


Genetic Predisposition and Mitochondrial Dysfunction in Sudden Cardiac Death: Role of MCU Complex Genetic Variations
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Populations in Case-Control Study
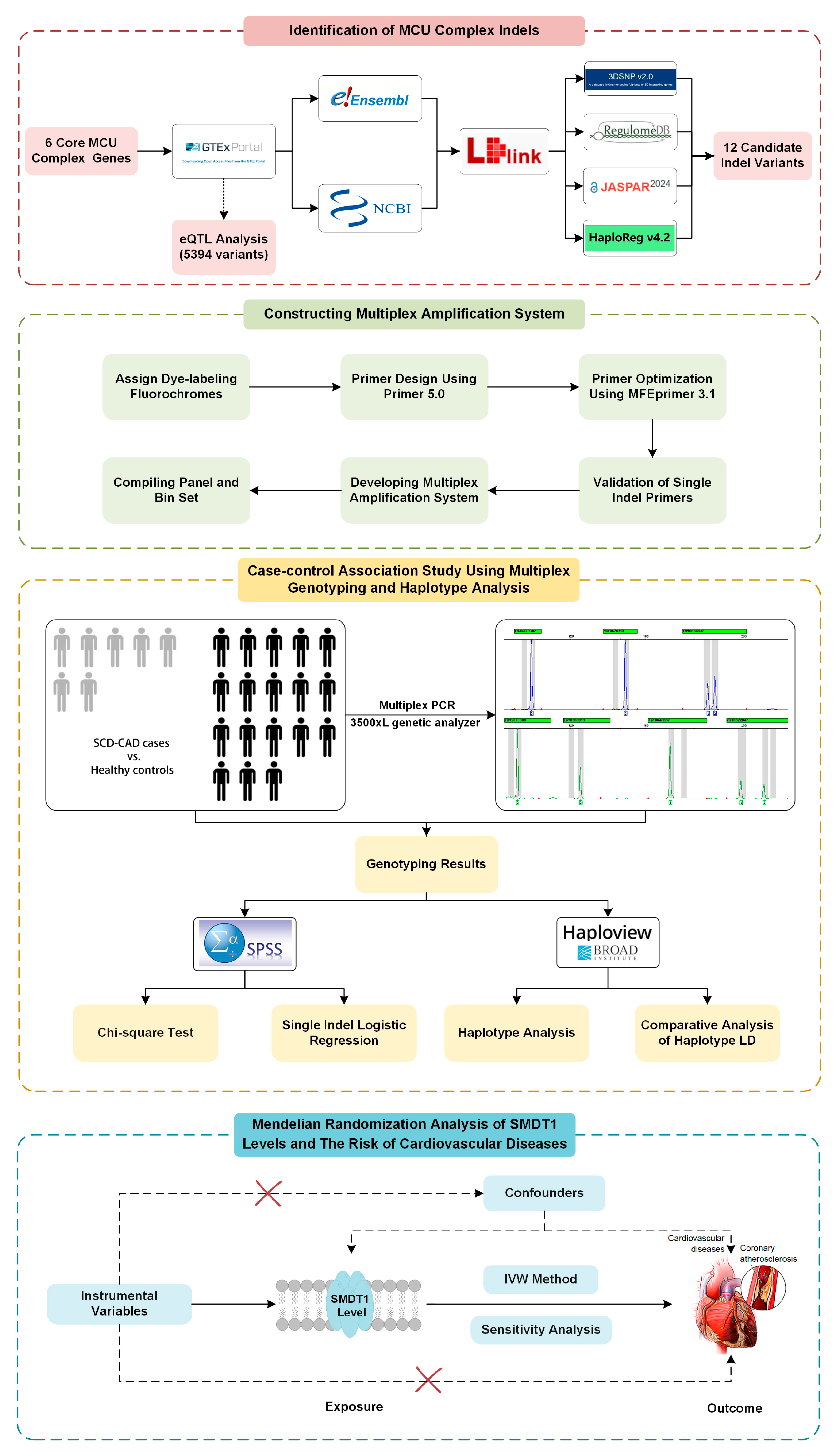
2.3. Selection of MCU Gene Variants
2.4. Multiplex Genotyping
2.5. Statistical Analysis of Case-Control Study and Haplotype Analysis
2.6. Bioinformatic Analysis of Positive Indel Variants
2.7. MR Analysis of SMDT1-Encoded Mitochondrial MCU Regulator Levels with Cardiovascular Diseases
3. Results
3.1. Selection of Candidate Indel Variants
3.2. Construction and Optimization of Multiplex Amplification System
3.3. Association Between MCU Indel Variants and SCD Susceptibilities
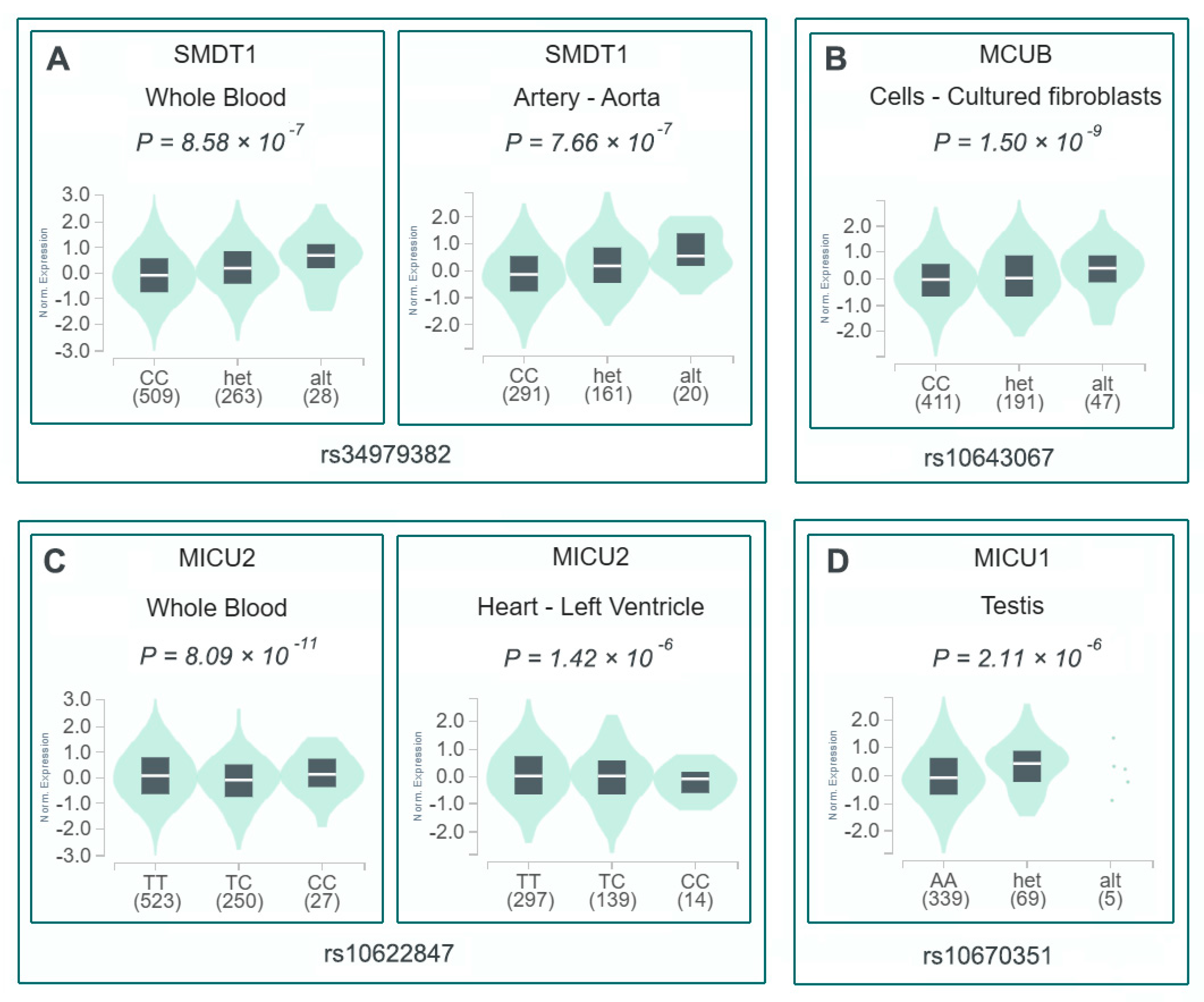
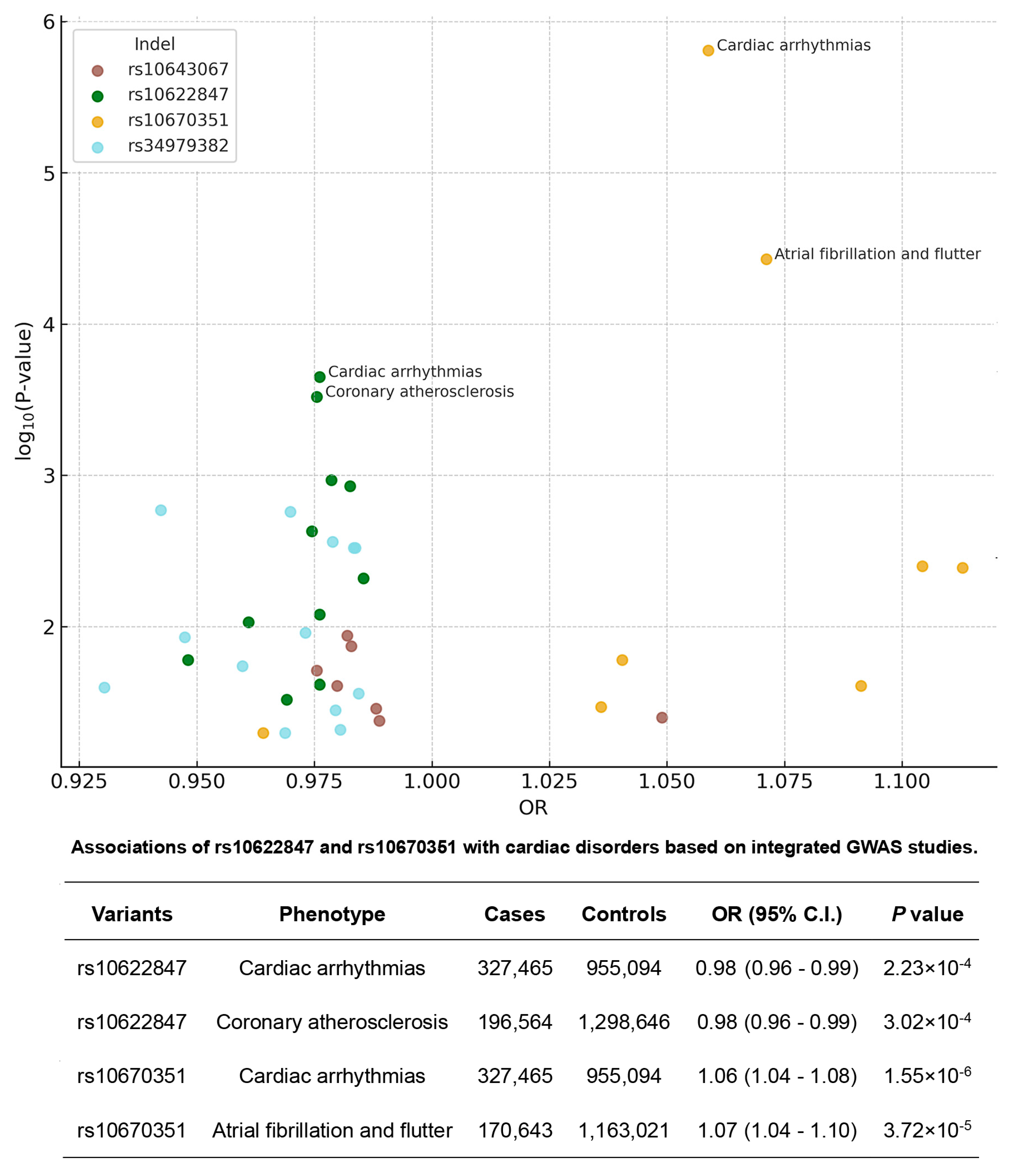
3.4. Bioinformatic Characterization of Four Risk-Associated Indel Variants
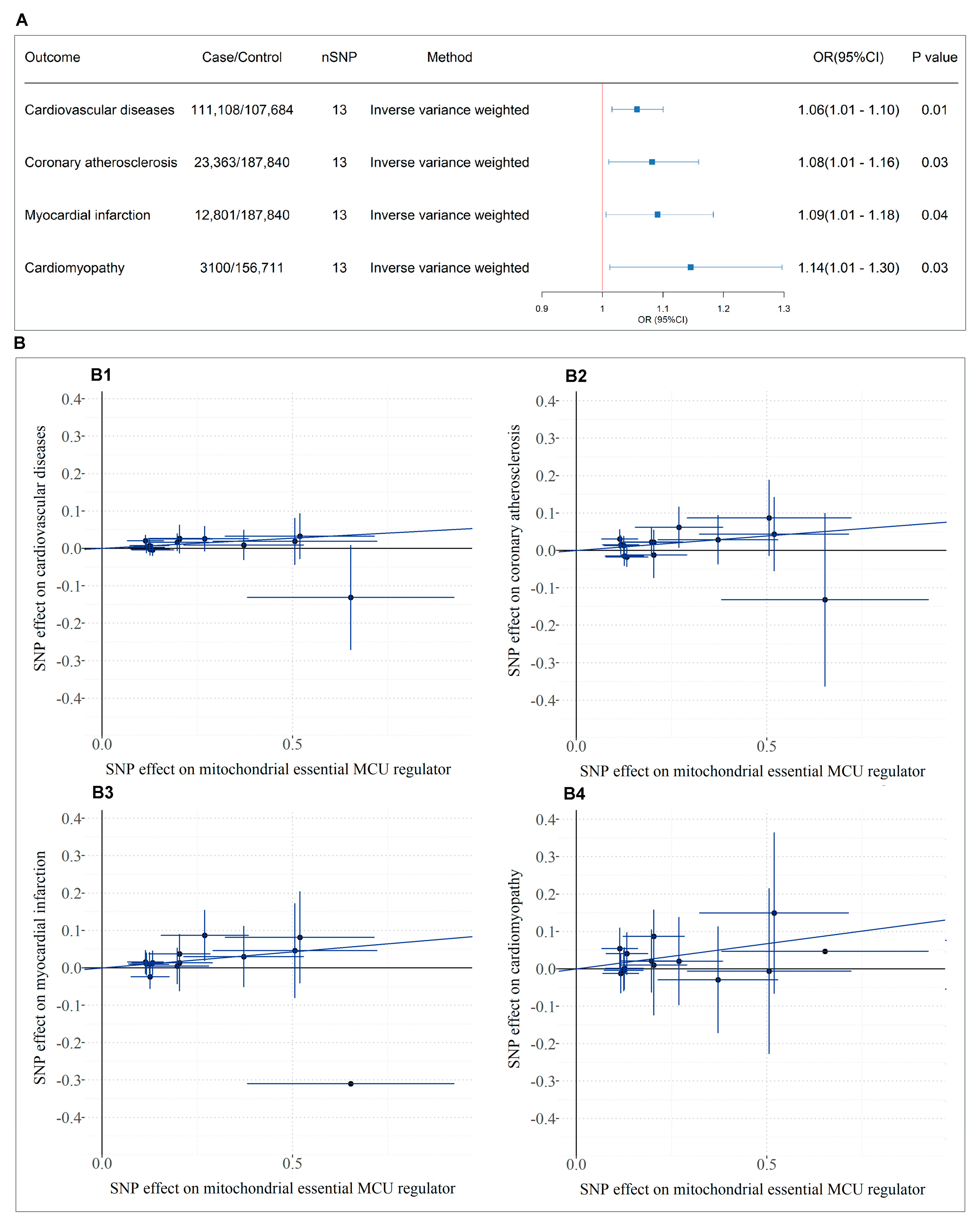
3.5. Causal Relationship Between SMDT1-Encoded Mitochondrial MCU Regulator Levels and Cardiovascular Diseases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCD | sudden cardiac death |
| SCD-CAD | coronary artery disease-related sudden cardiac death |
| MCU | mitochondrial calcium uniporter |
| IMM | the inner mitochondrial membrane |
| MCUB | the dominant-negative β-subunit |
| MICU | the mitochondrial calcium uptake |
| EMRE | the essential MCU regulator |
| MCUR1 | the MCU regulator 1 |
| ROS | reactive oxygen species |
| I/R | ischemia-reperfusion |
| CE | capillary electrophoresis |
| STR | short tandem repeat |
| Indel | insertions and deletion |
| SNP | single nucleotide polymorphisms |
| MR | Mendelian randomization |
| GWAS | genome-wide association studies |
| GTEx | the genotype-tissue expression |
| SMDT1 | single-pass membrane protein with aspartate rich tail 1 |
| eQTL | expression quantitative trait loci |
| MAF | minor allele frequency |
| PAGE | polyacrylamide gel electrophoresis |
| LD | Linkage disequilibrium |
| RFU | relative fluorescence units |
| HWE | Hardy–Weinberg equilibrium |
| OR | odds ratio |
| CI | confidence interval |
| IVW | inverse-variance weighted |
| MR-PRESSO | Mendelian randomization pleiotropy residual sum and outlier |
References
- Zipes, D.P.; Wellens, H.J. Sudden cardiac death. Circulation 1998, 98, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Wellens, H.J.; Schwartz, P.J.; Lindemans, F.W.; Buxton, A.E.; Goldberger, J.J.; Hohnloser, S.H.; Huikuri, H.V.; Kääb, S.; La Rovere, M.T.; Malik, M.; et al. Risk stratification for sudden cardiac death: Current status and challenges for the future. Eur. Heart J. 2014, 35, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Zhang, L.F.; Wu, Y.F.; Liu, X.Q.; Guo, D.S.; Zhou, H.L.; Gou, Z.P.; Zhao, L.C.; Niu, H.X.; Chen, K.P.; et al. Incidence of sudden cardiac death in China: Analysis of 4 regional populations. J. Am. Coll. Cardiol. 2009, 54, 1110–1118. [Google Scholar] [CrossRef]
- Corrado, D.; Schmied, C.; Basso, C.; Borjesson, M.; Schiavon, M.; Pelliccia, A.; Vanhees, L.; Thiene, G. Risk of sports: Do we need a pre-participation screening for competitive and leisure athletes? Eur. Heart J. 2011, 32, 934–944. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Xu, H.; Li, L.; Guo, Y.; Zhang, J.; Cheng, L.; Yu, H.; Dai, Y.; Yang, Q.; et al. Modulation of STIM1 by a risk insertion/deletion polymorphism underlying genetics susceptibility to sudden cardiac death originated from coronary artery disease. Forensic. Sci. Int. 2021, 328, 111010. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2018, 72, 1677–1749. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Barth, E.; Stämmler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Liu, J.C. Mitochondrial calcium and reactive oxygen species in cardiovascular disease. Cardiovasc. Res. 2023, 119, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Liu, M.; Chen, Y.; Wu, Y.; Li, Q.; Ma, T.; Gao, J.; Xia, Y.; Fan, M.; et al. Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury. Basic Res. Cardiol. 2021, 116, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, H.; Wang, S. Vitamin D3 Suppresses Human Cytomegalovirus-Induced Vascular Endothelial Apoptosis via Rectification of Paradoxical m6A Modification of Mitochondrial Calcium Uniporter mRNA, Which Is Regulated by METTL3 and YTHDF3. Front. Microbiol. 2022, 13, 861734. [Google Scholar] [CrossRef]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Fischer, B.; Dittmann, S.; Brodehl, A.; Unger, A.; Stallmeyer, B.; Paul, M.; Seebohm, G.; Kayser, A.; Peischard, S.; Linke, W.A.; et al. Functional characterization of novel alpha-helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int. J. Cardiol. 2021, 329, 167–174. [Google Scholar] [CrossRef]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef]
- Alves-Figueiredo, H.; Silva-Platas, C.; Estrada, M.; Oropeza-Almazán, Y.; Ramos-González, M.; Bernal-Ramírez, J.; Vázquez-Garza, E.; Tellez, A.; Salazar-Ramírez, F.; Méndez-Fernández, A.; et al. Mitochondrial Ca2+ Uniporter-Dependent Energetic Dysfunction Drives Hypertrophy in Heart Failure. JACC Basic Transl. Sci. 2024, 9, 496–518. [Google Scholar] [CrossRef] [PubMed]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stølen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, S.; Xu, J.; Xin, Y.; Lu, Y.; Zhang, H.; Zhou, B.; Xu, H.; Sheu, S.S.; Tian, R.; et al. Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling. Circulation 2022, 145, 1067–1083. [Google Scholar] [CrossRef]
- Pascali, J.P.; Bortolotti, F.; Tagliaro, F. Recent advances in the application of CE to forensic sciences, an update over years 2009–2011. Electrophoresis 2012, 33, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, J.M.; Mills, R.E.; Pittard, W.S.; Devine, S.E. Small insertions and deletions (INDELs) in human genomes. Hum. Mol. Genet. 2010, 19, R131–R136. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, Q.; Zhang, J.; Chen, X.; Zhou, W.; Yang, Z.; Yang, Q.; Yu, H.; Li, L.; He, Y.; et al. A common indel polymorphism of the Desmoglein-2 (DSG2) is associated with sudden cardiac death in Chinese populations. Forensic Sci. Int. 2019, 301, 382–387. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Yang, Y.; Wang, C.; Tao, R.; Hu, S.; Yin, Z.; Zhang, Q.; Li, L.; He, Y.; et al. An insertion/deletion polymorphism within 3′UTR of RYR2 modulates sudden unexplained death risk in Chinese populations. Forensic Sci. Int. 2017, 270, 165–172. [Google Scholar] [CrossRef]
- Allen, R.C.; Graves, G.; Budowle, B. Polymerase chain reaction amplification products separated on rehydratable polyacrylamide gels and stained with silver. Biotechniques 1989, 7, 736–744. [Google Scholar]
- Wang, K.; Li, H.; Xu, Y.; Shao, Q.; Yi, J.; Wang, R.; Cai, W.; Hang, X.; Zhang, C.; Cai, H.; et al. MFEprimer-3.0: Quality control for PCR primers. Nucleic Acids. Res. 2019, 47, W610–W613. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 2013, 178, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Dudbridge, F.; Thompson, S.G. Combining information on multiple instrumental variables in Mendelian randomization: Comparison of allele score and summarized data methods. Stat. Med. 2016, 35, 1880–1906. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Holmes, M.V. Meta-analysis and Mendelian randomization: A review. Res. Synth. Methods 2019, 10, 486–496. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Hemani, G.; Davey Smith, G. Invited Commentary: Detecting Individual and Global Horizontal Pleiotropy in Mendelian Randomization—A Job for the Humble Heterogeneity Statistic? Am. J. Epidemiol. 2018, 187, 2681–2685. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef]
- Sun, L.; Leng, R.; Liu, M.; Su, M.; He, Q.; Zhang, Z.; Liu, Z.; Wang, Z.; Jiang, H.; Wang, L.; et al. Endothelial MICU1 protects against vascular inflammation and atherosclerosis by inhibiting mitochondrial calcium uptake. J. Clin. Investig. 2025, 135, e181928. [Google Scholar] [CrossRef]
- Yang, Y.; Du, J.; Xu, R.; Shen, Y.; Yang, D.; Li, D.; Hu, H.; Pei, H.; Yang, Y. Melatonin alleviates angiotensin-II-induced cardiac hypertrophy via activating MICU1 pathway. Aging 2020, 13, 493–515. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, F.; Chen, D.; Tian, Y.; Liu, C.; Li, F. MICU1 alleviates hypobaric hypoxia-induced myocardial injury through regulating Ca2+ uptake to inhibit mitochondria-dependent apoptosis. Cell Signal. 2025, 125, 111524. [Google Scholar] [CrossRef]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP3R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154. [Google Scholar] [CrossRef]
- Chapoy Villanueva, H.; Sung, J.H.; Stevens, J.A.; Zhang, M.J.; Nelson, P.M.; Denduluri, L.S.; Feng, F.; O’Connell, T.D.; Townsend, D.; Liu, J.C. Distinct effects of cardiac mitochondrial calcium uniporter inactivation via EMRE deletion in the short and long term. J. Mol. Cell Cardiol. 2023, 181, 33–45. [Google Scholar] [CrossRef]
- Huo, J.; Lu, S.; Kwong, J.Q.; Bround, M.J.; Grimes, K.M.; Sargent, M.A.; Brown, M.E.; Davis, M.E.; Bers, D.M.; Molkentin, J.D. MCUb Induction Protects the Heart From Postischemic Remodeling. Circ. Res. 2020, 127, 379–390. [Google Scholar] [CrossRef]
- Kaludercic, N.; Scorrano, L. MCUB Hearts Mitochondria in Sickness, Less in Health. Circulation 2019, 140, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Faccenda, D.; Gorini, G.; Jones, A.; Thornton, C.; Baracca, A.; Solaini, G.; Campanella, M. The ATPase Inhibitory Factor 1 (IF1) regulates the expression of the mitochondrial Ca2+ uniporter (MCU) via the AMPK/CREB pathway. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118860. [Google Scholar] [CrossRef]
- Liao, Q.; Zhang, Y.; Pan, T.; Sun, Y.; Liu, S.; Zhang, Z.; Li, Y.; Yu, L.; Luo, Z.; Xiao, Y.; et al. Liver knockout of MCU leads to greater dysregulation of lipid metabolism in MAFLD. Sci. Rep. 2024, 14, 28167. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Li, Z.; Bu, K.; Li, T.; Ma, Z.; Wang, B.; Ma, L.; Lu, H.; Zhang, K.; et al. Pyk2/MCU Pathway as a New Target for Reversing Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 651579. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Wakimoto, H.; Kamer, K.J.; Sancak, Y.; Goldberger, O.; Axelsson, A.; DeLaughter, D.M.; Gorham, J.M.; Mootha, V.K.; Seidman, J.G.; et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E9096–E9104. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Indel ID | Chr: Position (GRCh38) | MAF (RGC—Million Exome Variant Browser) | Alleles |
|---|---|---|---|---|
| SMDT1 | rs34979382 | 22:42218818–42218821 | 0.35 | dupGTT |
| MICU1 | rs10670351 | 10:72985345–72985347 | 0.25 | insTGTT |
| MICU1 | rs10634037 | 10:73062986–73062989 | 0.33 | dupCCC |
| SMDT1 | rs139522 | 22:41266253–41266255 | 0.20 | insCCTGAAGCAACCACAGC |
| MICU3 | rs10680396 | 8:17120219–17120220 | 0.21 | insGATT |
| SMDT1 | rs140490511 | 22:42068821–42068844 | 0.14 | dupGGAATTAGC(A)4CTAACACCT |
| MICU2 | rs35073080 | 13:21422625–21422636 | 0.22 | delATT |
| MICU2 | rs10580013 | 13:21371494–21371499 | 0.23 | delAATA |
| MCUB | rs10643067 | 4:109572694–109572697 | 0.10 | insGACTT |
| MICU2 | rs10622847 | 13:21600794–21600812 | 0.39 | dup(ATT)3/dup(ATT)4 |
| MCUB | rs5860983 | 4:109630153–109630157 | 0.29 | del(T)5 |
| SMDT1 | rs59575728 | 22:42042421–42042442 | 0.43 | delACTTGAGTCA(TCT)2 |
| Characteristic | SCD-CAD | SCD Matched Controls | p-Value |
|---|---|---|---|
| No. of individuals | 229 | 598 | |
| Sex, No. | |||
| Male | 207 | 518 | 0.14 a |
| Female | 22 | 80 | |
| Age, mean ± SD (range) | |||
| Overall | 49.52 ± 13.07 (23–86) | 48.09 ± 14.04 (16–90) | 0.18 b |
| Males | 48.66 ± 12.48 (23–86) | 47.16 ± 13.28 (16–90) | 0.16 b |
| Females | 57.55 ± 15.88 (27–85) | 54.11 ± 17.20 (24–89) | 0.40 b |
| Events at sudden death (SD) | |||
| Nonspecific | 96 | ||
| Physical activity | 47 | ||
| Stress | 68 | ||
| Sleep | 18 | ||
| Symptoms before SD | |||
| None | 143 | ||
| Others | 86 | ||
| Megalothymus | |||
| Positive | 5 | ||
| Negative | 224 |
| Indel | Genetic Model | Genotype | Cases | (%) | Control | (%) | OR (95% C.I.) a | p-Value |
|---|---|---|---|---|---|---|---|---|
| rs34979382 | Codominant model | ins/ins | 34 | 15.35 | 58 | 9.70 | 1.00 (Reference) | |
| ins/del | 91 | 39.91 | 234 | 39.13 | 0.66 (0.41–1.08) | 0.098 | ||
| del/del | 102 | 44.74 | 306 | 51.17 | 0.57 (0.35–0.92) | 0.020 | ||
| Ptrend | 0.029 | |||||||
| Recessive model | ins/ins | 34 | 14.98 | 58 | 9.70 | 1.00 (Reference) | ||
| del/del + ins/del | 193 | 85.02 | 540 | 90.30 | 0.61 (0.39–0.96) | 0.031 | ||
| Additive model | ins allele | 159 | 35.02 | 350 | 29.26 | 1.00 (Reference) | ||
| del allele | 295 | 64.98 | 846 | 70.74 | 0.77 (0.61–0.97) | 0.024 | ||
| rs10670351 | Codominant model | ins/ins | 8 | 3.51 | 54 | 9.03 | 1.00 (Reference) | |
| ins/del | 95 | 41.67 | 244 | 40.80 | 2.63 (1.21–5.73) | 0.012 | ||
| del/del | 125 | 54.82 | 300 | 50.17 | 2.81 (1.30–6.08) | 0.006 | ||
| Ptrend | 0.038 | |||||||
| Recessive model | ins/ins | 8 | 3.51 | 54 | 9.03 | 1.00 (Reference) | ||
| del/del + ins/del | 220 | 96.49 | 544 | 90.97 | 2.73 (1.28–5.83) | 0.007 | ||
| Additive model | ins allele | 111 | 24.34 | 352 | 29.43 | 1.00 (Reference) | ||
| del allele | 345 | 75.66 | 844 | 70.57 | 1.30 (1.01–1.66) | 0.040 | ||
| rs10634037 | Codominant model | ins/ins | 19 | 8.52 | 55 | 9.20 | 1.00 (Reference) | |
| ins/del | 91 | 40.81 | 254 | 42.47 | 1.04 (0.58–1.84) | 0.901 | ||
| del/del | 113 | 50.67 | 289 | 48.33 | 1.13 (0.64–1.99) | 0.667 | ||
| Ptrend | 0.552 | |||||||
| Recessive model | ins/ins | 19 | 8.52 | 55 | 9.20 | 1.00 (Reference) | ||
| del/del + ins/del | 204 | 91.48 | 543 | 90.80 | 1.09 (0.63–1.88) | 0.763 | ||
| Additive model | ins allele | 129 | 28.92 | 364 | 30.43 | 1.00 (Reference) | ||
| del allele | 317 | 71.08 | 832 | 69.57 | 1.08 (0.85–1.37) | 0.552 | ||
| rs10680396 | Codominant model | ins/ins | 14 | 6.42 | 30 | 5.03 | 1.00 (Reference) | |
| ins/del | 73 | 33.49 | 210 | 35.24 | 0.75 (0.37–1.48) | 0.400 | ||
| del/del | 131 | 60.09 | 356 | 59.73 | 0.79 (0.41–1.53) | 0.483 | ||
| Ptrend | 0.828 | |||||||
| Recessive model | ins/ins | 14 | 6.42 | 30 | 5.03 | 1.00 (Reference) | ||
| del/del + ins/del | 204 | 93.58 | 566 | 94.97 | 0.77 (0.40–1.49) | 0.438 | ||
| Additive model | ins allele | 101 | 23.17 | 270 | 22.65 | 1.00 (Reference) | ||
| del allele | 335 | 76.83 | 922 | 77.35 | 0.97 (0.75–1.26) | 0.827 | ||
| rs140490511 | Codominant model | ins/ins | 163 | 74.43 | 425 | 71.19 | 1.00 (Reference) | |
| ins/del | 49 | 22.37 | 156 | 26.13 | 0.82 (0.57–1.18) | 0.287 | ||
| del/del | 7 | 3.20 | 16 | 2.68 | 1.14 (0.46–2.82) | 0.776 | ||
| Ptrend | 0.507 | |||||||
| Recessive model | ins/ins | 163 | 74.43 | 425 | 71.19 | 1.00 (Reference) | ||
| del/del + ins/del | 56 | 25.57 | 172 | 28.81 | 0.85 (0.60–1.21) | 0.361 | ||
| Additive model | ins allele | 375 | 85.62 | 1006 | 84.25 | 1.00 (Reference) | ||
| del allele | 63 | 14.38 | 188 | 15.75 | 0.90 (0.66–1.22) | 0.499 | ||
| rs35073080 | Codominant model | ins/ins | 104 | 47.71 | 279 | 47.45 | 1.00 (Reference) | |
| ins/del | 87 | 39.91 | 246 | 41.84 | 0.95 (0.68–1.32) | 0.756 | ||
| del/del | 27 | 12.38 | 63 | 10.71 | 1.15 (0.70–1.90) | 0.587 | ||
| Ptrend | 0.791 | |||||||
| Recessive model | ins/ins | 104 | 47.71 | 279 | 47.45 | 1.00 (Reference) | ||
| del/del + ins/del | 114 | 52.29 | 309 | 52.55 | 0.99 (0.73–1.35) | 0.948 | ||
| Additive model | ins allele | 295 | 67.66 | 804 | 68.37 | 1.00 (Reference) | ||
| del allele | 141 | 32.34 | 372 | 31.63 | 1.03 (0.82–1.31) | 0.787 | ||
| rs10580013 | Codominant model | ins/ins | 109 | 48.66 | 281 | 47.23 | 1.00 (Reference) | |
| ins/del | 89 | 39.73 | 252 | 42.35 | 0.91 (0.66–1.26) | 0.575 | ||
| del/del | 26 | 11.61 | 62 | 10.42 | 1.08 (0.65–1.80) | 0.764 | ||
| Ptrend | 0.962 | |||||||
| Recessive model | ins/ins | 109 | 48.66 | 281 | 47.23 | 1.00 (Reference) | ||
| del/del + ins/del | 115 | 51.34 | 314 | 52.77 | 0.94 (0.69–1.28) | 0.714 | ||
| Additive model | ins allele | 307 | 68.53 | 814 | 68.40 | 1.00 (Reference) | ||
| del allele | 141 | 31.47 | 376 | 31.60 | 0.99 (0.79–1.26) | 0.962 | ||
| rs10643067 | Codominant model | ins/ins | 5 | 2.21 | 3 | 0.50 | 1.00 (Reference) | |
| ins/del | 45 | 19.91 | 91 | 15.24 | 0.30 (0.07–1.30) | 0.089 | ||
| del/del | 176 | 77.88 | 503 | 84.26 | 0.21 (0.05–0.89) | 0.020 | ||
| Ptrend | 0.012 | |||||||
| Recessive model | ins/ins | 5 | 2.21 | 3 | 0.50 | 1.00 (Reference) | ||
| del/del + ins/del | 221 | 97.79 | 594 | 99.50 | 0.22 (0.05–0.94) | 0.026 | ||
| Additive model | ins allele | 55 | 12.17 | 97 | 8.12 | |||
| del allele | 395 | 87.83 | 1097 | 91.88 | 0.64 (0.45–0.90) | 0.011 | ||
| rs5860983 | Codominant model | ins/ins | 20 | 9.01 | 39 | 6.54 | 1.00 (Reference) | |
| ins/del | 71 | 31.98 | 237 | 39.77 | 0.58 (0.32–1.07) | 0.077 | ||
| del/del | 131 | 59.01 | 320 | 53.69 | 0.80 (0.45–1.42) | 0.443 | ||
| Ptrend | 0.563 | |||||||
| Recessive model | ins/ins | 20 | 9.01 | 39 | 6.54 | 1.00 (Reference) | ||
| del/del + ins/del | 202 | 90.99 | 557 | 93.46 | 0.71 (0.40–1.24) | 0.226 | ||
| Additive model | ins allele | 111 | 25.00 | 315 | 26.43 | 1.00 (Reference) | ||
| del allele | 333 | 75.00 | 877 | 73.57 | 1.08 (0.84–1.38) | 0.559 | ||
| rs59575728 | Codominant model | ins/ins | 48 | 21.92 | 146 | 24.46 | 1.00 (Reference) | |
| ins/del | 102 | 46.58 | 287 | 48.07 | 1.08 (0.73–1.61) | 0.700 | ||
| del/del | 69 | 31.50 | 164 | 27.47 | 1.28 (0.83–1.97) | 0.261 | ||
| Ptrend | 0.249 | |||||||
| Recessive model | ins/ins | 48 | 21.92 | 146 | 24.46 | 1.00 (Reference) | ||
| del/del + ins/del | 171 | 78.08 | 451 | 75.54 | 1.15 (0.80–1.67) | 0.450 | ||
| Additive model | ins allele | 198 | 45.21 | 579 | 48.49 | 1.00 (Reference) | ||
| del allele | 240 | 54.79 | 615 | 51.51 | 1.14 (0.92–1.42) | 0.239 | ||
| rs10622847 | Codominant model | A(ATT)9/A(ATT)9 + A(ATT)9/A(ATT)10 | 107 | 47.56 | 209 | 35.13 | 1.00 (Reference) | |
| A(ATT)6/A(ATT)9 + A(ATT)6/A(ATT)10 | 82 | 36.44 | 299 | 50.25 | 0.54 (0.38–0.75) | 2 × 10−4 | ||
| A(ATT)6/A(ATT)6 | 36 | 16.00 | 87 | 14.62 | 0.81 (0.51–1.27) | 0.356 | ||
| Ptrend | 0.042 | |||||||
| Recessive model | A(ATT)9/A(ATT)9 + A(ATT)9/A(ATT)10 | 107 | 47.56 | 209 | 35.13 | 1.00 (Reference) | ||
| A(ATT)6/A(ATT)9 + A(ATT)6/A(ATT)10 + A(ATT)6/A(ATT)6 | 118 | 52.44 | 386 | 64.87 | 0.60 (0.44–0.82) | 0.001 | ||
| Additive model | A(ATT)9 + A(ATT)10 | 296 | 65.78 | 717 | 60.25 | 1.00 (Reference) | ||
| A(ATT)6 | 154 | 34.22 | 471 | 39.75 | 0.79 (0.63–0.99) | 0.044 |
| Indel Variants Haplotype | Freq (Case) | Freq (Control) | χ2 | p-Value | ||||
|---|---|---|---|---|---|---|---|---|
| MICU1:rs10670351|rs10634037 | ||||||||
| DEL | DEL | 62.63 | 61.31 | 0.244 | 0.6211 | |||
| INS | INS | 16.07 | 21.18 | 5.424 | 0.020 | |||
| DEL | INS | 13.03 | 9.26 | 5.072 | 0.024 | |||
| INS | DEL | 8.27 | 8.25 | 0.001 | 0.992 | |||
| MCUB:rs10643067|rs5860983 | ||||||||
| DEL | DEL | 64.40 | 66.55 | 0.670 | 0.413 | |||
| DEL | INS | 23.43 | 25.33 | 0.636 | 0.425 | |||
| INS | DEL | 10.58 | 7.03 | 5.642 | 0.018 | |||
| INS | INS | 1.59 | 1.10 | 0.634 | 0.426 | |||
| MICU2:rs10580013|rs35073080|rs10622847 | ||||||||
| INS | INS | INS | 58.25 | 55.47 | 1.022 | 0.312 | ||
| DEL | DEL | DEL | 24.25 | 26.66 | 0.991 | 0.320 | ||
| INS | INS | DEL | 9.65 | 12.67 | 2.854 | 0.091 | ||
| DEL | DEL | INS | 6.26 | 4.59 | 1.926 | 0.165 | ||
| SMDT1:rs59575728|rs140490511|rs34979382 | ||||||||
| INS | INS | DEL | 29.24 | 31.57 | 0.826 | 0.363 | ||
| DEL | INS | INS | 32.91 | 27.88 | 3.991 | 0.046 | ||
| DEL | INS | DEL | 21.88 | 23.44 | 0.452 | 0.502 | ||
| INS | DEL | DEL | 14.28 | 15.50 | 0.382 | 0.537 | ||
| INS | INS | INS | 1.64 | 1.39 | 0.141 | 0.707 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, H.; He, Y.; Rui, Y.; Cai, M.; Fu, D.; Bi, W.; Luo, B.; Gao, Y. Genetic Predisposition and Mitochondrial Dysfunction in Sudden Cardiac Death: Role of MCU Complex Genetic Variations. Cells 2025, 14, 728. https://doi.org/10.3390/cells14100728
Meng H, He Y, Rui Y, Cai M, Fu D, Bi W, Luo B, Gao Y. Genetic Predisposition and Mitochondrial Dysfunction in Sudden Cardiac Death: Role of MCU Complex Genetic Variations. Cells. 2025; 14(10):728. https://doi.org/10.3390/cells14100728
Chicago/Turabian StyleMeng, Haoliang, Yan He, Yukun Rui, Mengqi Cai, Dongke Fu, Wanli Bi, Bin Luo, and Yuzhen Gao. 2025. "Genetic Predisposition and Mitochondrial Dysfunction in Sudden Cardiac Death: Role of MCU Complex Genetic Variations" Cells 14, no. 10: 728. https://doi.org/10.3390/cells14100728
APA StyleMeng, H., He, Y., Rui, Y., Cai, M., Fu, D., Bi, W., Luo, B., & Gao, Y. (2025). Genetic Predisposition and Mitochondrial Dysfunction in Sudden Cardiac Death: Role of MCU Complex Genetic Variations. Cells, 14(10), 728. https://doi.org/10.3390/cells14100728