Google Scholar] [CrossRef]
- Yamada-Hunter, S.A.; Theruvath, J.; McIntosh, B.J.; Freitas, K.A.; Lin, F.; Radosevich, M.T.; Leruste, A.; Dhingra, S.; Martinez-Velez, N.; Xu, P.; et al. Engineered CD47 protects T cells for enhanced antitumour immunity. Nature 2024, 630, 457–465. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, S.; Qi, Z.; Xu, X.; Long, X.; Huang, A.; Liu, J.; Cheng, P. Mitochondrial metabolic reprogramming of macrophages and T cells enhances CD47 antibody-engineered oncolytic virus antitumor immunity. J. Immunother. Cancer 2024, 12, e009768. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef]
- Tang, F.; Wang, Y.; Zeng, Y.; Xiao, A.; Tong, A.; Xu, J. Tumor-associated macrophage-related strategies for glioma immunotherapy. NPJ Precis. Oncol. 2023, 7, 78. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, K.; Chen, Y.; Ge, X.; Wu, J.; Xu, P.; Yao, J. Progress of single-cell RNA sequencing combined with spatial transcriptomics in tumour microenvironment and treatment of pancreatic cancer. J. Transl. Med. 2024, 22, 563. [Google Scholar] [CrossRef]
- Lu, X.; Green, B.L.; Xie, C.; Liu, C.; Chen, X. Preclinical and clinical studies of immunotherapy for the treatment of cholangiocarcinoma. JHEP Rep. 2023, 5, 100723. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Ren, Y.; Wang, R.; Zhang, Y.; Weng, S.; Zhou, Z.; Luo, P.; Cheng, Q.; Xu, H.; et al. Macrophage-derived exosomes in cancer: A double-edged sword with therapeutic potential. J. Nanobiotechnol. 2025, 23, 319. [Google Scholar] [CrossRef]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Qian, L.; Sun, R.; Aebersold, R.; Bühlmann, P.; Sander, C.; Guo, T. AI-empowered perturbation proteomics for complex biological systems. Cell Genom. 2024, 4, 100691. [Google Scholar] [CrossRef] [PubMed]
- Fendl, B.; Berghoff, A.S.; Preusser, M.; Maier, B. Macrophage and monocyte subsets as new therapeutic targets in cancer immunotherapy. ESMO Open 2023, 8, 100776. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: From molecular mechanisms to targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef]
- Chaudagar, K.; Hieromnimon, H.M.; Khurana, R.; Labadie, B.; Hirz, T.; Mei, S.; Hasan, R.; Shafran, J.; Kelley, A.; Apostolov, E.; et al. Reversal of Lactate and PD-1-mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin. Cancer Res. 2023, 29, 1952–1968. [Google Scholar] [CrossRef]
- Lopresti, L.; Tatangelo, V.; Baldari, C.T.; Patrussi, L. Rewiring the T cell-suppressive cytokine landscape of the tumor microenvironment: A new frontier for precision anti-cancer therapy. Front. Immunol. 2024, 15, 1418527. [Google Scholar] [CrossRef]
- Liu, L.; Chen, G.; Gong, S.; Huang, R.; Fan, C. Targeting tumor-associated macrophage: An adjuvant strategy for lung cancer therapy. Front. Immunol. 2023, 14, 1274547. [Google Scholar] [CrossRef]
- Su, P.; Li, O.; Ke, K.; Jiang, Z.; Wu, J.; Wang, Y.; Mou, Y.; Jin, W. Targeting tumor-associated macrophages: Critical players in tumor progression and therapeutic strategies. Int. J. Oncol. 2024, 64, 60. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Features | M1-like TAMs | M2-like TAMs |
|---|---|---|
| Primary metabolism | Glycolysis | Mixed glycolysis/OXPHOS |
| Key pathways | HIF-1α, PKM2 | mTORC2, FAO |
| Functional output | Pro-inflammatory (limited) | Immunosuppression, tissue repair |
| Drug | Phase | Cancer Type | Combination Therapy | NCT Identifier |
|---|---|---|---|---|
| Chemokine inhibitors | ||||
| Carlumab (anti-CCL2 antibodies; Centocor) | Phase II (completed) | Prostate cancer | NA | NCT00992186 |
| BMS-813160 (CCR2/CCR5 antagonist; Bristol Myers Squibb) | Phase II (completed) | Renal cell carcinoma | Nivolumab (OPDIVO) in conjunction with ipilimumab (Yervoy) | NCT02996110 |
| Phase I/II (completed) | Pancreatic cancer, colorectal cancer, non-small cell lung cancer | Nab-paclitaxel with nivolumab | NCT03184870 | |
| Phase II (ongoing) | Hepatocellular carcinoma | Nivolumab | NCT04123379 | |
| PF-4136309 (CCR2 antagonist; Pfizer) | Phase II (completed) | PDAC | Nab-paclitaxel, gemcitabine | NCT01413022 |
| CSF1R inhibitors | ||||
| PLX3397 (Plexxikon) | Phase I/II (ongoing) | Tumors of the nerve sheath and sarcoma | Sirolimus (Rapamune) | NCT02584647 |
| Phase I/II (terminated) | Melanoma and solid tumors, both at advanced stages | Pembrolizumab (Keytruda) | NCT02452424 | |
| Phase I/II (completed) | Breast carcinoma | Eribulin (Halaven) | NCT01596751 | |
| Phase I/II (completed) | Glioblastoma | Radiotherapy, temozolomide (TMZ) | NCT01790503 | |
| BLZ945 (Novartis) | Phase I/II (terminated) | Solid tumors | PDR001 (anti-PD1) | NCT02829723 |
| Antibodies targeting CSF1R | ||||
| LY3022855 (Eli Lilly’s IMC-C S4) | Phase I/II (completed) | Melanoma | MEK/BRAF inhibitors | NCT03101254 |
| Emactuzumab (RO5509554/RG7155; Roche) | Phase II (terminated) | Gynecological neoplasms and OC | Gynecological neoplasms and OC | NCT02923739 |
| Phase I/II (ongoing) | PDAC | Nab-paclitaxel, gemcitabine | NCT03193190 | |
| AMG820 (Amgen) | Phase I/II (completed) | Pancreatic cancer, CRC, NSCLC | Pembrolizumab | NCT02713529 |
| ARRAY-382 (Pfizer) | Phase I/II (terminated) | Solid tumors | Solid tumors | NCT02880371 |
| Agonist anti-CD40 antibodies (cont.) | ||||
| APX005M (Apexigen) | Phase II (completed) | Esophageal cancer | Radiation, paclitaxel, carboplatin | NCT03214250 |
| Phase I/II (completed) | Pancreatic cancer | Nab-paclitaxel, gemcitabine, nivolumab | NCT03214250 | |
| Factor | Impact on Depletion vs. Reprogramming | Ref |
|---|---|---|
| Tumor Type | CSF1R inhibitors succeeded in CSF1R-addicted tumors but failed in others (e.g., pancreatic cancer). | [52] |
| TME Heterogeneity | TAM subsets exhibit divergent functions (e.g., angiogenesis vs. immunosuppression), complicating targeting. | [61] |
| Therapeutic Timing | Post-surgery was depleted to prevent recurrence, while reprogramming was synergized with immunotherapy. | [86,329] |
| Combination Therapies | Reprogramming + checkpoint inhibitors improved responses in resistant tumors, whereas depletion alone rarely sufficed. | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, A.F. Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy. Cells 2025, 14, 741. https://doi.org/10.3390/cells14100741
Saeed AF. Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy. Cells. 2025; 14(10):741. https://doi.org/10.3390/cells14100741
Chicago/Turabian StyleSaeed, Abdullah Farhan. 2025. "Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy" Cells 14, no. 10: 741. https://doi.org/10.3390/cells14100741
APA StyleSaeed, A. F. (2025). Tumor-Associated Macrophages: Polarization, Immunoregulation, and Immunotherapy. Cells, 14(10), 741. https://doi.org/10.3390/cells14100741