
Coupling of Intracellular Calcium Homeostasis and Formation and Secretion of Matrix Vesicles: Their Role in the Mechanism of Biomineralization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
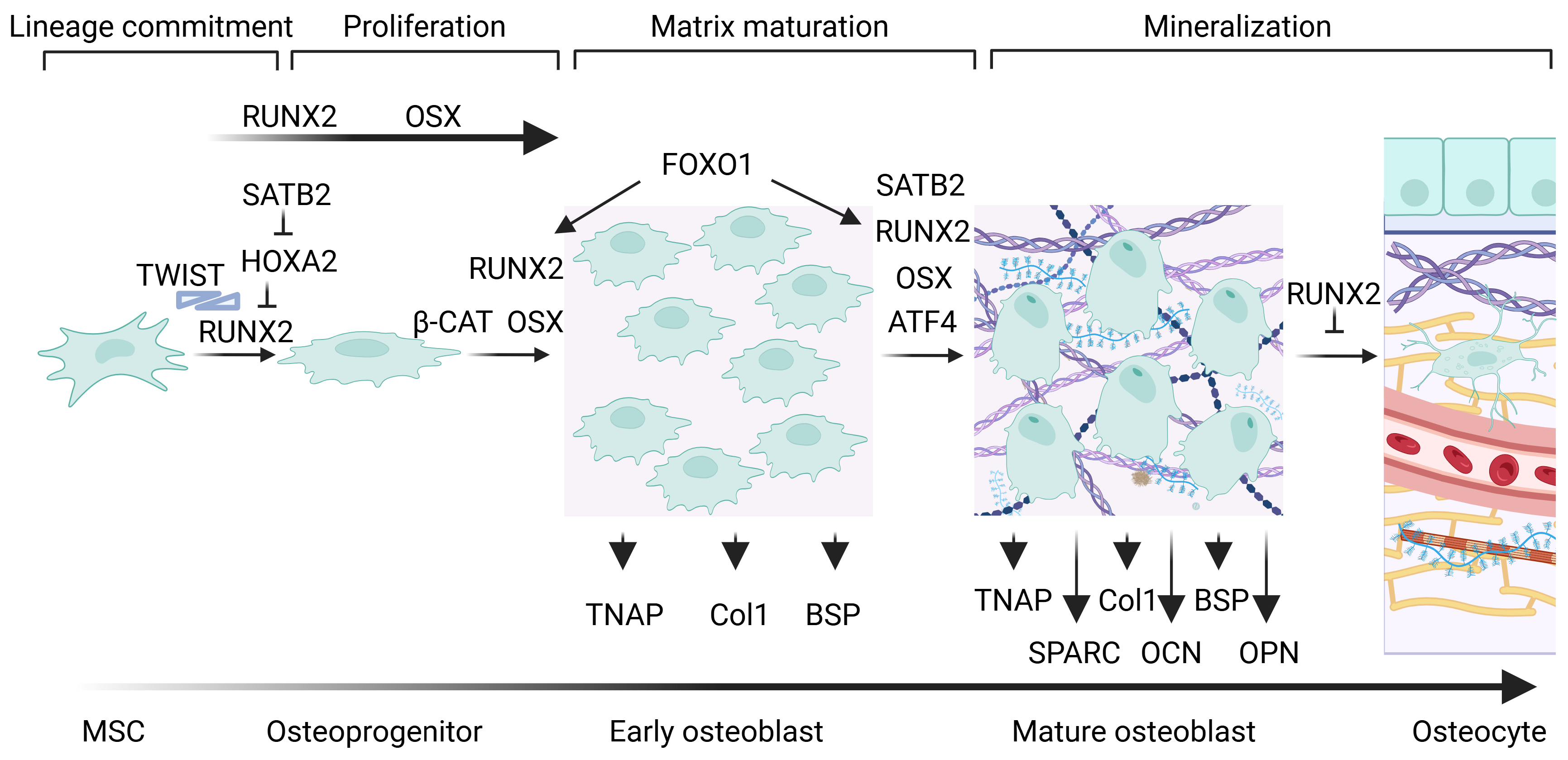
1. Cellular, Molecular, and Genetic Aspects of Bone Formation
1.1. The “Master Regulator of Osteogenesis”: Runx2
1.2. Osterix Regulates Osteogenesis
1.3. Other Factors Mediate Osteoblastogenesis and Osteogenesis
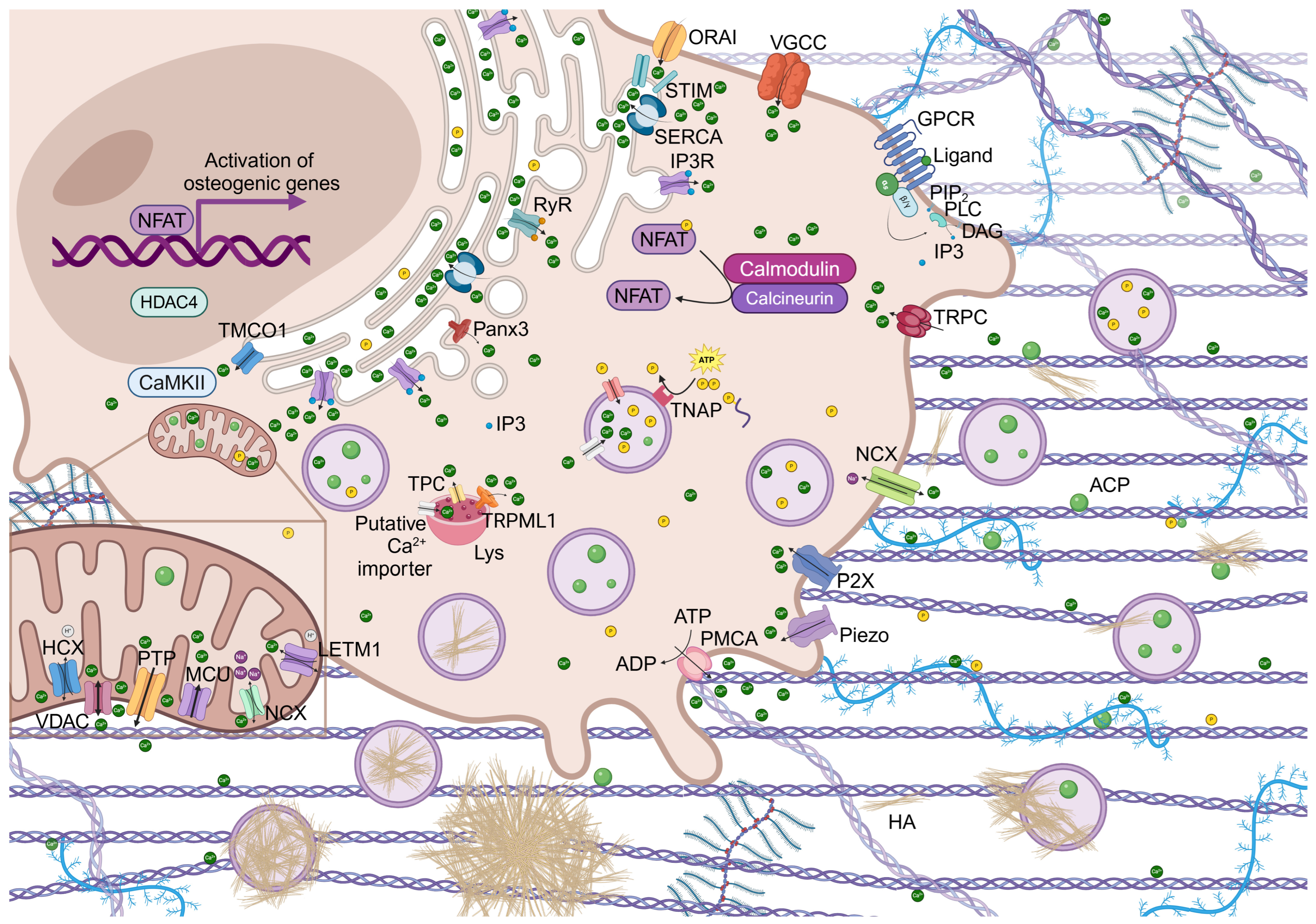
2. Cellular Calcium Signaling and Transcription Activity
Role of NFAT Signaling in Bone Homeostasis
3. Regulation of Intracellular Calcium Homeostasis
4. Formation of the Prebone and Bone Matrix
4.1. Osteoid
4.2. Bone Formation and Mineralization
4.3. Regulation of Phosphate Ions Homeostasis
4.4. Mechanisms Driving Mineralization and Resorption
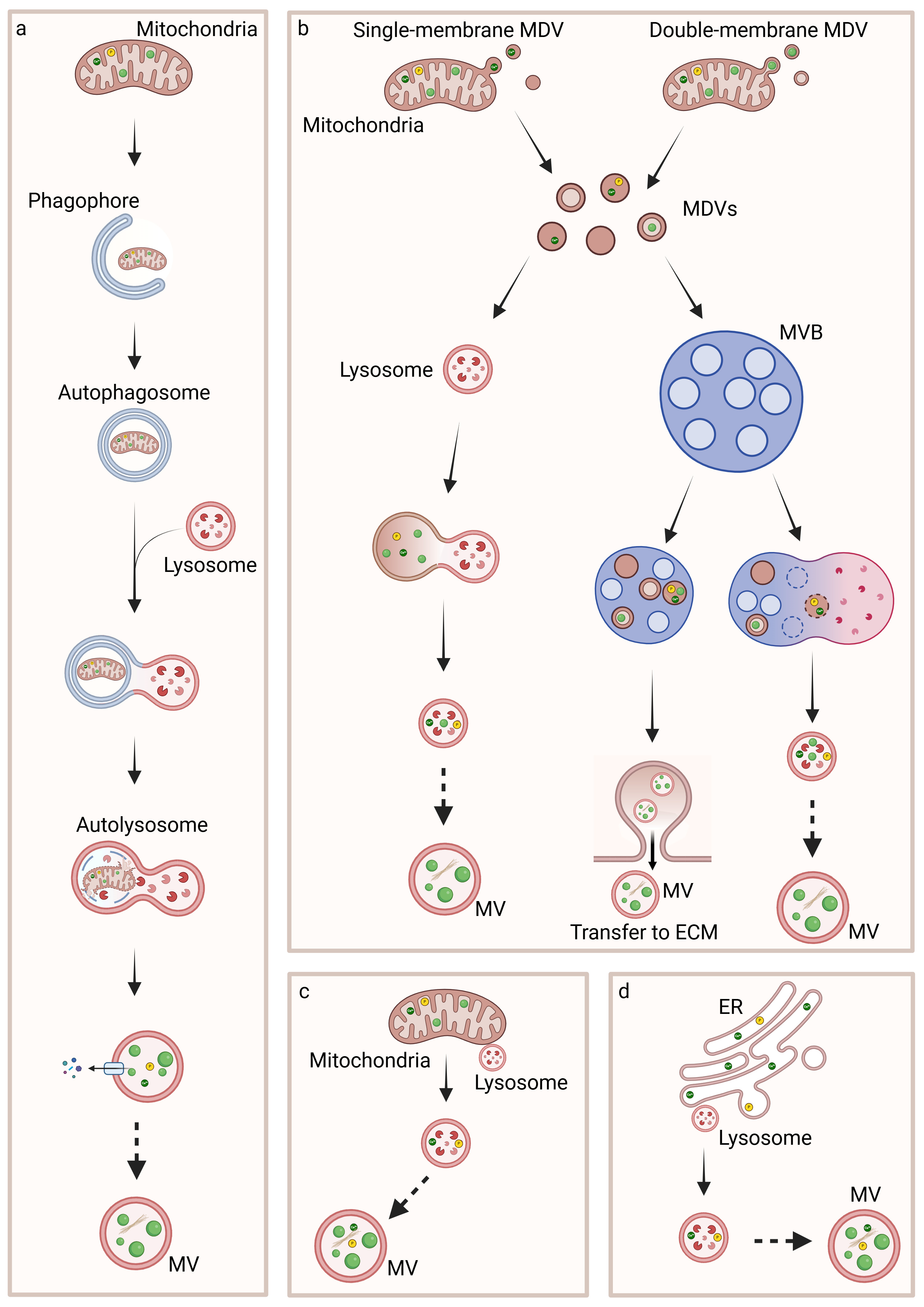
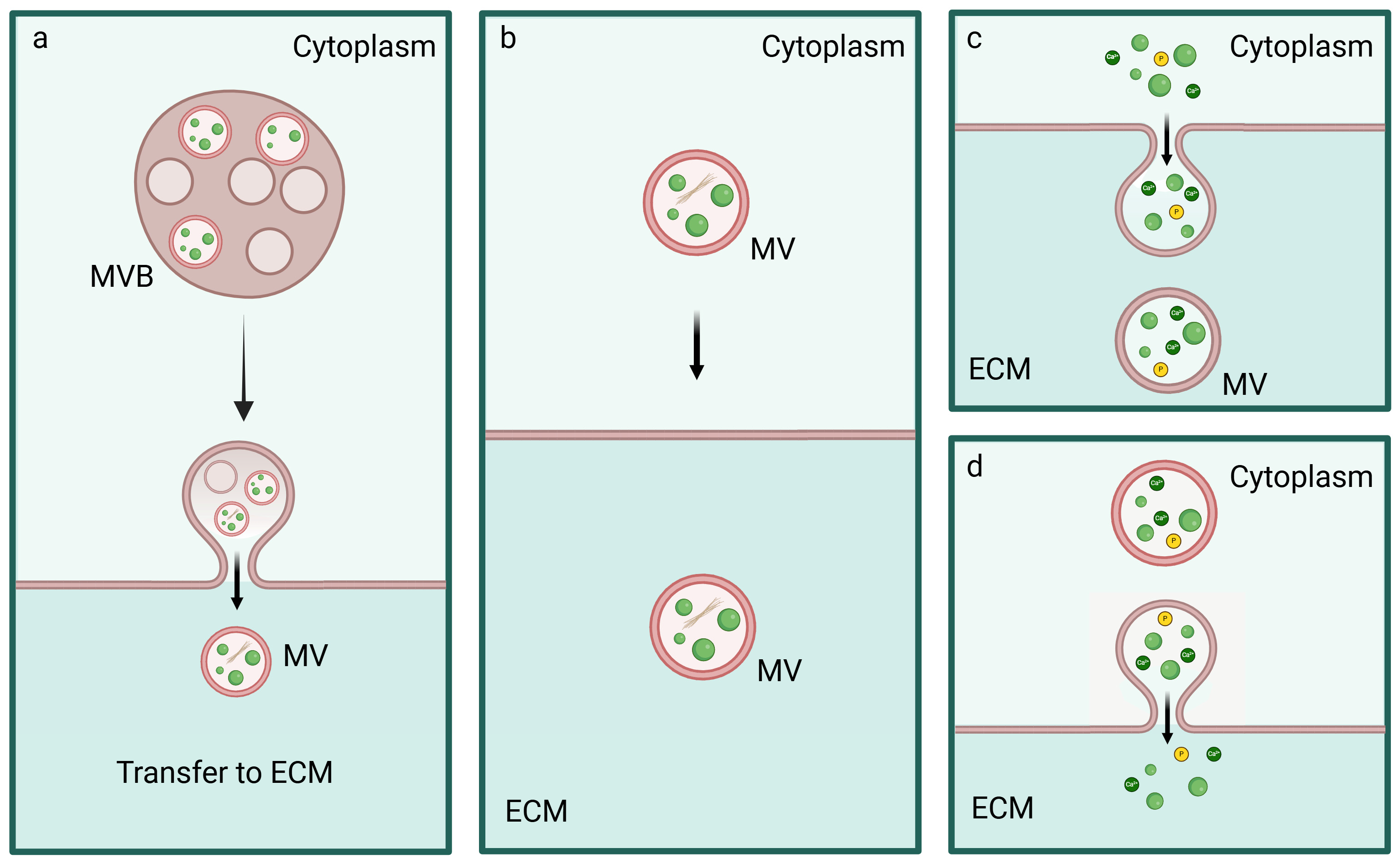
5. Biogenesis, Trafficking and Release of MVs
5.1. Inter-Organelle Communication and Physicochemical Structure Development of CaP
5.2. Regulation of MVs Biogenesis and Intracellular Trafficking
6. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sommerfeldt, D.W.; Rubin, C.T. Biology of bone and how it orchestrates the form and function of the skeleton. Eur. Spine J. 2001, 10 (Suppl. S2), S86–S95. [Google Scholar] [CrossRef]
- Ren, R.; Guo, J.; Chen, Y.; Zhang, Y.; Chen, L.; Xiong, W. The role of Ca2+/Calcineurin/NFAT signalling pathway in osteoblastogenesis. Cell Prolif. 2021, 54, e13122. [Google Scholar] [CrossRef]
- Bartold, M.; Gronthos, S.; Haynes, D.; Ivanovski, S. Mesenchymal stem cells and biologic factors leading to bone formation. J. Clin. Periodontol. 2019, 46 (Suppl. S21), 12–32. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, N.S. RUNX1 as a Novel Molecular Target for Breast Cancer. Clin. Breast Cancer 2022, 22, 499–506. [Google Scholar] [CrossRef]
- Krasnova, O.; Neganova, I. Assembling the Puzzle Pieces. Insights for in Vitro Bone Remodeling. Stem Cell Rev. Rep. 2023, 19, 1635–1658. [Google Scholar] [CrossRef]
- Chan, W.C.W.; Tan, Z.; To, M.K.T.; Chan, D. Regulation and Role of Transcription Factors in Osteogenesis. Int. J. Mol. Sci. 2021, 22, 5445. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [PubMed]
- Khotib, J.; Marhaeny, H.D.; Miatmoko, A.; Budiatin, A.S.; Ardianto, C.; Rahmadi, M.; Pratama, Y.A.; Tahir, M. Differentiation of osteoblasts: The links between essential transcription factors. J. Biomol. Struct. Dyn. 2023, 41, 10257–10276. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef]
- Yoshida, C.A.; Furuichi, T.; Fujita, T.; Fukuyama, R.; Kanatani, N.; Kobayashi, S.; Satake, M.; Takada, K.; Komori, T. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat. Genet. 2002, 32, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef]
- Meyer, M.B.; Benkusky, N.A.; Pike, J.W. The RUNX2 cistrome in osteoblasts: Characterization, down-regulation following differentiation, and relationship to gene expression. J. Biol. Chem. 2014, 289, 16016–16031. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Whitfield, T.W.; Gordon, J.A.; Dobson, J.R.; Tai, P.W.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol. 2014, 15, R52. [Google Scholar] [CrossRef]
- Weng, J.J.; Su, Y. Nuclear matrix-targeting of the osteogenic factor Runx2 is essential for its recognition and activation of the alkaline phosphatase gene. Biochim. Biophys. Acta 2013, 1830, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Saito, T.; He, X.; Guo, Q.; Onodera, S.; Azuma, T.; Koebis, M.; Nakao, K.; Aiba, A.; Seki, M.; et al. Runx2 regulates chromatin accessibility to direct the osteoblast program at neonatal stages. Cell Rep. 2022, 40, 111315. [Google Scholar] [CrossRef]
- Schroeder, T.M.; Jensen, E.D.; Westendorf, J.J. Runx2: A master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res. C Embryo Today 2005, 75, 213–225. [Google Scholar] [CrossRef]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or separate entities?—Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef]
- Makowski, A.J.; Uppuganti, S.; Wadeer, S.A.; Whitehead, J.M.; Rowland, B.J.; Granke, M.; Mahadevan-Jansen, A.; Yang, X.; Nyman, J.S. The loss of activating transcription factor 4 (ATF4) reduces bone toughness and fracture toughness. Bone 2014, 62, 1–9. [Google Scholar] [CrossRef]
- Xiao, G.; Jiang, D.; Ge, C.; Zhao, Z.; Lai, Y.; Boules, H.; Phimphilai, M.; Yang, X.; Karsenty, G.; Franceschi, R.T. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J. Biol. Chem. 2005, 280, 30689–30696. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef]
- Gomathi, K.; Akshaya, N.; Srinaath, N.; Moorthi, A.; Selvamurugan, N. Regulation of Runx2 by post-translational modifications in osteoblast differentiation. Life Sci. 2020, 245, 117389. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H. Emerging RUNX2-Mediated Gene Regulatory Mechanisms Consisting of Multi-Layered Regulatory Networks in Skeletal Development. Int. J. Mol. Sci. 2023, 24, 2979. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, C.; Li, Y.; Zheng, Q.; Xu, Y.; Liu, B.; Sun, W.; Ji, S.; Liu, M.; Zhang, J.; et al. TMCO1-mediated Ca2+ leak underlies osteoblast functions via CaMKII signaling. Nat. Commun. 2019, 10, 1589. [Google Scholar] [CrossRef]
- Adhami, M.; Ghori-Javed, F.Y.; Chen, H.; Gutierrez, S.E.; Javed, A. Runx2 regulates the gene network associated with insulin signaling and energy homeostasis. Cells Tissues Organs 2011, 194, 232–237. [Google Scholar] [CrossRef]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Lezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef]
- Liu, L.; Xie, H.; Zhao, S.; Huang, X. The GLUT1-mTORC1 axis affects odontogenic differentiation of human dental pulp stem cells. Tissue Cell 2022, 76, 101766. [Google Scholar] [CrossRef]
- Masuda, T.; Kubota, H.; Sakuramoto, N.; Hada, A.; Horiuchi, A.; Sasaki, A.; Takeda, K.; Takeda, M.; Matsuo, H.; Sugiyama, H.; et al. RUNX-NFAT Axis As a Novel Therapeutic Target for AML and T Cell Immunity. Blood 2020, 136 (Suppl. S1), 25–26. [Google Scholar] [CrossRef]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front. Cell Dev. Biol. 2020, 8, 601224. [Google Scholar] [CrossRef]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 2005, 11, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Schilling, L.; Eller, T.; Yu, J. Nuclear factor of activated T cells 1 and 2 are required for vertebral homeostasis. J. Cell. Physiol. 2020, 235, 8520–8532. [Google Scholar] [CrossRef]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hao, Z.; Wang, Y.; Zhu, H.; Hu, Y.; Chen, T.; Zhang, P.; Li, J. Mesenchymal Stem Cell-Immune Cell Interaction and Related Modulations for Bone Tissue Engineering. Stem Cells Int. 2022, 2022, 7153584. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Yang, N.; Sun, M.; Yang, S.; Chen, X. The role of calcium channels in osteoporosis and their therapeutic potential. Front Endocrinol. 2024, 15, 1450328. [Google Scholar] [CrossRef]
- Kito, H.; Ohya, S. Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions. Int. J. Mol. Sci. 2021, 22, 10459. [Google Scholar] [CrossRef]
- Chen, S.; Pan, M. NFAT Signaling and Bone Homeostasis. J. Hematol. Thrombo Dis. 2013, 1, 102. [Google Scholar]
- Lee, H.L.; Bae, O.Y.; Baek, K.H.; Kwon, A.; Hwang, H.R.; Qadir, A.S.; Park, H.J.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. High extracellular calcium-induced NFATc3 regulates the expression of receptor activator of NF-κB ligand in osteoblasts. Bone 2011, 49, 242–249. [Google Scholar] [CrossRef]
- Hu, F.; Pan, L.; Zhang, K.; Xing, F.; Wang, X.; Lee, I.; Zhang, X.; Xu, J. Elevation of extracellular Ca2+ induces store-operated calcium entry via calcium-sensing receptors: A pathway contributes to the proliferation of osteoblasts. PLoS ONE 2014, 9, e107217. [Google Scholar] [CrossRef]
- Zayzafoon, M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell. Biochem. 2006, 97, 56–70. [Google Scholar] [CrossRef]
- Shaw, J.P.; Utz, P.J.; Durand, D.B.; Toole, J.J.; Emmel, E.A.; Crabtree, G.R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241, 202–205. [Google Scholar] [CrossRef]
- Winslow, M.M.; Pan, M.; Starbuck, M.; Gallo, E.M.; Deng, L.; Karsenty, G.; Crabtree, G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 2006, 10, 771–782. [Google Scholar] [CrossRef]
- Fromigué, O.; Haÿ, E.; Barbara, A.; Marie, P.J. Essential role of nuclear factor of activated T cells (NFAT)-mediated Wnt signaling in osteoblast differentiation induced by strontium ranelate. J. Biol. Chem. 2010, 285, 25251–25258. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.K.; Yeo, H.; Zayzafoon, M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone 2009, 45, 579–589. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, X.; Siu, R.K.; Chen, F.; Shen, J.; Zara, J.N.; Culiat, C.T.; Tetradis, S.; Ting, K.; Soo, C. Nfatc2 is a primary response gene of Nell-1 regulating chondrogenesis in ATDC5 cells. J. Bone Miner. Res. 2011, 26, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.G.; Xiong, Y.; Chen, F. NFAT gene family in inflammation and cancer. Curr. Mol. Med. 2013, 13, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem. 2004, 279, 26475–26480. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef]
- Cao, X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. 2018, 6, 35. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, X.; Terauchi, M.; Li, J.Y.; Grassi, F.; Galley, S.; Yang, X.; Weitzmann, M.N.; Pacifici, R. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab. 2008, 8, 132–145. [Google Scholar] [CrossRef]
- Sarode, A.Y.; Jha, M.K.; Zutshi, S.; Ghosh, S.K.; Mahor, H.; Sarma, U.; Saha, B. Residue-Specific Message Encoding in CD40-Ligand. iScience 2020, 23, 101441. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhu, H.; Yan, Q.; Shen, X.; Lu, X.; Wang, J.; Li, J.; Chen, L. TRPV2-induced Ca2+-calcineurin-NFAT signaling regulates differentiation of osteoclast in multiple myeloma. Cell Commun. Signal. 2018, 16, 68. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Baek, K.; Baek, J.H.; Kim, H.R. TNFα Increases RANKL Expression via PGE₂-Induced Activation of NFATc1. Int. J. Mol. Sci. 2017, 18, 495. [Google Scholar] [CrossRef]
- Sitara, D.; Aliprantis, A.O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 2010, 233, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Okabe, K.; Tanaka, S. Finely-Tuned Calcium Oscillations in Osteoclast Differentiation and Bone Resorption. Int. J. Mol. Sci. 2020, 22, 180. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Ishikawa, M.; Williams, G.; Forcinito, P.; Petrie, R.J.; Saito, K.; Fukumoto, S.; Yamada, Y. Pannexin 3 ER Ca2+ channel gating is regulated by phosphorylation at the Serine 68 residue in osteoblast differentiation. Sci. Rep. 2019, 9, 18759. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, Y.; Alharbi, A.; Hanbashi, A.; Alhoshani, A.; Parrington, J. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol. Sci. 2020, 41, 582–594. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Boyman, L.; Williams, G.S.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef]
- Carraro, M.; Bernardi, P. The mitochondrial permeability transition pore in Ca2+ homeostasis. Cell Calcium 2023, 111, 102719. [Google Scholar] [CrossRef]
- Bischof, H.; Burgstaller, S.; Waldeck-Weiermair, M.; Rauter, T.; Schinagl, M.; Ramadani-Muja, J.; Graier, W.F.; Malli, R. Live-Cell Imaging of Physiologically Relevant Metal Ions Using Genetically Encoded FRET-Based Probes. Cells 2019, 8, 492. [Google Scholar] [CrossRef] [PubMed]
- García-Sancho, J. The coupling of plasma membrane calcium entry to calcium uptake by endoplasmic reticulum and mitochondria. J. Physiol. 2014, 592, 261–268. [Google Scholar] [CrossRef]
- Pei, D.D.; Sun, J.L.; Zhu, C.H.; Tian, F.C.; Jiao, K.; Anderson, M.R.; Yiu, C.; Huang, C.; Jin, C.X.; Bergeron, B.E.; et al. Contribution of Mitophagy to Cell-Mediated Mineralization: Revisiting a 50-Year-Old Conundrum. Adv. Sci. 2018, 5, 1800873. [Google Scholar] [CrossRef] [PubMed]
- Reith, E.J. The binding of calcium within the Golgi saccules of the rat odontoblast. Am. J. Anat. 1976, 147, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Báthori, G.; Csordás, G.; Garcia-Perez, C.; Davies, E.; Hajnóczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Stenzinger, A.; Schreiner, D.; Koch, P.; Hofer, H.W.; Wimmer, M. Cell and molecular biology of the novel protein tyrosine-phosphatase-interacting protein 51. Int. Rev. Cell Mol. Biol. 2009, 275, 183–246. [Google Scholar] [CrossRef]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca2+ Homeostasis and Signaling in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium homeostasis and cancer: Insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Guerra, F.; Bucci, C.; Marzetti, E.; Picca, A. Mitochondria break free: Mitochondria-derived vesicles in aging and associated conditions. Ageing Res. Rev. 2024, 102, 102549. [Google Scholar] [CrossRef]
- Iwayama, T.; Bhongsatiern, P.; Takedachi, M.; Murakami, S. Matrix Vesicle-Mediated Mineralization and Potential Applications. J. Dent. Res. 2022, 101, 1554–1562. [Google Scholar] [CrossRef]
- Iwayama, T.; Okada, T.; Ueda, T.; Tomita, K.; Matsumoto, S.; Takedachi, M.; Wakisaka, S.; Noda, T.; Ogura, T.; Okano, T.; et al. Osteoblastic lysosome plays a central role in mineralization. Sci. Adv. 2019, 5, eaax0672. [Google Scholar] [CrossRef]
- Boonrungsiman, S.; Gentleman, E.; Carzaniga, R.; Evans, N.D.; McComb, D.W.; Porter, A.E.; Stevens, M.M. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14170–14175. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Kirsch, T.; Harrison, G.; Golub, E.E.; Nah, H.D. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J. Biol. Chem. 2000, 275, 35577–35583. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Landis, W.J.; Risbud, M.V. Matrix vesicles: Are they anchored exosomes? Bone 2015, 79, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part. B Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Dean, D.; Hornicek, F.J.; Chen, Z.; Duan, Z. The role of extracelluar matrix in osteosarcoma progression and metastasis. J. Exp. Clin. Cancer Res. 2020, 39, 178. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Skelton, A.M.; Cohen, D.J.; Boyan, B.D.; Schwartz, Z. Osteoblast-Derived Matrix Vesicles Exhibit Exosomal Traits and a Unique Subset of microRNA: Their Caveolae-Dependent Endocytosis Results in Reduced Osteogenic Differentiation. Int. J. Mol. Sci. 2023, 24, 12770. [Google Scholar] [CrossRef]
- Bhadada, S.K.; Rao, S.D. Role of Phosphate in Biomineralization. Calcif. Tissue Int. 2021, 108, 32–40. [Google Scholar] [CrossRef]
- Newman, W.F.; Newman, M.W. The Chemical Dynamics of Bone Mineral. In Arthritis & Rheumatism; The University of Chicago Press: Chicago, IL, USA, 1958; Volume 1, pp. 473–474. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, Q.; Ma, X.; Zhong, Y.; Tang, H.; Mai, S. The mechanism of biomineralization: Progress in mineralization from intracellular generation to extracellular deposition. Jpn. Dent. Sci. Rev. 2023, 59, 181–190. [Google Scholar] [CrossRef]
- Merametdjian, L.; Beck-Cormier, S.; Bon, N.; Couasnay, G.; Sourice, S.; Guicheux, J.; Gaucher, C.; Beck, L. Expression of Phosphate Transporters during Dental Mineralization. J. Dent. Res. 2018, 97, 209–217. [Google Scholar] [CrossRef]
- Adams, C.S.; Mansfield, K.; Perlot, R.L.; Shapiro, I.M. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J. Biol. Chem. 2001, 276, 20316–20322. [Google Scholar] [CrossRef]
- Hu, P.; Lacruz, R.S.; Smith, C.E.; Smith, S.M.; Kurtz, I.; Paine, M.L. Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts. Cells Tissues Organs 2012, 196, 501–509. [Google Scholar] [CrossRef]
- Parry, D.A.; Poulter, J.A.; Logan, C.V.; Brookes, S.J.; Jafri, H.; Ferguson, C.H.; Anwari, B.M.; Rashid, Y.; Zhao, H.; Johnson, C.A.; et al. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am. J. Hum. Genet. 2013, 92, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Goretti Penido, M.; Alon, U.S. Phosphate homeostasis and its role in bone health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J.; Glimcher, M.J. Electron optical and analytical observations of rat growth plate cartilage prepared by ultracryomicrotomy: The failure to detect a mineral phase in matrix vesicles and the identification of heterodispersed particles as the initial solid phase of calcium phosphate deposited in the extracellular matrix. J. Ultrastruct. Res. 1982, 78, 227–268. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.; Lyon, R.; Swindin, K.J.; Chesters, J. Ultrastructure and energy-dispersive x-ray microanalysis of cartilage after rapid freezing, low temperature freeze drying, and embedding in Spurr’s resin. J. Histochem. Cytochem. 1985, 33, 1073–1079. [Google Scholar] [CrossRef]
- Hsu, H.H.; Morris, D.C.; Davis, L.; Moylan, P.; Anderson, C.H. In vitro Ca deposition by rat matrix vesicles: Is the membrane association of alkaline phosphatase essential for matrix vesicle-mediated calcium deposition? Int. J. Biochem. 1993, 25, 1737–1742. [Google Scholar] [CrossRef]
- Mackie, E.J. Osteoblasts: Novel roles in orchestration of skeletal architecture. Int. J. Biochem. Cell Biol. 2003, 35, 1301–1305. [Google Scholar] [CrossRef]
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell Sci. 2000, 113 Pt 3, 377–381. [Google Scholar] [CrossRef]
- Mahamid, J.; Aichmayer, B.; Shimoni, E.; Ziblat, R.; Li, C.; Siegel, S.; Paris, O.; Fratzl, P.; Weiner, S.; Addadi, L. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc. Natl. Acad. Sci. USA 2010, 107, 6316–6321. [Google Scholar] [CrossRef]
- Boraldi, F.; Lofaro, F.D.; Quaglino, D. Apoptosis in the Extraosseous Calcification Process. Cells 2021, 10, 131. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Mayer, H. Exocytotic process as a novel model for mineralization by osteoblasts in vitro and in vivo determined by electron microscopic analysis. Calcif. Tissue Int. 2007, 80, 323–336. [Google Scholar] [CrossRef]
- Tang, C.; Wei, Y.; Gu, L.; Zhang, Q.; Li, M.; Yuan, G.; He, Y.; Huang, L.; Liu, Y.; Zhang, Y. Biomineral Precursor Formation Is Initiated by Transporting Calcium and Phosphorus Clusters from the Endoplasmic Reticulum to Mitochondria. Adv. Sci. 2020, 7, 1902536. [Google Scholar] [CrossRef] [PubMed]
- Akisaka, T.; Kawaguchi, H.; Subita, G.P.; Shigenaga, Y.; Gay, C.V. Ultrastructure of matrix vesicles in chick growth plate as revealed by quick freezing and freeze substitution. Calcif. Tissue Int. 1988, 42, 383–393. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Greenawalt, J.W.; Rossi, C.S.; Lehninger, A.L. Effect of active accumulation of calcium and phosphate ions on the structure of rat liver mitochondria. J. Cell Biol. 1964, 23, 21–38. [Google Scholar] [CrossRef]
- Anderson, H.C. Electron microscopic studies of induced cartilage development and calcification. J. Cell Biol. 1967, 35, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E. Fine structure of early cartilage calcification. J. Ultrastruct. Res. 1967, 20, 33–50. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bucci, C.; Marzetti, E. Mitochondrial-Derived Vesicles: The Good, the Bad, and the Ugly. Int. J. Mol. Sci. 2023, 24, 13835. [Google Scholar] [CrossRef]
- Docampo, R. The origin and evolution of the acidocalcisome and its interactions with other organelles. Mol. Biochem. Parasitol. 2016, 209, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Liou, W.; Geuze, H.J.; Geelen, M.J.; Slot, J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997, 136, 61–70. [Google Scholar] [CrossRef]
- Mahamid, J.; Sharir, A.; Gur, D.; Zelzer, E.; Addadi, L.; Weiner, S. Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: A cryo-electron microscopy study. J. Struct. Biol. 2011, 174, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, E.; Petersson, U.; Hultenby, K.; Wendel, M. Calreticulin—An endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol. 2003, 22, 179–191. [Google Scholar] [CrossRef]
- Petersson, U.; Somogyi, E.; Reinholt, F.P.; Karlsson, T.; Sugars, R.V.; Wendel, M. Nucleobindin is produced by bone cells and secreted into the osteoid, with a potential role as a modulator of matrix maturation. Bone 2004, 34, 949–960. [Google Scholar] [CrossRef]
- Lavoie, C.; Meerloo, T.; Lin, P.; Farquhar, M.G. Calnuc, an EF-hand Ca(2+)-binding protein, is stored and processed in the Golgi and secreted by the constitutive-like pathway in AtT20 cells. Mol. Endocrinol. 2002, 16, 2462–2474. [Google Scholar] [CrossRef]
- Bonucci, E. Fine structure and histochemistry of "calcifying globules" in epiphyseal cartilage. Z. Zellforsch. Mikrosk. Anat. 1970, 103, 192–217. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.Y.; Sajdera, S.W.; Anderson, H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA 1970, 67, 1513–1520. [Google Scholar] [CrossRef]
- Anderson, H.C.; Matsuzawa, T.; Sajdera, S.W.; Ali, S.Y. Membranous particles in calcifying cartilage matrix. Trans. N. Y. Acad. Sci. 1970, 32, 619–630. [Google Scholar] [CrossRef]
- Anderson, H.C. Molecular biology of matrix vesicles. Clin. Orthop. Relat. Res. 1995, 314, 266–280. [Google Scholar] [CrossRef]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef]
- Rosenthal, A.K.; Gohr, C.M.; Ninomiya, J.; Wakim, B.T. Proteomic analysis of articular cartilage vesicles from normal and osteoarthritic cartilage. Arthritis Rheum. 2011, 63, 401–411. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Investig. 2016, 126, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Bento-Lopes, L.; Ramalho, J.S.; Barral, D.C. Rab11 is required for lysosome exocytosis through the interaction with Rab3a, Sec15 and GRAB. J. Cell Sci. 2021, 134, jcs246694. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef]
- Komarova, S.V.; Ataullakhanov, F.I.; Globus, R.K. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am. J. Physiol. Cell Physiol. 2000, 279, C1220–C1229. [Google Scholar] [CrossRef]
- De Rooij, J.F.; Heughebaert, J.C.; Nancollas, G.H. A ph study of calcium phosphate seeded precipitation. J. Colloid Interface Sci. 1984, 100, 350–358. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Kerschnitzki, M.; Akiva, A.; Ben Shoham, A.; Asscher, Y.; Wagermaier, W.; Fratzl, P.; Addadi, L.; Weiner, S. Bone mineralization pathways during the rapid growth of embryonic chicken long bones. J. Struct. Biol. 2016, 195, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Fang, F.; Yuan, H.; Yang, D.; Chen, Y.; Williams, L.; Goldstein, S.A.; Krebsbach, P.H.; Guan, J.L. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J. Bone Miner. Res. 2013, 28, 2414–2430. [Google Scholar] [CrossRef] [PubMed]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Shen, M.; Sui, B.; Lu, W.; Han, X.; Wan, Q.; Liu, Y.; Kang, J.; Qin, W.; Zhang, Z.; et al. Autophagic LC3. Sci. Adv. 2022, 8, eabn1556. [Google Scholar] [CrossRef]
- Hale, J.E.; Wuthier, R.E. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J. Biol. Chem. 1987, 262, 1916–1925. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margiotta, A. Coupling of Intracellular Calcium Homeostasis and Formation and Secretion of Matrix Vesicles: Their Role in the Mechanism of Biomineralization. Cells 2025, 14, 733. https://doi.org/10.3390/cells14100733
Margiotta A. Coupling of Intracellular Calcium Homeostasis and Formation and Secretion of Matrix Vesicles: Their Role in the Mechanism of Biomineralization. Cells. 2025; 14(10):733. https://doi.org/10.3390/cells14100733
Chicago/Turabian StyleMargiotta, Azzurra. 2025. "Coupling of Intracellular Calcium Homeostasis and Formation and Secretion of Matrix Vesicles: Their Role in the Mechanism of Biomineralization" Cells 14, no. 10: 733. https://doi.org/10.3390/cells14100733
APA StyleMargiotta, A. (2025). Coupling of Intracellular Calcium Homeostasis and Formation and Secretion of Matrix Vesicles: Their Role in the Mechanism of Biomineralization. Cells, 14(10), 733. https://doi.org/10.3390/cells14100733