
Misregulation of the Ubiquitin–Proteasome System and Autophagy in Muscular Dystrophies Associated with the Dystrophin–Glycoprotein Complex



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Duchenne Muscular Dystrophy
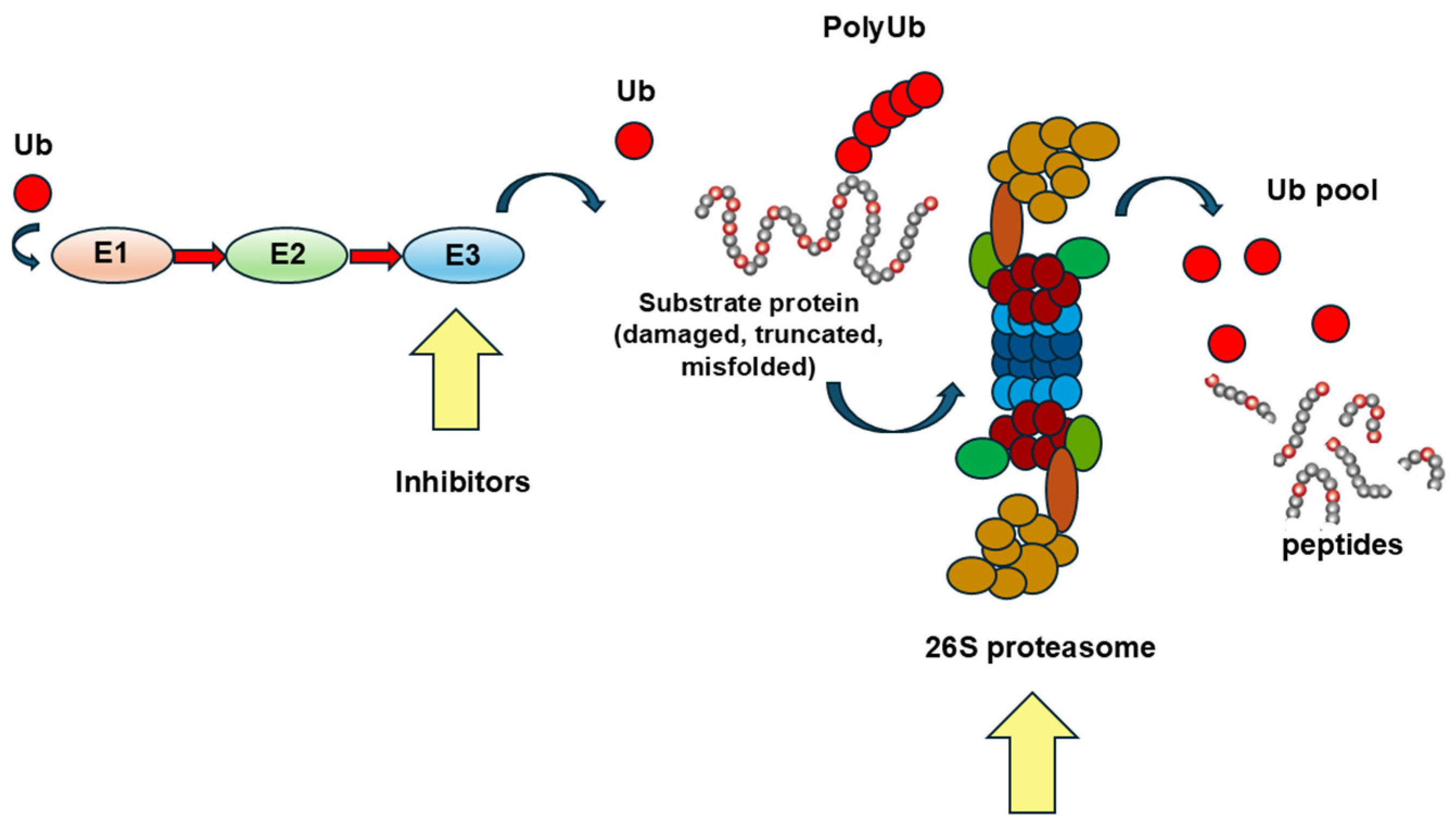
2.1. The Upregulation of the UPS and Its Therapeutic Modulation/Inhibition
2.1.1. Inhibition of Muscle Specific Ubiquitin-Conjugating Enzymes
2.1.2. The Inhibition of the Immunoproteasome
2.1.3. The Limits of a Therapeutic Approach Based on the Inhibition of Proteasome-Driven Protein Degradation
2.2. Defective Autophagy and Signaling in DMD
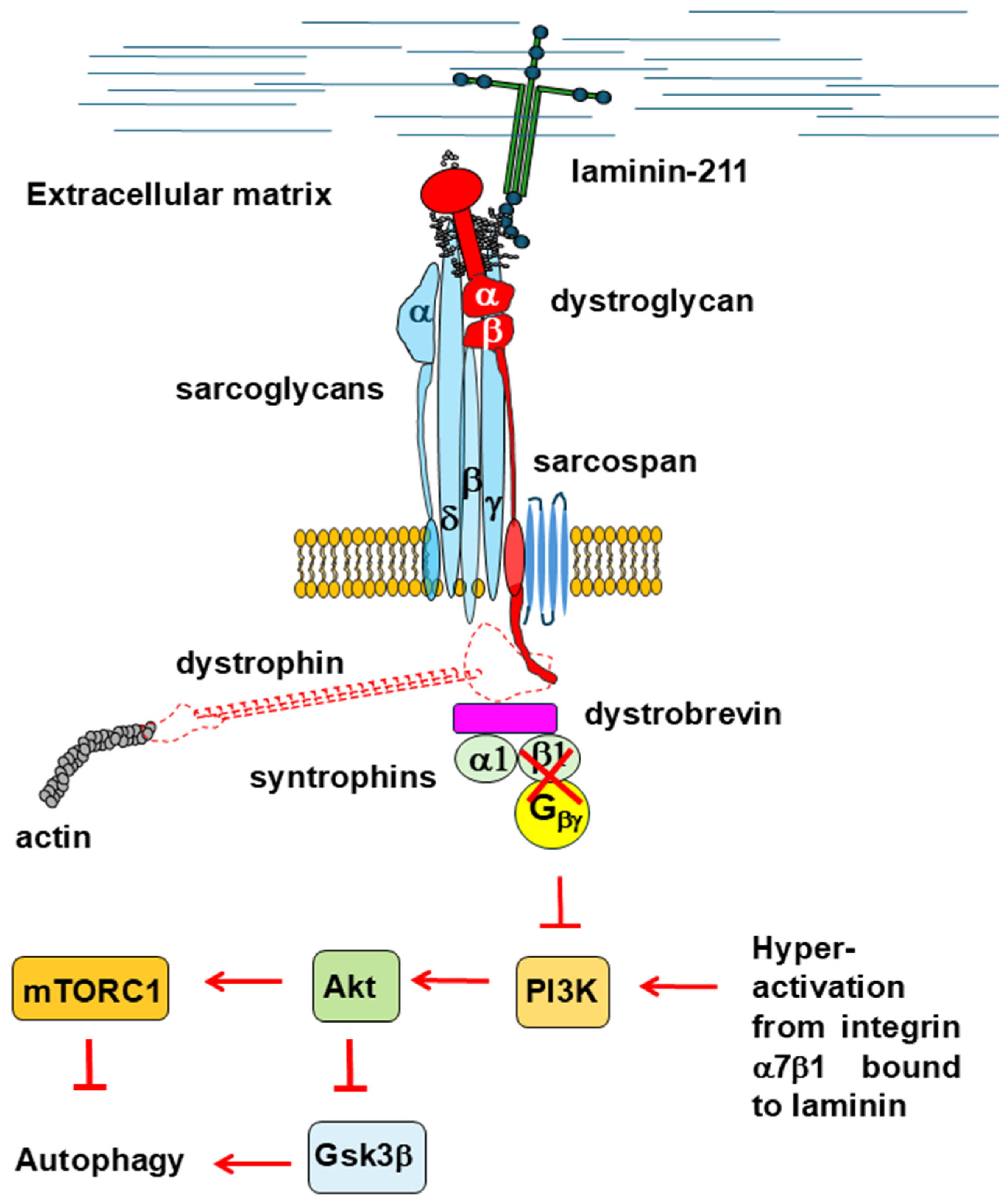
2.2.1. Akt/mTOR Signaling
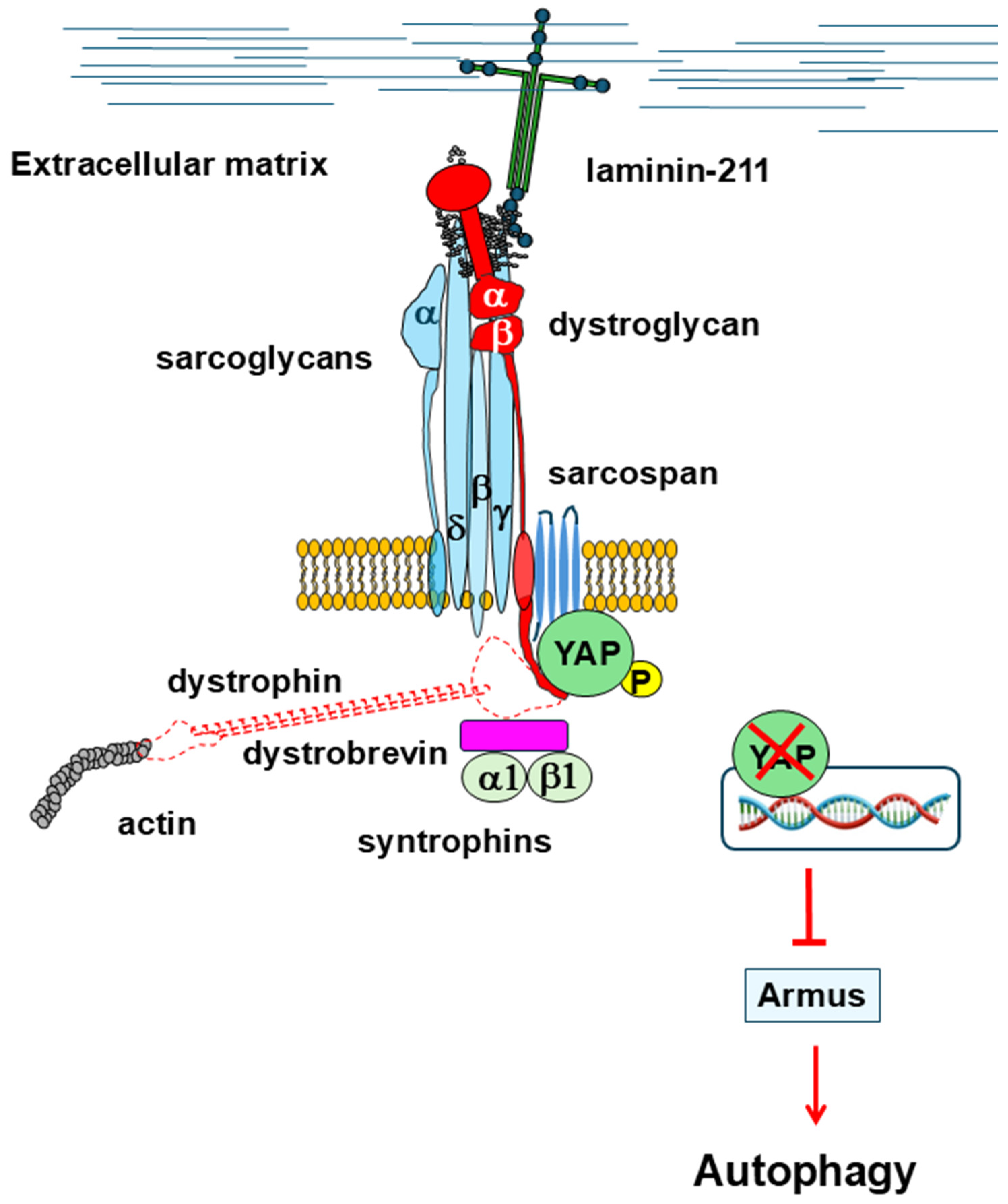
2.2.2. Hyppo Pathway
2.2.3. RhoA/ROCK Signaling
2.2.4. MEK/ERK Signaling
2.2.5. The Actin Cytoskeleton Scaffold
3. Dystroglycanopathies Associated with Fukutin and Fukutin-Related Protein
3.1. The Upregulation of the Ubiquitin–Proteasome System
3.2. Defective Autophagy
3.2.1. Akt/mTOR Signaling
3.2.2. Potential Involvement of Other Signal Transduction Pathways
4. Sarcoglycanopathies
5. Congenital Muscular Dystrophies Associated with Laminin α2 Deficiency
5.1. The Upregulation of the Ubiquitin–Proteasome System
5.2. Defective Autophagy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DGC | Dystrophin–Glycoprotein Complex |
| DMD | Duchenne Muscular Dystrophy |
| BMD | Becker Muscular Dystrophy |
| UPS | Ubiquitin–Proteasome System |
References
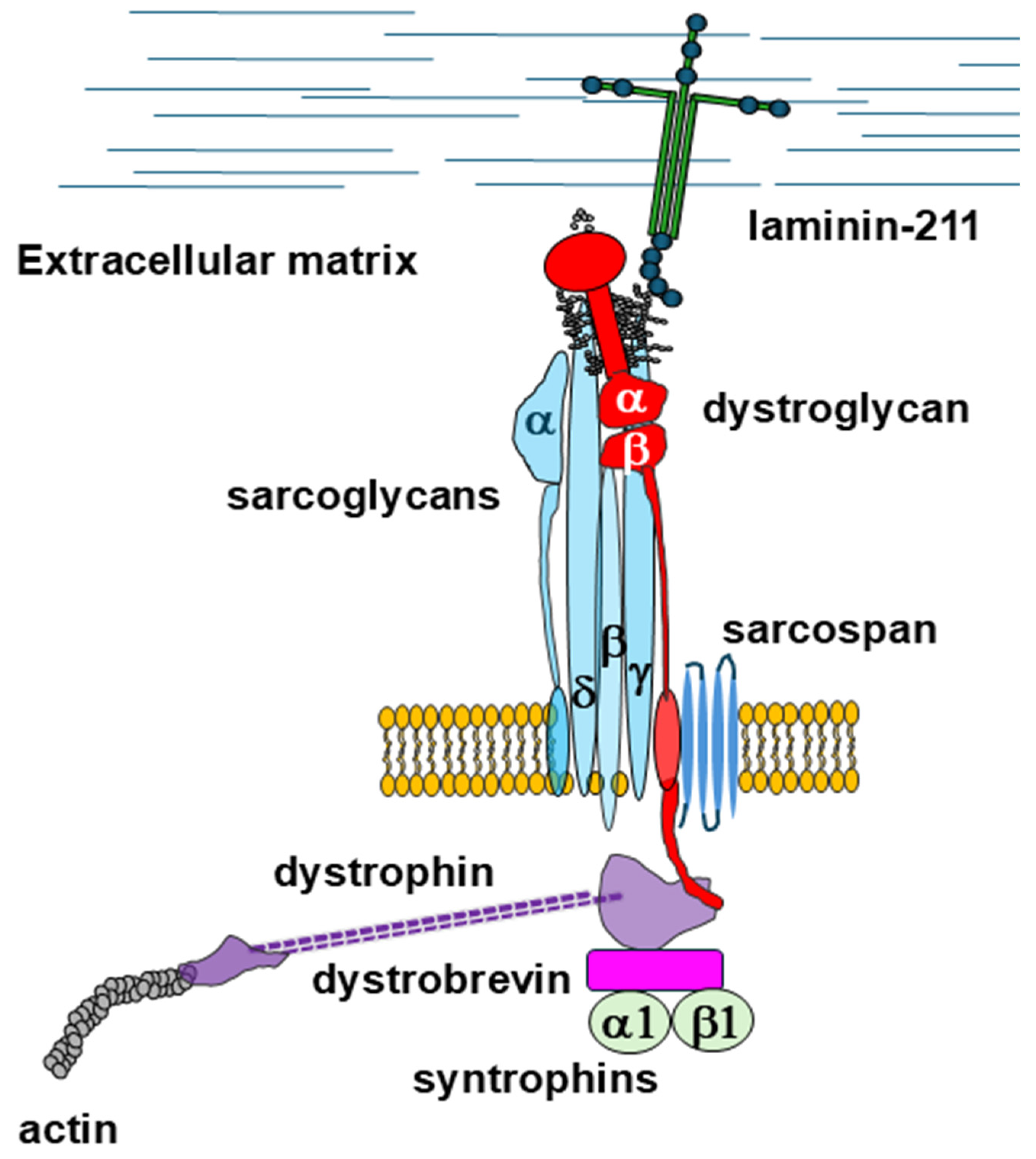
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the Striated Muscle Dystrophin-Glycoprotein Complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Jung, D.; Yang, B.; Meyer, J.; Chamberlain, J.S.; Campbell, K.P. Identification and Characterization of the Dystrophin Anchoring Site on β-Dystroglycan. J. Biol. Chem. 1995, 270, 27305–27310. [Google Scholar] [CrossRef]
- Sciandra, F.; Schneider, M.; Giardina, B.; Baumgartner, S.; Petrucci, T.C.; Brancaccio, A. Identification of the β-Dystroglycan Binding Epitope within the C-Terminal Region of α-Dystroglycan. Eur. J. Biochem. 2001, 268, 4590–4597. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary Structure of Dystrophin-Associated Glycoproteins Linking Dystrophin to the Extracellular Matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.C.; Yoshida-Moriguchi, T.; Zheng, T.; Venzke, D.; Anderson, M.E.; Strazzulli, A.; Moracci, M.; Yu, L.; Hohenester, E.; Campbell, K.P. Structural Basis of Laminin Binding to the LARGE Glycans on Dystroglycan. Nat. Chem. Biol. 2016, 12, 810–814. [Google Scholar] [CrossRef]
- Liu, S.; Su, T.; Xia, X.; Zhou, Z.H. Native DGC Structure Rationalizes Muscular Dystrophy-Causing Mutations. Nature 2025, 637, 1261–1271. [Google Scholar] [CrossRef]
- Wan, L.; Ge, X.; Xu, Q.; Huang, G.; Yang, T.; Campbell, K.P.; Yan, Z.; Wu, J. Structure and Assembly of the Dystrophin Glycoprotein Complex. Nature 2025, 637, 1252–1260. [Google Scholar] [CrossRef]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The Role of the Dystrophin Glycoprotein Complex in Muscle Cell Mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef]
- Sciandra, F.; Bozzi, M.; Bigotti, M.G. From Adhesion Complex to Signaling Hub: The Dual Role of Dystroglycan. Front. Mol. Biosci. 2023, 10, 1325284. [Google Scholar] [CrossRef]
- Brancaccio, A. A Molecular Overview of the Primary Dystroglycanopathies. J. Cell Mol. Med. 2019, 23, 3058–3062. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Tatara, K.; Kawai, M. Changes in Clinical Condition and Causes of Death of Inpatients with Duchenne Muscular Dystrophy in Japan from 1999 to 2012. Rinsho Shinkeigaku 2014, 54, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loire, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L.; et al. Characterization of Dystrophin in Muscle-Biopsy Specimens from Patients with Duchenne’s or Becker’s Muscular Dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, O.; Yokota, T. Advances in Genetic Characterization and Genotype-Phenotype Correlation of Duchenne and Becker Muscular Dystrophy in the Personalized Medicine Era. J. Pers. Med. 2020, 10, 111. [Google Scholar] [CrossRef]
- Barresi, R.; Campbell, K.P. Dystroglycan: From Biosynthesis to Pathogenesis of Human Disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef]
- Rennie, M.J.; Edwards, R.H.T.; Millward, D.J.; Wolman, S.L.; Halliday, D.; Matthews, D.E. Effects of Duchenne Muscular Dystrophy on Muscle Protein Synthesis. Nature 1982, 296, 165–167. [Google Scholar] [CrossRef]
- Kämper, A.; Rodemann, H.P. Alterations of Protein Degradation and 2-D Protein Pattern in Muscle Cells of mdx and DMD Origin. Biochem. Biophys. Res. Commun. 1992, 189, 1484–1490. [Google Scholar] [CrossRef]
- Spencer, M.J.; Croall, D.E.; Tidball, J.G. Calpains Are Activated in Necrotic Fibers from mdx Dystrophic Mice. J. Biol. Chem. 1995, 270, 10909–10914. [Google Scholar] [CrossRef]
- Gailly, P.; De Backer, F.; Van Schoor, M.; Gillis, J.M. In Situ Measurements of Calpain Activity in Isolated Muscle Fibres from Normal and Dystrophin-Lacking mdx Mice. J. Physiol. 2007, 582, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Fujimoto, S.; Ito, T.; Horinouchi, H.; Ueyama, H.; Tsuda, T. Proteasome Expression in the Skeletal Muscles of Patients with Muscular Dystrophy. Acta Neuropathol. 2000, 100, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Badalamente, M.A.; Stracher, A. Delay of Muscle Degeneration and Necrosis in mdx Mice by Calpain Inhibition. Muscle Nerve 2000, 23, 106–111. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Sotgia, F.; Schubert, W.; Park, D.S.; Frank, P.G.; Woodman, S.E.; Insabato, L.; Cammer, M.; Minetti, C.; Lisanti, M.P. Proteasome Inhibitor (MG-132) Treatment of mdx Mice Rescues the Expression and Membrane Localization of Dystrophin and Dystrophin-Associated Proteins. Am. J. Pathol. 2003, 163, 1663–1675. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Sotgia, F.; Capozza, F.; Gazzerro, E.; Minetti, C.; Lisanti, M.P. Localized Treatment with a Novel FDA-Approved Proteasome Inhibitor Blocks the Degradation of Dystrophin and Dystrophin-Associated Proteins in mdx Mice. Cell Cycle 2007, 6, 1242–1248. [Google Scholar] [CrossRef]
- Briguet, A.; Erb, M.; Courdier-Fruh, I.; Barzaghi, P.; Santos, G.; Herzner, H.; Lescop, C.; Siendt, H.; Henneboehle, M.; Weyermann, P.; et al. Effect of Calpain and Proteasome Inhibition on Ca2+-Dependent Proteolysis and Muscle Histopathology in the mdx Mouse. FASEB J. 2008, 22, 4190–4200. [Google Scholar] [CrossRef]
- Burdi, R.; Didonna, M.P.; Pignol, B.; Nico, B.; Mangieri, D.; Rolland, J.F.; Camerino, C.; Zallone, A.; Ferro, P.; Andreetta, F.; et al. First Evaluation of the Potential Effectiveness in Muscular Dystrophy of a Novel Chimeric Compound, BN 82270, Acting as Calpain-Inhibitor and Anti-Oxidant. Neuromuscul. Disord. 2006, 16, 237–248. [Google Scholar] [CrossRef]
- Araujo, K.P.C.; Bonuccelli, G.; Duarte, C.N.; Gaiad, T.P.; Moreira, D.F.; Feder, D.; Belizario, J.E.; Miglino, M.A.; Lisanti, M.P.; Ambrosio, C.E. Bortezomib (PS-341) Treatment Decreases Inflammation and Partially Rescues the Expression of the Dystrophin-Glycoprotein Complex in GRMD Dogs. PLoS ONE 2013, 8, e61367. [Google Scholar] [CrossRef]
- Micheletto, M.L.J.; Hermes, T.D.A.; Bertassoli, B.M.; Petri, G.; Perez, M.M.; Fonseca, F.L.A.; Carvalho, A.A.D.S.; Feder, D. Ixazomib, an Oral Proteasome Inhibitor, Exhibits Potential Effect in Dystrophin-Deficient mdx Mice. Int. J. Exp. Pathol. 2021, 102, 11–21. [Google Scholar] [CrossRef]
- Gazzerro, E.; Assereto, S.; Bonetto, A.; Sotgia, F.; Scarfi, S.; Pistorio, A.; Bonuccelli, G.; Cilli, M.; Bruno, C.; Zara, F.; et al. Therapeutic Potential of Proteasome Inhibition in Duchenne and Becker Muscular Dystrophies. Am. J. Pathol. 2010, 176, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Selsby, J.; Morris, C.; Morris, L.; Sweeney, L. A Proteasome Inhibitor Fails to Attenuate Dystrophic Pathology in mdx Mice. PLoS Curr. 2012, 4, e4f84a944d8930. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, K.; Selsby, J.T. The Physiological Response of Protease Inhibition in Dystrophic Muscle. Acta Physiol. 2013, 208, 234–244. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of Autophagy and Protein Degradation Systems in Myopathies and Muscular Dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef] [PubMed]
- Colson, K. Treatment-Related Symptom Management in Patients with Multiple Myeloma: A Review. Support. Care Cancer 2015, 23, 1431–1445. [Google Scholar] [CrossRef]
- Maddocks, K.; Blum, K.A. Treatment Strategies in Mantle Cell Lymphoma. Cancer Treat. Res. 2015, 165, 251–270. [Google Scholar] [CrossRef]
- McBride, A.; Klaus, J.O.; Stockerl-Goldstein, K. Carfilzomib: A Second-Generation Proteasome Inhibitor for the Treatment of Multiple Myeloma. Am. J. Health Syst. Pharm. 2015, 72, 353–360. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Li, L.; RodríGuez, J.E.; Min, J.N.; Bogan, D.; Gonzalez, J.; Patterson, C.; Kornegay, J.N.; Willis, M. Regulation of the Calpain and Ubiquitin-Proteasome Systems in a Canine Model of Muscular Dystrophy. Muscle Nerve 2011, 44, 553–562. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a Muscle-Specific F-Box Protein Highly Expressed during Muscle Atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Assereto, S.; Piccirillo, R.; Baratto, S.; Scudieri, P.; Fiorillo, C.; Massacesi, M.; Traverso, M.; Galietta, L.J.; Bruno, C.; Minetti, C.; et al. The Ubiquitin Ligase Tripartite-Motif-Protein 32 Is Induced in Duchenne Muscular Dystrophy. Lab. Investig. 2016, 96, 862–871. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Choi, J.H.; Kim, J.; Woo, J.S.; Lee, E.H. Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle? Cells 2023, 12, 2104. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.L.; Talsness, D.M.; Lindsay, A.; Arpke, R.W.; Chatterton, P.D.; Nelson, D.M.; Chamberlain, C.M.; Olthoff, J.T.; Belanto, J.J.; McCourt, P.M.; et al. Mouse Models of Two Missense Mutations in Actin-Binding Domain 1 of Dystrophin Associated with Duchenne or Becker Muscular Dystrophy. Hum. Mol. Genet. 2018, 27, 451–462. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Hu, Q.; Xi, Y.; Xing, Z.; Zhang, Z.; Huang, L.; Wu, J.; Liang, K.; Nguyen, T.K.; et al. The LncRNA H19 Alleviates Muscular Dystrophy by Stabilizing Dystrophin. Nat. Cell Biol. 2020, 22, 1332–1345. [Google Scholar] [CrossRef]
- Lenk, U.; Oexle, K.; Voit, T.; Ancker, U.; Hellner, K.A.; Speer, A.; Hübner, C. A Cysteine 3340 Substitution in the Dystroglycan-Binding Domain of Dystrophin Associated with Duchenne Muscular Dystrophy, Mental Retardation and Absence of the ERG b-Wave. Hum. Mol. Genet. 1996, 5, 973–975. [Google Scholar] [CrossRef]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ Promotes Muscle Damage in the mdx Mouse Model of Duchenne Muscular Dystrophy by Suppressing M2 Macrophage Activation and Inhibiting Muscle Cell Proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [PubMed]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy-Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef]
- Cui, Z.; Hwang, S.M.; Gomes, A.V. Identification of the Immunoproteasome as a Novel Regulator of Skeletal Muscle Differentiation. Mol. Cell Biol. 2014, 34, 96–109. [Google Scholar] [CrossRef]
- Gomes, A.V.; Zong, C.; Edmondson, R.D.; Berhane, B.T.; Wang, G.W.; Le, S.; Young, G.; Zhang, J.; Vondriska, T.M.; Whitelegge, J.P.; et al. The Murine Cardiac 26S Proteasome: An Organelle Awaiting Exploration. Ann. N. Y. Acad. Sci. 2005, 1047, 197–207. [Google Scholar] [CrossRef]
- Basler, M.; Kirk, C.J.; Groettrup, M. The Immunoproteasome in Antigen Processing and Other Immunological Functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef]
- Liu, H.M.; Ferrington, D.A.; Baumann, C.W.; Thompson, L.V. Denervation-Induced Activation of the Standard Proteasome and Immunoproteasome. PLoS ONE 2016, 11, e0166831. [Google Scholar] [CrossRef]
- Chen, C.N.J.; Graber, T.G.; Bratten, W.M.; Ferrington, D.A.; Thompson, L.V. Immunoproteasome in Animal Models of Duchenne Muscular Dystrophy. J. Muscle Res. Cell Motil. 2014, 35, 191–201. [Google Scholar] [CrossRef]
- Farini, A.; Sitzia, C.; Cassani, B.; Cassinelli, L.; Rigoni, R.; Colleoni, F.; Fusco, N.; Gatti, S.; Bella, P.; Villa, C.; et al. Therapeutic Potential of Immunoproteasome Inhibition in Duchenne Muscular Dystrophy. Mol. Ther. 2016, 24, 1898–1912. [Google Scholar] [CrossRef] [PubMed]
- Kwak, D.; Wei, G.; Thompson, L.V.; Kim, J.H. Short-Term ONX-0914 Administration: Performance and Muscle Phenotype in mdx Mice. Int. J. Environ. Res. Public. Health 2020, 17, 5211. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Molinaro, D.; Fortunato, F.; Mella, C.; Cassani, B.; Torrente, Y.; Farini, A. Immunoproteasome Inhibition Ameliorates Aged Dystrophic Mouse Muscle Environment. Int. J. Mol. Sci. 2022, 23, 14657. [Google Scholar] [CrossRef]
- Teramoto, N.; Sugihara, H.; Yamanouchi, K.; Nakamura, K.; Kimura, K.; Okano, T.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Nagata, T.; et al. Pathological Evaluation of Rats Carrying In-Frame Mutations in the Dystrophin Gene: A New Model of Becker Muscular Dystrophy. DMM Dis. Models Mech. 2020, 13, dmm044701. [Google Scholar] [CrossRef]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the Small Heat Shock Proteins αB-Crystallin, MKBP, HSP25, HSP20, and CvHSP in Heart and Skeletal Muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef]
- Singh, B.N.; Rao, K.S.; Ramakrishna, T.; Rangaraj, N.; Rao, C.M. Association of αB-Crystallin, a Small Heat Shock Protein, with Actin: Role in Modulating Actin Filament Dynamics in Vivo. J. Mol. Biol. 2007, 366, 756–767. [Google Scholar] [CrossRef]
- Wattin, M.; Gaweda, L.; Muller, P.; Baritaud, M.; Scholtes, C.; Lozano, C.; Gieseler, K.; Kretz-Remy, C. Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2018, 19, 178. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Gatto, F.; Benemei, S.; Piluso, G.; Bello, L. The Complex Landscape of DMD Mutations: Moving towards Personalized Medicine. Front. Genet. 2024, 15, 1360224. [Google Scholar] [CrossRef]
- Talsness, D.M.; Belanto, J.J.; Ervasti, J.M. Disease-Proportional Proteasomal Degradation of Missense Dystrophins. Proc. Natl. Acad. Sci. USA 2015, 112, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Eghtesad, S.; Jhunjhunwala, S.; Little, S.R.; Clemens, P.R. Rapamycin Ameliorates Dystrophic Phenotype in mdx Mouse Skeletal Muscle. Mol. Med. 2011, 17, 917–924. [Google Scholar] [CrossRef]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a New Therapeutic Target in Duchenne Muscular Dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Grumati, P.; Hiller, M.; Chrisam, M.; Aartsma-Rus, A.; Bonaldo, P. Autophagy Is Impaired in the Tibialis Anterior of Dystrophin Null Mice. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the Dystrophin-ATP Connection: How Half a Century of Research Still Implicates Mitochondrial Dysfunction in Duchenne Muscular Dystrophy Aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial Stress Responses in Duchenne Muscular Dystrophy: Metabolic Dysfunction or Adaptive Reprogramming? Am. J. Physiol. Cell Physiol. 2022, 323, C718–C730. [Google Scholar] [CrossRef]
- Higashitani, A.; Teranishi, M.; Nakagawa, Y.; Itoh, Y.; Sudevan, S.; Szewczyk, N.J.; Kubota, Y.; Abe, T.; Kobayashi, T. Increased Mitochondrial Ca2+ Contributes to Health Decline with Age and Duchene Muscular Dystrophy in C. elegans. FASEB J. 2023, 37, e22851. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Luan, P.; Amico, D.D.; Andreux, P.A.; Laurila, P.P.; Wohlwend, M.; Li, H.; Lima, T.I.; Place, N.; Rinsch, C.; Zanou, N.; et al. Urolithin A Improves Muscle Function by Inducing Mitophagy in Muscular Dystrophy. Sci. Transl. Med. 2021, 13, eabb0319. [Google Scholar] [CrossRef]
- Bibee, K.P.; Cheng, Y.J.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin Nanoparticles Target Defective Autophagy in Muscular Dystrophy to Enhance Both Strength and Cardiac Function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef]
- Sebori, R.; Kuno, A.; Hosoda, R.; Hayashi, T.; Horio, Y. Resveratrol Decreases Oxidative Stress by Restoring Mitophagy and Improves the Pathophysiology of Dystrophin-Deficient mdx Mice. Oxid. Med. Cell Longev. 2018, 2018, 9179270. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, H.; He, J.; Liang, J.; Liu, Y.; Zhang, W. TRIM72 Alleviates Muscle Inflammation in mdx Mice via Promoting Mitophagy-Mediated NLRP3 Inflammasome Inactivation. Oxid. Med. Cell Longev. 2023, 2023, 8408574. [Google Scholar] [CrossRef]
- Buonomo, V.; Lohachova, K.; Reggio, A.; Cano-Franco, S.; Cillo, M.; Santorelli, L.; Venditti, R.; Polishchuk, E.; Peluso, I.; Brunello, L.; et al. Two FAM134B Isoforms Differentially Regulate ER Dynamics during Myogenesis. EMBO J. 2025, 44, 1039–1073. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Changotra, H.; Wergedal, J.E.; Kumar, A. Regulation of Phosphatidylinositol 3-Kinase (PI3K)/Akt and Nuclear Factor-Kappa B Signaling Pathways in Dystrophin-Deficient Skeletal Muscle in Response to Mechanical Stretch. J. Cell Physiol. 2006, 208, 575–585. [Google Scholar] [CrossRef]
- Peter, A.K.; Crosbie, R.H. Hypertrophic Response of Duchenne and Limb-Girdle Muscular Dystrophies Is Associated with Activation of Akt Pathway. Exp. Cell Res. 2006, 312, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-Induced Skeletal Myotube Hypertrophy by PI(3)K/Akt/MTOR and PI(3)K/Akt/GSK3 Pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/MTOR Pathway Is a Crucial Regulator of Skeletal Muscle Hypertrophy and Can Prevent Muscle Atrophy in Vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Peter, A.K.; Ko, C.Y.; Kim, M.H.; Hsu, N.; Ouchi, N.; Rhie, S.; Izumiya, Y.; Zeng, L.; Walsh, K.; Crosbie, R.H. Myogenic Akt Signaling Upregulates the Utrophin-Glycoprotein Complex and Promotes Sarcolemma Stability in Muscular Dystrophy. Hum. Mol. Genet. 2009, 18, 318–327. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Watt, P.W.; Shaw, M.; Van Der Kaay, J.; Downes, C.P.; Holder, J.C.; Cohen, P. Insulin Activates Protein Kinase B, Inhibits Glycogen Synthase Kinase-3 and Activates Glycogen Synthase by Rapamycin-Insensitive Pathways in Skeletal Muscle and Adipose Tissue. FEBS Lett. 1997, 406, 211–215. [Google Scholar] [CrossRef]
- Jiang, H.; Xiao, J.; Kang, B.; Zhu, X.; Xin, N.; Wang, Z. PI3K/SGK1/GSK3β Signaling Pathway Is Involved in Inhibition of Autophagy in Neonatal Rat Cardiomyocytes Exposed to Hypoxia/Reoxygenation by Hydrogen Sulfide. Exp. Cell Res. 2016, 345, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK Promotes Skeletal Muscle Autophagy through Activation of Forkhead FoxO3a and Interaction with Ulk1. J. Cell Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK Activation Stimulates Autophagy and Ameliorates Muscular Dystrophy in the mdx Mouse Diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Khogali, S.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK Stimulation Attenuates Adaptive Signaling in Dystrophic Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2012, 302, C110–C121. [Google Scholar] [CrossRef]
- Spaulding, H.R.; Kelly, E.M.; Quindry, J.C.; Sheffield, J.B.; Hudson, M.B.; Selsby, J.T. Autophagic Dysfunction and Autophagosome Escape in the mdx Mus musculus Model of Duchenne Muscular Dystrophy. Acta Physiol. 2018, 222, e12944. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of Autophagy and the Ubiquitin-Proteasome System by the FoxO Transcriptional Network during Muscle Atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Tang, E.D.; Nuñez, G.; Barr, F.G.; Guan, K.L. Negative Regulation of the Forkhead Transcription Factor FKHR by Akt. J. Biol. Chem. 1999, 274, 16741–16746. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 Functions as a Transcriptional Regulator of Autophagy by Preventing Nuclear Transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubin-de-Celis, S.; Peña-Llopis, S.; Konda, M.; Brugarolas, J. Multistep Regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Hosoda, R.; Tatekoshi, Y.; Iwahara, N.; Saga, Y.; Kuno, A. Transcriptional Dysregulation of Autophagy in the Muscle of a Mouse Model of Duchenne Muscular Dystrophy. Sci. Rep. 2024, 14, 1365. [Google Scholar] [CrossRef]
- Boppart, M.D.; Burkin, D.J.; Kaufman, S.J. Activation of AKT Signaling Promotes Cell Growth and Survival in A7β1 Integrin-Mediated Alleviation of Muscular Dystrophy. Biochim. Biophys. Acta 2011, 1812, 439–446. [Google Scholar] [CrossRef]
- You, J.S.; Karaman, K.; Reyes-Ordoñez, A.; Lee, S.; Kim, Y.; Bashir, R.; Chen, J. Leucyl-TRNA Synthetase Contributes to Muscle Weakness through Mammalian Target of Rapamycin Complex 1 Activation and Autophagy Suppression in a Mouse Model of Duchenne Muscular Dystrophy. Am. J. Pathol. 2024, 194, 1571–1580. [Google Scholar] [CrossRef]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a). Mol. Cell Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FoxO Transcription Factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear Trapping of the Forkhead Transcription Factor FoxO1 via Sirt-Dependent Deacetylation Promotes Expression of Glucogenetic Genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef]
- Rimmelé, P.; Bigarella, C.L.; Liang, R.; Izac, B.; Dieguez-Gonzalez, R.; Barbet, G.; Donovan, M.; Brugnara, C.; Blander, J.M.; Sinclair, D.A.; et al. Aging-like Phenotype and Defective Lineage Specification in SIRT1-Deleted Hematopoietic Stem and Progenitor Cells. Stem Cell Rep. 2014, 3, 44–59. [Google Scholar] [CrossRef]
- Kuno, A.; Hosoda, R.; Sebori, R.; Hayashi, T.; Sakuragi, H.; Tanabe, M.; Horio, Y. Resveratrol Ameliorates Mitophagy Disturbance and Improves Cardiac Pathophysiology of Dystrophin-Deficient mdx Mice. Sci. Rep. 2018, 8, 15555. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yu, T.; Huh, J.Y.; Cai, Y.; Yoon, S.; Javaid, H.M.A. Aminoguanidine Hemisulfate Improves Mitochondrial Autophagy, Oxidative Stress, and Muscle Force in Duchenne Muscular Dystrophy via the AKT/FOXO1 Pathway in mdx Mice. Skelet. Muscle 2025, 15, 2. [Google Scholar] [CrossRef]
- Eid Mutlak, Y.; Aweida, D.; Volodin, A.; Ayalon, B.; Dahan, N.; Parnis, A.; Cohen, S. A Signaling Hub of Insulin Receptor, Dystrophin Glycoprotein Complex and Plakoglobin Regulates Muscle Size. Nat. Commun. 2020, 11, 1381. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, K.J.; Rando, T.A. Inhibition of Dystroglycan Binding to Laminin Disrupts the PI3K/AKT Pathway and Survival Signaling in Muscle Cells. Muscle Nerve 2002, 26, 644–653. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhou, Y.; Jarrett, H.W. Dystrophin Glycoprotein Complex-Associated Gβγ Subunits Activate Phosphatidylinositol-3-Kinase/Akt Signaling in Skeletal Muscle in a Laminin-Dependent Manner. J. Cell Physiol. 2009, 219, 402–414. [Google Scholar] [CrossRef]
- Burkin, D.J.; Wallace, G.Q.; Nicol, K.J.; Kaufman, D.J.; Kaufman, S.J. Enhanced Expression of the α7β1 Integrin Reduces Muscular Dystrophy and Restores Viability in Dystrophic Mice. J. Cell Biol. 2001, 152, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Hodges, B.L.; Hayashi, Y.K.; Nonaka, I.; Wang, W.; Arahata, K.; Kaufman, S.J. Altered Expression of the α7β1 Integrin in Human and Murine Muscular Dystrophies. J. Cell Sci. 1997, 110 Pt 22, 2873–2881. [Google Scholar] [CrossRef]
- Li, J.; Rao, H.; Burkin, D.; Kaufman, S.J.; Wu, C. The Muscle Integrin Binding Protein (MIBP) Interacts with α7β1 Integrin and Regulates Cell Adhesion and Laminin Matrix Deposition. Dev. Biol. 2003, 261, 209–219. [Google Scholar] [CrossRef]
- Hayashi, Y.K.; Tezak, Z.; Momoi, T.; Nonaka, I.; Garcia, C.A.; Hoffman, E.P.; Arahata, K. Massive Muscle Cell Degeneration in the Early Stage of Merosin-Deficient Congenital Muscular Dystrophy. Neuromuscul. Disord. 2001, 11, 350–359. [Google Scholar] [CrossRef]
- Miyagoe, Y.; Hanaoka, K.; Nonaka, I.; Hayasaka, M.; Nabeshima, Y.; Arahata, K.; Nabeshima, Y.I.; Takeda, S. Laminin α2 Chain-Null Mutant Mice by Targeted Disruption of the Lama2 Gene: A New Model of Merosin (Laminin 2)-Deficient Congenital Muscular Dystrophy. FEBS Lett. 1997, 415, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Boppart, M.D.; Burkin, D.J.; Kaufman, S.J. α7β1-Integrin Regulates Mechanotransduction and Prevents Skeletal Muscle Injury. Am. J. Physiol. Cell Physiol. 2006, 290, C1660-5. [Google Scholar] [CrossRef]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ Functions and Their Regulation at a Glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ Upstream Signals and Downstream Responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo Pathway Effector YAP Is a Critical Regulator of Skeletal Muscle Fibre Size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Shah, S.B.; Ward, C.W.; Stains, J.P.; Spangenburg, E.E.; Folker, E.S.; Lovering, R.M. Differential YAP Nuclear Signaling in Healthy and Dystrophic Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2019, 317, C48–C57. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.H.; Ma, H.; Pierre, P.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Muscle Protein Synthesis, MTORC1/MAPK/Hippo Signaling, and Capillary Density Are Altered by Blocking of Myostatin and Activins. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E41–E50. [Google Scholar] [CrossRef]
- Vita, G.L.; Polito, F.; Oteri, R.; Arrigo, R.; Ciranni, A.M.; Musumeci, O.; Messina, S.; Rodolico, C.; Di Giorgio, R.M.; Vita, G.; et al. Hippo Signaling Pathway Is Altered in Duchenne Muscular Dystrophy. PLoS ONE 2018, 13, e0205514. [Google Scholar] [CrossRef]
- Ramirez, M.P.; Anderson, M.J.M.; Kelly, M.D.; Sundby, L.J.; Hagerty, A.R.; Wenthe, S.J.; Odde, D.J.; Ervasti, J.M.; Gordon, W.R. Dystrophin Missense Mutations Alter Focal Adhesion Tension and Mechanotransduction. Proc. Natl. Acad. Sci. USA 2022, 119, e2205536119. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Park, S.; Sung, S.E.; Lee, J.; Kim, D.S.; Lee, J.; Lee, J.R.; Kim, N.S.; Lee, D.Y. Altered Gene Expression Profiles in Neural Stem Cells Derived from Duchenne Muscular Dystrophy Patients with Intellectual Disability. Exp. Neurobiol. 2021, 30, 263–274. [Google Scholar] [CrossRef]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell Phenotypic Plasticity Requires Autophagic Flux Driven by YAP/TAZ Mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef]
- Carroll, B.; Mohd-Naim, N.; Maximiano, F.; Frasa, M.A.; McCormack, J.; Finelli, M.; Thoresen, S.B.; Perdios, L.; Daigaku, R.; Francis, R.E.; et al. The TBC/RabGAP Armus Coordinates Rac1 and Rab7 Functions during Autophagy. Dev. Cell 2013, 25, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Morimoto, K.; Sasawatari, S.; Kumanogoh, A. Leucine-Rich Repeat Kinase 1 Regulates Autophagy through Turning On TBC1D2-Dependent Rab7 Inactivation. Mol. Cell Biol. 2015, 35, 3044–3058. [Google Scholar] [CrossRef]
- James, M.; Nuttall, A.; Ilsley, J.L.; Ottersbach, K.; Tinsley, J.M.; Sudol, M.; Winder, S.J. Adhesion-Dependent Tyrosine Phosphorylation of β-Dystroglycan Regulates Its Interaction with Utrophin. J. Cell Sci. 2000, 113, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Heallen, T.; Leach, J.; Xiao, Y.; Martin, J.F. Dystrophin-Glycoprotein Complex Sequesters Yap to Inhibit Cardiomyocyte Proliferation. Nature 2017, 547, 227–231. [Google Scholar] [CrossRef]
- Sotgia, F.; Bonuccelli, G.; Bedford, M.; Brancaccio, A.; Mayer, U.; Wilson, M.T.; Campos-Gonzalez, R.; Brooks, J.W.; Sudol, M.; Lisanti, M.P. Localization of Phospho-β-Dystroglycan (PY892) to an Intracellular Vesicular Compartment in Cultured Cells and Skeletal Muscle Fibers in Vivo. Biochemistry 2003, 42, 7110–7123. [Google Scholar] [CrossRef]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The Extracellular Matrix Protein Agrin Promotes Heart Regeneration in Mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Bozzi, M.; Sciandra, F.; Brancaccio, A. Role of Gelatinases in Pathological and Physiological Processes Involving the Dystrophin-Glycoprotein Complex. Matrix Biol. 2015, 44–46, 130–137. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase Signalling in Cell Migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.I.; Blaabjerg, M.; Freude, K.; Meyer, M. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11, 1520. [Google Scholar] [CrossRef]
- Liu, F.T.; Yang, Y.J.; Wu, J.J.; Li, S.; Tang, Y.L.; Zhao, J.; Liu, Z.Y.; Xiao, B.G.; Zuo, J.; Liu, W.; et al. Fasudil, a Rho Kinase Inhibitor, Promotes the Autophagic Degradation of A53T α-Synuclein by Activating the JNK 1/Bcl-2/Beclin 1 Pathway. Brain Res. 2016, 1632, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The Role of PI3K/AKT/MTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Uehata, M.; Himeno, M. Small Guanosine Triphosphatase Rho/Rho-Associated Kinase as a Novel Regulator of Intracellular Redistribution of Lysosomes in Invasive Tumor Cells. Cell Tissue Res. 2000, 301, 341–351. [Google Scholar] [CrossRef]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Ikeda, K.; Himeno, M. A Role for Small GTPase RhoA in Regulating Intracellular Membrane Traffic of Lysosomes in Invasive Rat Hepatoma Cells. Histochem. J. 2002, 34, 189–213. [Google Scholar] [CrossRef]
- You, J.S.; Kim, Y.; Lee, S.; Bashir, R.; Chen, J. RhoA/ROCK Signalling Activated by ARHGEF3 Promotes Muscle Weakness via Autophagy in Dystrophic mdx Mice. J. Cachexia Sarcopenia Muscle 2023, 14, 1880–1893. [Google Scholar] [CrossRef]
- Mu, X.; Usas, A.; Tang, Y.; Lu, A.; Wang, B.; Weiss, K.; Huard, J. RhoA Mediates Defective Stem Cell Function and Heterotopic Ossification in Dystrophic Muscle of Mice. FASEB J. 2013, 27, 3619–3631. [Google Scholar] [CrossRef]
- Fernández-Simón, E.; Suárez-Calvet, X.; Carrasco-Rozas, A.; Piñol-Jurado, P.; López-Fernández, S.; Pons, G.; Bech Serra, J.J.; de la Torre, C.; de Luna, N.; Gallardo, E.; et al. RhoA/ROCK2 Signalling Is Enhanced by PDGF-AA in Fibro-Adipogenic Progenitor Cells: Implications for Duchenne Muscular Dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1373–1384. [Google Scholar] [CrossRef]
- Yang, B.; Jung, D.; Motto, D.; Meyer, J.; Koretzky, G.; Campbell, K.P. SH3 Domain-Mediated Interaction of Dystroglycan and Grb2. J. Biol. Chem. 1995, 270, 11711–11714. [Google Scholar] [CrossRef]
- Chockalingam, P.S.; Cholera, R.; Oak, S.A.; Zheng, Y.; Jarrett, H.W.; Thomason, D.B. Dystrophin-Glycoprotein Complex and Ras and Rho GTPase Signaling Are Altered in Muscle Atrophy. Am. J. Physiol. Cell Physiol. 2002, 283, C500–C511. [Google Scholar] [CrossRef] [PubMed]
- Oak, S.A.; Zhou, Y.W.; Jarrett, H.W. Skeletal Muscle Signaling Pathway through the Dystrophin Glycoprotein Complex and Rac1. J. Biol. Chem. 2003, 278, 39287–39295. [Google Scholar] [CrossRef]
- Hall, A. Rho Family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef]
- Eliazer, S.; Muncie, J.M.; Christensen, J.; Sun, X.; D’Urso, R.S.; Weaver, V.M.; Brack, A.S. Wnt4 from the Niche Controls the Mechano-Properties and Quiescent State of Muscle Stem Cells. Cell Stem Cell 2019, 25, 654–665.e4. [Google Scholar] [CrossRef] [PubMed]
- Hnia, K.; Gayraud, J.; Hugon, G.; Ramonatxo, M.; De La Porte, S.; Matecki, S.; Mornet, D. L-Arginine Decreases Inflammation and Modulates the Nuclear Factor-KappaB/Matrix Metalloproteinase Cascade in mdx Muscle Fibers. Am. J. Pathol. 2008, 172, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Khandelwal, N.; Malya, R.; Reid, M.B.; Boriek, A.M. Loss of Dystrophin Causes Aberrant Mechanotransduction in Skeletal Muscle Fibers. FASEB J. 2004, 18, 102–113. [Google Scholar] [CrossRef]
- Lang, J.M.; Esser, K.A.; Dupont-Versteegden, E.E. Altered Activity of Signaling Pathways in Diaphragm and Tibialis Anterior Muscle of Dystrophic Mice. Exp. Biol. Med. 2004, 229, 503–511. [Google Scholar] [CrossRef]
- Kolodziejczyk, S.M.; Walsh, G.S.; Balazsi, K.; Seale, P.; Sandoz, J.; Hierlihy, A.M.; Rudnicki, M.A.; Chamberlain, J.S.; Miller, F.D.; Megeney, L.A. Activation of JNK1 Contributes to Dystrophic Muscle Pathogenesis. Curr. Biol. 2001, 11, 1278–1282. [Google Scholar] [CrossRef]
- St-Pierre, S.J.G.; Chakkalakal, J.V.; Kolodziejczyk, S.M.; Knudson, J.C.; Jasmin, B.J.; Megeney, L.A. Glucocorticoid Treatment Alleviates Dystrophic Myofiber Pathology by Activation of the Calcineurin/NF-AT Pathway. FASEB J. 2004, 18, 1937–1939. [Google Scholar] [CrossRef]
- Nakamura, A.; Yoshida, K.; Ueda, H.; Takeda, S.; Ikeda, S.I. Up-Regulation of Mitogen Activated Protein Kinases in mdx Skeletal Muscle Following Chronic Treadmill Exercise. Biochim. Biophys. Acta 2005, 1740, 326–331. [Google Scholar] [CrossRef]
- Spence, H.J.; Dhillon, A.S.; James, M.; Winder, S.J. Dystroglycan, a Scaffold for the ERK-MAP Kinase Cascade. EMBO Rep. 2004, 5, 484–489. [Google Scholar] [CrossRef]
- Huang, Y.; Zhen, Y.; Chen, Y.; Sui, S.; Zhang, L. Unraveling the Interplay between RAS/RAF/MEK/ERK Signaling Pathway and Autophagy in Cancer: From Molecular Mechanisms to Targeted Therapy. Biochem. Pharmacol. 2023, 217, 115842. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A Non-Canonical MEK/ERK Signaling Pathway Regulates Autophagy via Regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef]
- Wei, Z.; Hu, X.; Wu, Y.; Zhou, L.; Zhao, M.; Lin, Q. Molecular Mechanisms Underlying Initiation and Activation of Autophagy. Biomolecules 2024, 14, 1517. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective Autophagy Elicited by RAF→MEK→ERK Inhibition Suggests a Treatment Strategy for RAS-Driven Cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Chang, W.S.W.; Cheung, C.H.A.; Lin, C.C.; Huang, C.C.; Yang, Y.N.; Kuo, C.P.; Kuo, C.C.; Chang, Y.H.; Liu, K.J.; et al. Targeting Cathepsin S Induces Tumor Cell Autophagy via the EGFR-ERK Signaling Pathway. Cancer Lett. 2012, 317, 89–98. [Google Scholar] [CrossRef]
- Kim, J.H.; Hong, S.K.; Wu, P.K.; Richards, A.L.; Jackson, W.T.; Park, J.I. Raf/MEK/ERK Can Regulate Cellular Levels of LC3B and SQSTM1/P62 at Expression Levels. Exp. Cell Res. 2014, 327, 340–352. [Google Scholar] [CrossRef]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A. Cytoskeletal Elements Are Required for the Formation and Maturation of Autophagic Vacuoles. J. Cell Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Shintani, T.; Klionsky, D.J. The Actin Cytoskeleton Is Required for Selective Types of Autophagy, but Not Nonspecific Autophagy, in the Yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 5843–5856. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The Actin Cytoskeleton Participates in the Early Events of Autophagosome Formation upon Starvation Induced Autophagy. Autophagy 2012, 8, 1590–1603. [Google Scholar] [CrossRef]
- Zhuo, C.; Ji, Y.; Chen, Z.; Kitazato, K.; Xiang, Y.; Zhong, M.; Wang, Q.; Pei, Y.; Ju, H.; Wang, Y. Proteomics Analysis of Autophagy-Deficient Atg7−/− MEFs Reveals a Close Relationship between F-Actin and Autophagy. Biochem. Biophys. Res. Commun. 2013, 437, 482–488. [Google Scholar] [CrossRef]
- Dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin Binding Proteins: Regulation of Cytoskeletal Microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. α-Synuclein Impairs Autophagosome Maturation through Abnormal Actin Stabilization. PLoS Genet. 2021, 17, e1009359. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.T.; Schinaman, J.M.; Liu-Abramowicz, N.; Williams, K.S.; Walker, D.W. Accumulation of F-Actin Drives Brain Aging and Limits Healthspan in Drosophila. Nat. Commun. 2024, 15, 9238. [Google Scholar] [CrossRef]
- Marotta, M.; Ruiz-Roig, C.; Sarria, Y.; Peiro, J.L.; Nuñez, F.; Ceron, J.; Munell, F.; Roig-Quilis, M. Muscle Genome-Wide Expression Profiling during Disease Evolution in mdx Mice. Physiol. Genom. 2009, 37, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Mi, N.; Chen, Y.; Wang, S.; Chen, M.; Zhao, M.; Yang, G.; Ma, M.; Su, Q.; Luo, S.; Shi, J.; et al. CapZ Regulates Autophagosomal Membrane Shaping by Promoting Actin Assembly inside the Isolation Membrane. Nat. Cell Biol. 2015, 17, 1112–1123. [Google Scholar] [CrossRef]
- Holland, P.; Simonsen, A. Actin Shapes the Autophagosome. Nat. Cell Biol. 2015, 17, 1094–1096. [Google Scholar] [CrossRef]
- Xu, J.; Kozlov, G.; McPherson, P.S.; Gehring, K. A PH-like Domain of the Rab12 Guanine Nucleotide Exchange Factor DENND3 Binds Actin and Is Required for Autophagy. J. Biol. Chem. 2018, 293, 4566–4574. [Google Scholar] [CrossRef]
- Hasegawa, J.; Iwamoto, R.; Otomo, T.; Nezu, A.; Hamasaki, M.; Yoshimori, T. Autophagosome-Lysosome Fusion in Neurons Requires INPP5E, a Protein Associated with Joubert Syndrome. EMBO J. 2016, 35, 1853–1867. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 Controls Autophagosome Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Kruppa, A.J.; Kendrick-Jones, J.; Buss, F. Myosins, Actin and Autophagy. Traffic 2016, 17, 878–890. [Google Scholar] [CrossRef]
- Coutts, A.S.; La Thangue, N.B. Actin Nucleation by WH2 Domains at the Autophagosome. Nat. Commun. 2015, 6, 7888. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Ghislat, G.; Hochfeld, W.; Renna, M.; Zavodszky, E.; Runwal, G.; Puri, C.; Lee, S.; Siddiqi, F.; Menzies, F.M.; et al. Transcriptional Regulation of Annexin A2 Promotes Starvation-Induced Autophagy. Nat. Commun. 2015, 6, 8045. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Du, Y.; Zhao, Z.; Shi, L.; Sun, L.; Huang, G.; Ye, B.; Li, C.; Dai, Z.; et al. WASH Inhibits Autophagy through Suppression of Beclin 1 Ubiquitination. EMBO J. 2013, 32, 2685–2696. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 Associated with Parkinson’s Disease Impairs WASH Complex Association and Inhibits Autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Rotty, J.D.; Wu, C.; Bear, J.E. New Insights into the Regulation and Cellular Functions of the ARP2/3 Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. WHAMM Links Actin Assembly via the Arp2/3 Complex to Autophagy. Autophagy 2015, 11, 1702–1704. [Google Scholar] [CrossRef]
- Dominguez, R. The WH2 Domain and Actin Nucleation: Necessary but Insufficient. Trends Biochem. Sci. 2016, 41, 478–490. [Google Scholar] [CrossRef]
- Kast, D.J.; Zajac, A.L.; Holzbaur, E.L.F.; Ostap, E.M.; Dominguez, R. WHAMM Directs the Arp2/3 Complex to the ER for Autophagosome Biogenesis through an Actin Comet Tail Mechanism. Curr. Biol. 2015, 25, 1791–1797. [Google Scholar] [CrossRef]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The Dystrophin Complex Forms a Mechanically Strong Link between the Sarcolemma and Costameric Actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Amann, K.J.; Ervasti, J.M. A New Model for the Interaction of Dystrophin with F-Actin. J. Cell Biol. 1996, 135, 661–672. [Google Scholar] [CrossRef]
- Amann, K.J.; Renley, B.A.; Ervasti, J.M. A Cluster of Basic Repeats in the Dystrophin Rod Domain Binds F-Actin through an Electrostatic Interaction. J. Biol. Chem. 1998, 273, 28419–28423. [Google Scholar] [CrossRef]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin Is a Microtubule-Associated Protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.W.; Bloch, R.J. Extensive but Coordinated Reorganization of the Membrane Skeleton in Myofibers of Dystrophic (mdx) Mice. J. Cell Biol. 1999, 144, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Hanft, L.M.; Rybakova, I.N.; Patel, J.R.; Rafael-Fortney, J.A.; Ervasti, J.M. Cytoplasmic Gamma-Actin Contributes to a Compensatory Remodeling Response in Dystrophin-Deficient Muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 5385–5390. [Google Scholar] [CrossRef]
- Batchelor, C.L.; Higginson, J.R.; Chen, Y.J.; Vanni, C.; Eva, A.; Winder, S.J. Recruitment of Dbl by Ezrin and Dystroglycan Drives Membrane Proximal Cdc42 Activation and Filopodia Formation. Cell Cycle 2007, 6, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Spence, H.J.; Chen, Y.J.; Batchelor, C.L.; Higginson, J.R.; Suila, H.; Carpen, O.; Winder, S.J. Ezrin-Dependent Regulation of the Actin Cytoskeleton by β -Dystroglycan. Hum. Mol. Genet. 2004, 13, 1657–1668. [Google Scholar] [CrossRef]
- Chen, Y.J.; Spence, H.J.; Cameron, J.M.; Jess, T.; Ilsley, J.L.; Winder, S.J. Direct Interaction of β-Dystroglycan with F-Actin. Biochem. J. 2003, 375, 329–337. [Google Scholar] [CrossRef]
- Palmieri, V.; Bozzi, M.; Signorino, G.; Papi, M.; De Spirito, M.; Brancaccio, A.; Maulucci, G.; Sciandra, F. α-Dystroglycan Hypoglycosylation Affects Cell Migration by Influencing β-Dystroglycan Membrane Clustering and Filopodia Length: A Multiscale Confocal Microscopy Analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2182–2191. [Google Scholar] [CrossRef]
- Thompson, O.; Kleino, I.; Crimaldi, L.; Gimona, M.; Saksela, K.; Winder, S.J. Dystroglycan, Tks5 and Src Mediated Assembly of Podosomes in Myoblasts. PLoS ONE 2008, 3, e3638. [Google Scholar] [CrossRef] [PubMed]
- Sciandra, F.; Desiderio, C.; Vincenzoni, F.; Viscuso, S.; Bozzi, M.; Hübner, W.; Jimenez-Gutierrez, G.E.; Cisneros, B.; Brancaccio, A. Analysis of the GFP-Labelled β-Dystroglycan Interactome in HEK-293 Transfected Cells Reveals Novel Intracellular Networks. Biochem. Biophys. Res. Commun. 2024, 703, 149656. [Google Scholar] [CrossRef]
- Gerin, I.; Ury, B.; Breloy, I.; Bouchet-Seraphin, C.; Bolsée, J.; Halbout, M.; Graff, J.; Vertommen, D.; Muccioli, G.G.; Seta, N.; et al. ISPD Produces CDP-Ribitol Used by FKTN and FKRP to Transfer Ribitol Phosphate onto α-Dystroglycan. Nat. Commun. 2016, 7, 11534. [Google Scholar] [CrossRef]
- Kanagawa, M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int. J. Mol. Sci. 2021, 22, 13162. [Google Scholar] [CrossRef] [PubMed]
- Praissman, J.L.; Willer, T.; Osman Sheikh, M.; Toi, A.; Chitayat, D.; Lin, Y.Y.; Lee, H.; Stalnaker, S.H.; Wang, S.; Prabhakar, P.K.; et al. The Functional O-Mannose Glycan on α-Dystroglycan Contains a Phospho-Ribitol Primed for Matriglycan Addition. eLife 2016, 5, e14473. [Google Scholar] [CrossRef] [PubMed]
- Praissman, J.L.; Live, D.H.; Wang, S.; Ramiah, A.; Chinoy, Z.S.; Boons, G.J.; Moremen, K.W.; Wells, L. B4GAT1 Is the Priming Enzyme for the LARGE-Dependent Functional Glycosylation of α-Dystroglycan. eLife 2014, 3, e03943. [Google Scholar] [CrossRef]
- Inamori, K.I.; Yoshida-Moriguchi, T.; Hara, Y.; Anderson, M.E.; Yu, L.; Campbell, K.P. Dystroglycan Function Requires Xylosyl- and Glucuronyltransferase Activities of LARGE. Science 2012, 335, 93–96. [Google Scholar] [CrossRef]
- Manya, H.; Yamaguchi, Y.; Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Akasaka-Manya, K.; Kawakami, H.; Mizuno, M.; Wada, Y.; Toda, T.; et al. The Muscular Dystrophy Gene TMEM5 Encodes a Ribitol Β1,4-Xylosyltransferase Required for the Functional Glycosylation of Dystroglycan. J. Biol. Chem. 2016, 291, 24618–24627. [Google Scholar] [CrossRef]
- Willer, T.; Inamori, K.I.; Venzke, D.; Harvey, C.; Morgensen, G.; Hara, Y.; Beltrán Valero de Bernabé, D.; Yu, L.; Wright, K.M.; Campbell, K.P. The Glucuronyltransferase B4GAT1 Is Required for Initiation of LARGE-Mediated α-Dystroglycan Functional Glycosylation. eLife 2014, 3, e03941. [Google Scholar] [CrossRef]
- Dempsey, C.E.; Bigotti, M.G.; Adams, J.C.; Brancaccio, A. Analysis of α-Dystroglycan/LG Domain Binding Modes: Investigating Protein Motifs That Regulate the Affinity of Isolated LG Domains. Front. Mol. Biosci. 2019, 6, 18. [Google Scholar] [CrossRef]
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E.; et al. Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin α2 Deficiency and Abnormal Glycosylation of α-Dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. [Google Scholar] [CrossRef]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.B.; Bashir, R.; Burgunder, J.M.; et al. Mutations in the Fukutin-Related Protein Gene (FKRP) Identify Limb Girdle Muscular Dystrophy 2I as a Milder Allelic Variant of Congenital Muscular Dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An Ancient Retrotransposal Insertion Causes Fukuyama-Type Congenital Muscular Dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Valero De Bernabé, D.; Currier, S.; Steinbrecher, A.; Celli, J.; Van Beusekom, E.; Van Der Zwaag, B.; Lya Kayserili, H.; Merlini, L.; Chitayat, D.; Dobyns, W.B.; et al. Mutations in the O-Mannosyltransferase Gene POMT1 Give Rise to the Severe Neuronal Migration Disorder Walker-Warburg Syndrome. Am. J. Hum. Genet. 2002, 71, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Franekova, V.; Storjord, H.I.; Leivseth, G.; Nilssen, Ø. Protein Homeostasis in LGMDR9 (LGMD2I)—The Role of Ubiquitin–Proteasome and Autophagy–Lysosomal System. Neuropathol. Appl. Neurobiol. 2021, 47, 519–531. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Esapa, C.T.; Benson, M.A.; Schröder, J.E.; Martin-Rendon, E.; Brockington, M.; Brown, S.C.; Muntoni, F.; Kröger, S.; Blake, D.J. Functional Requirements for Fukutin-Related Protein in the Golgi Apparatus. Hum. Mol. Genet. 2002, 11, 3319–3331. [Google Scholar] [CrossRef]
- Esapa, C.T.; McIlhinney, R.A.J.; Blake, D.J. Fukutin-Related Protein Mutations That Cause Congenital Muscular Dystrophy Result in ER-Retention of the Mutant Protein in Cultured Cells. Hum. Mol. Genet. 2005, 14, 295–305. [Google Scholar] [CrossRef]
- Esapa, C.T.; McIlhinney, R.A.J.; Waite, A.J.; Benson, M.A.; Mirzayan, J.; Piko, H.; Herczegfalvi, Á.; Horvath, R.; Karcagi, V.; Walter, M.C.; et al. Misfolding of Fukutin-Related Protein (FKRP) Variants in Congenital and Limb Girdle Muscular Dystrophies. Front. Mol. Biosci. 2023, 10, 1279700. [Google Scholar] [CrossRef]
- Tan, R.L.; Sciandra, F.; Hübner, W.; Bozzi, M.; Reimann, J.; Schoch, S.; Brancaccio, A.; Blaess, S. The Missense Mutation C667F in Murine β-Dystroglycan Causes Embryonic Lethality, Myopathy and Blood-Brain Barrier Destabilization. Dis. Model. Mech. 2024, 17, dmm050594. [Google Scholar] [CrossRef]
- Henriques, S.F.; Patissier, C.; Bourg, N.; Fecchio, C.; Sandona, D.; Marsolier, J.; Richard, I. Different Outcome of Sarcoglycan Missense Mutation between Human and Mouse. PLoS ONE 2018, 13, e0191274. [Google Scholar] [CrossRef] [PubMed]
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-Week Rapamycin Treatment Improves Muscular Dystrophy in a Fukutin-Deficient Mouse Model of Dystroglycanopathy. Skelet. Muscle 2016, 6, 20. [Google Scholar] [CrossRef]
- Ortiz-Cordero, C.; Bincoletto, C.; Dhoke, N.R.; Selvaraj, S.; Magli, A.; Zhou, H.; Kim, D.H.; Bang, A.G.; Perlingeiro, R.C.R. Defective Autophagy and Increased Apoptosis Contribute toward the Pathogenesis of FKRP-Associated Muscular Dystrophies. Stem Cell Rep. 2021, 16, 2752–2767. [Google Scholar] [CrossRef] [PubMed]
- Alhamidi, M.; Brox, V.; Stensland, E.; Liset, M.; Lindal, S.; Nilssen, Ø. Limb Girdle Muscular Dystrophy Type 2I: No Correlation between Clinical Severity, Histopathology and Glycosylated α-Dystroglycan Levels in Patients Homozygous for Common FKRP Mutation. Neuromuscul. Disord. 2017, 27, 619–626. [Google Scholar] [CrossRef]
- Henriques, S.F.; Gicquel, E.; Marsolier, J.; Richard, I. Functional and Cellular Localization Diversity Associated with Fukutin-Related Protein Patient Genetic Variants. Hum. Mutat. 2019, 40, 1874–1885. [Google Scholar] [CrossRef]
- Lee, A.J.; Jones, K.A.; Butterfield, R.J.; Cox, M.O.; Konersman, C.G.; Grosmann, C.; Abdenur, J.E.; Boyer, M.; Beson, B.; Wang, C.; et al. Clinical, Genetic, and Pathologic Characterization of FKRP Mexican Founder Mutation c.1387A>G. Neurol. Genet. 2019, 5, e315. [Google Scholar] [CrossRef]
- Rojek, J.M.; Moraz, M.L.; Pythoud, C.; Rothenberger, S.; Van der Goot, F.G.; Campbell, K.P.; Kunz, S. Binding of Lassa Virus Perturbs Extracellular Matrix-Induced Signal Transduction via Dystroglycan. Cell Microbiol. 2012, 14, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.H.; Campbell, K.P. Assembly of the Sarcoglycan Complex. Insights for Muscular Dystrophy. J. Biol. Chem. 1998, 273, 34667–34670. [Google Scholar] [CrossRef]
- Sandonà, D.; Betto, R. Sarcoglycanopathies: Molecular Pathogenesis and Therapeutic Prospects. Expert. Rev. Mol. Med. 2009, 11, e28. [Google Scholar] [CrossRef]
- Guimarães-Costa, R.; Fernández-Eulate, G.; Wahbi, K.; Leturcq, F.; Malfatti, E.; Behin, A.; Leonard-Louis, S.; Desguerre, I.; Barnerias, C.; Nougues, M.C.; et al. Clinical Correlations and Long-Term Follow-up in 100 Patients with Sarcoglycanopathies. Eur. J. Neurol. 2021, 28, 660–669. [Google Scholar] [CrossRef]
- Alonso-Pérez, J.; González-Quereda, L.; Bruno, C.; Panicucci, C.; Alavi, A.; Nafissi, S.; Nilipour, Y.; Zanoteli, E.; Isihi, L.M.D.A.; Melegh, B.; et al. Clinical and Genetic Spectrum of a Large Cohort of Patients with δ-Sarcoglycan Muscular Dystrophy. Brain 2022, 145, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Gicquel, E.; Barrault, L.; Soheili, T.; Malissen, M.; Malissen, B.; Vincent-lacaze, N.; Perez, N.; Udd, B.; Danos, O.; et al. Mannosidase I Inhibition Rescues the Human α-Sarcoglycan R77C Recurrent Mutation. Hum. Mol. Genet. 2008, 17, 1214–1221. [Google Scholar] [CrossRef]
- Soheili, T.; Gicquel, E.; Poupiot, J.; N’Guyen, L.; Le Roy, F.; Bartoli, M.; Richard, I. Rescue of Sarcoglycan Mutations by Inhibition of Endoplasmic Reticulum Quality Control Is Associated with Minimal Structural Modifications. Hum. Mutat. 2012, 33, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; D’Angelo, S.; Franzoso, S.; Fanin, M.; Angelini, C.; Betto, R.; Sandonà, D. Inhibition of Proteasome Activity Promotes the Correct Localization of Disease-Causing α-Sarcoglycan Mutants in HEK-293 Cells Constitutively Expressing β-, γ-, and δ-Sarcoglycan. Am. J. Pathol. 2008, 173, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, E.; Fanin, M.; Mamchaoui, K.; Betto, R.; Sandonà, D. Unveiling the Degradative Route of the V247M α-Sarcoglycan Mutant Responsible for LGMD-2D. Hum. Mol. Genet. 2014, 23, 3746–3758. [Google Scholar] [CrossRef]
- Scano, M.; Benetollo, A.; Dalla Barba, F.; Sandonà, D. Advanced Therapeutic Approaches in Sarcoglycanopathies. Curr Opin Pharmacol 2024, 76, 102459. [Google Scholar] [CrossRef]
- Carotti, M.; Scano, M.; Fancello, I.; Richard, I.; Risato, G.; Bensalah, M.; Soardi, M.; Sandonà, D. Combined Use of CFTR Correctors in LGMD2D Myotubes Improves Sarcoglycan Complex Recovery. Int. J. Mol. Sci. 2020, 21, 1813. [Google Scholar] [CrossRef]
- Scano, M.; Benetollo, A.; Nogara, L.; Bondì, M.; Dalla Barba, F.; Soardi, M.; Furlan, S.; Akyurek, E.E.; Caccin, P.; Carotti, M.; et al. CFTR Corrector C17 Is Effective in Muscular Dystrophy, in Vivo Proof of Concept in LGMDR3. Hum. Mol. Genet. 2022, 31, 499–509. [Google Scholar] [CrossRef]
- Helbling-Leclerc, A.; Zhang, X.; Topaloglu, H.; Cruaud, C.; Tesson, F.; Weissenbach, J.; Tomé, F.M.S.; Schwartz, K.; Fardeau, M.; Tryggvason, K.; et al. Mutations in the Laminin α2-Chain Gene (LAMA2) Cause Merosin-Deficient Congenital Muscular Dystrophy. Nat. Genet. 1995, 11, 216–218. [Google Scholar] [CrossRef]
- Kubota, A.; Ishiura, H.; Mitsui, J.; Sakuishi, K.; Iwata, A.; Yamamoto, T.; Nishino, I.; Tsuji, S.; Shimizu, J. A Homozygous LAMA2 Mutation of c.818G>A Caused Partial Merosin Deficiency in a Japanese Patient. Intern. Med. 2018, 57, 877–882. [Google Scholar] [CrossRef]
- Allamand, V.; Guicheney, P. Merosin-Deficient Congenital Muscular Dystrophy, Autosomal Recessive (MDC1A, MIM#156225, LAMA2 Gene Coding for α2 Chain of Laminin). Eur. J. Hum. Genet. 2002, 10, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Quéré, R.; Durbeej, M. Proteasome Inhibition Improves the Muscle of Laminin α2 Chain-Deficient Mice. Hum. Mol. Genet. 2011, 20, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Körner, Z.; Fontes-Oliveira, C.C.; Holmberg, J.; Carmignac, V.; Durbeej, M. Bortezomib Partially Improves Laminin α2 Chain-Deficient Muscular Dystrophy. Am. J. Pathol. 2014, 184, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Svensson, M.; Körner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy Is Increased in Laminin α2 Chain-Deficient Muscle and Its Inhibition Improves Muscle Morphology in a Mouse Model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902. [Google Scholar] [CrossRef]
- Mastrapasqua, M.; Rossi, R.; De Cosmo, L.; Resta, A.; Errede, M.; Bizzoca, A.; Zampatti, S.; Resta, N.; Giardina, E.; Ruggieri, M.; et al. Autophagy Increase in Merosin-Deficient Congenital Muscular Dystrophy Type 1A. Eur. J. Transl. Myol. 2023, 33, 11501. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzi, M.; Sciandra, F.; Bigotti, M.G.; Brancaccio, A. Misregulation of the Ubiquitin–Proteasome System and Autophagy in Muscular Dystrophies Associated with the Dystrophin–Glycoprotein Complex. Cells 2025, 14, 721. https://doi.org/10.3390/cells14100721
Bozzi M, Sciandra F, Bigotti MG, Brancaccio A. Misregulation of the Ubiquitin–Proteasome System and Autophagy in Muscular Dystrophies Associated with the Dystrophin–Glycoprotein Complex. Cells. 2025; 14(10):721. https://doi.org/10.3390/cells14100721
Chicago/Turabian StyleBozzi, Manuela, Francesca Sciandra, Maria Giulia Bigotti, and Andrea Brancaccio. 2025. "Misregulation of the Ubiquitin–Proteasome System and Autophagy in Muscular Dystrophies Associated with the Dystrophin–Glycoprotein Complex" Cells 14, no. 10: 721. https://doi.org/10.3390/cells14100721
APA StyleBozzi, M., Sciandra, F., Bigotti, M. G., & Brancaccio, A. (2025). Misregulation of the Ubiquitin–Proteasome System and Autophagy in Muscular Dystrophies Associated with the Dystrophin–Glycoprotein Complex. Cells, 14(10), 721. https://doi.org/10.3390/cells14100721