
Epithelial Dysfunction in Congenital Diaphragmatic Hernia: Mechanisms, Models and Emerging Therapies

Abstract
1. Introduction
2. Emerging Technologies and Concepts Unravel Lung Cellular Heterogeneity in CDH
2.1. Single-Cell RNA Sequencing and Spatial Transcriptomics
2.2. Proteomic and Metabolomic Profiling
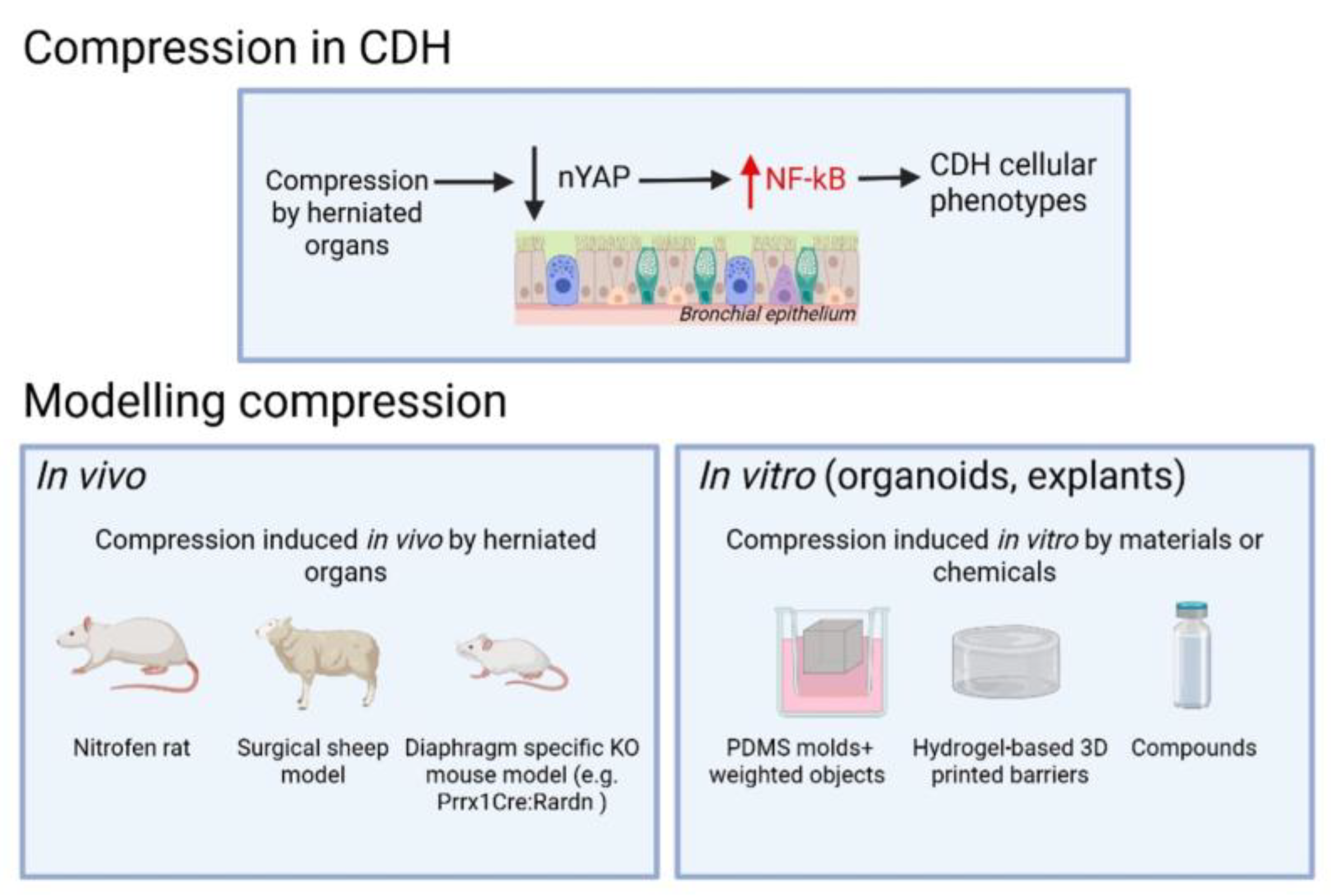
2.3. Mechanical Disruption as a Driver of Differentiation Defects
3. Revisiting and Refining Experimental Models of CDH
3.1. New Insights from the Nitrofen Rat Model
3.2. Air–Liquid Interface Culture Models
3.3. Organoid Culture for Modeling the CDH Lung
3.4. Ex Vivo Lung Explants
4. Novel Therapeutic Strategies Targeting Epithelial Dysfunction
4.1. Stem Cell and Regenerative Therapies
4.2. Future Directions: Precision Medicine and Defining CDH Endotypes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zepp, J.A.; Morley, M.P.; Loebel, C.; Kremp, M.M.; Chaudhry, F.N.; Basil, M.C.; Leach, J.P.; Liberti, D.C.; Niethamer, T.K.; Ying, Y.; et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021, 371, eabc3172. [Google Scholar] [CrossRef] [PubMed]
- Zepp, J.A.; Morrisey, E.E. Cellular crosstalk in the development and regeneration of the respiratory system. Nat. Rev. Mol. Cell Biol. 2019, 20, 551–566. [Google Scholar] [CrossRef]
- Miyake, Y.; Jank, M.; Patel, D.; Ozturk, A.; Aubert, O.; Ai, X.; Yamataka, A.; Keijzer, R. Yes-associated protein is dysregulated in human congenital diaphragmatic hernia patients during mid and end gestation. Pediatr. Surg. Int. 2024, 41, 16. [Google Scholar] [CrossRef] [PubMed]
- Aubert, O.; Miyake, Y.; Amonkar, G.M.; Dinwoodie, O.M.; Varisco, B.M.; Marotta, M.; Zhao, C.; Wagner, R.; Chen, Y.-W.; Moscatello, A.; et al. Fetal Tracheal Occlusion Corelates with Normalized YAP Expression and Alveolar Epithelial Differentiation in CDH. Am. J. Respir. Cell Mol. Biol. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Wagner, R.; Amonkar, G.M.; Wang, W.; Shui, J.E.; Bankoti, K.; Tse, W.H.; High, F.A.; Zalieckas, J.M.; Buchmiller, T.L.; Zani, A.; et al. A Tracheal Aspirate-derived Airway Basal Cell Model Reveals a Proinflammatory Epithelial Defect in Congenital Diaphragmatic Hernia. Am. J. Respir. Crit. Care Med. 2023, 207, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Dylong, F.; Riedel, J.; Amonkar, G.M.; Peukert, N.; Lieckfeldt, P.; Sturm, K.; Höxter, B.; Tse, W.H.; Miyake, Y.; Moormann, M.; et al. Overactivated Epithelial NF-κB Disrupts Lung Development in Congenital Diaphragmatic Hernia. Am. J. Respir. Cell Mol. Biol. 2023, 69, 545–555. [Google Scholar] [CrossRef]
- Antounians, L.; Catania, V.D.; Montalva, L.; Liu, B.D.; Hou, H.; Chan, C.; Matei, A.C.; Tzanetakis, A.; Li, B.; Figueira, R.L.; et al. Fetal lung underdevelopment is rescued by administration of amniotic fluid stem cell extracellular vesicles in rodents. Sci. Transl. Med. 2021, 13, eaax5941. [Google Scholar] [CrossRef]
- Longoni, M.; High, F.A.; Russell, M.K.; Kashani, A.; Tracy, A.A.; Coletti, C.M.; Hila, R.; Shamia, A.; Wells, J.; Ackerman, K.G.; et al. Molecular pathogenesis of congenital diaphragmatic hernia revealed by exome sequencing, developmental data, and bioinformatics. Proc. Natl. Acad. Sci. USA 2014, 111, 12450–12455. [Google Scholar] [CrossRef]
- Russell, M.K.; Longoni, M.; Wells, J.; Maalouf, F.I.; Tracy, A.A.; Loscertales, M.; Ackerman, K.G.; Pober, B.R.; Lage, K.; Bult, C.J.; et al. Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc. Natl. Acad. Sci. USA 2012, 109, 2978–2983. [Google Scholar] [CrossRef]
- Antounians, L.; Figueira, R.L.; Kukreja, B.; Litvack, M.L.; Zani-Ruttenstock, E.; Khalaj, K.; Montalva, L.; Doktor, F.; Obed, M.; Blundell, M.; et al. Fetal hypoplastic lungs have multilineage inflammation that is reversed by amniotic fluid stem cell extracellular vesicle treatment. Sci. Adv. 2024, 10, eadn5405. [Google Scholar] [CrossRef]
- Aubert, O.; Amonkar, G.M.; Varelas, X.; Tilston-Lunel, A.; Lerou, P.H.; Zalieckas, J.M.; Buchmiller, T.L.; Marotta, M.; Peiro, J.L.; Ai, X. YAP Deficiency Drives NF-κB Hyperactivation to Disrupt Airway Epithelium Differentiation in Congenital Diaphragmatic Hernia. Am. J. Respir. Cell Mol. Biol. 2025, 72, 112–115. [Google Scholar] [CrossRef]
- Mahoney, J.E.; Mori, M.; Szymaniak, A.D.; Varelas, X.; Cardoso, W.V. The Hippo Pathway Effector Yap Controls Patterning and Differentiation of Airway Epithelial Progenitors. Dev. Cell 2014, 30, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Tilston-Lunel, A.M.; Simonetti, J.; Hicks-Berthet, J.; Matschulat, A.; Pfefferkorn, R.; Spira, A.; Edwards, M.; Mazzilli, S.; Lenburg, M.E.; et al. Convergence of YAP/TAZ, TEAD and TP63 activity is associated with bronchial premalignant severity and progression. J. Exp. Clin. Cancer Res. 2023, 42, 116. [Google Scholar] [CrossRef]
- Gerli, M.F.M.; Calà, G.; Beesley, M.A.; Sina, B.; Tullie, L.; Sun, K.Y.; Panariello, F.; Michielin, F.; Davidson, J.R.; Russo, F.M.; et al. Single-cell guided prenatal derivation of primary fetal epithelial organoids from human amniotic and tracheal fluids. Nat. Med. 2024, 30, 875–887. [Google Scholar] [CrossRef]
- Robertson, J.O.; Bazeley, P.; Erzurum, S.C.; Asosingh, K. Single-cell transcriptomic profiling of microvascular endothelial cell heterogeneity in congenital diaphragmatic hernia. Sci. Rep. 2023, 13, 9851. [Google Scholar] [CrossRef]
- Lingappan, K.; Olutoye, O.O.; Cantu, A.; Cantu Gutierrez, M.E.; Cortes-Santiago, N.; Hammond, J.D.; Gilley, J.; Quintero, J.R.; Li, H.; Polverino, F.; et al. Molecular insights using spatial transcriptomics of the distal lung in congenital diaphragmatic hernia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2023, 325, L477–L486. [Google Scholar] [CrossRef]
- McCulley, D.J.; Wienhold, M.D.; Hines, E.A.; Hacker, T.A.; Rogers, A.; Pewowaruk, R.J.; Zewdu, R.; Chesler, N.C.; Selleri, L.; Sun, X. PBX transcription factors drive pulmonary vascular adaptation to birth. J. Clin. Investig. 2017, 128, 655–667. [Google Scholar] [CrossRef]
- Stokes, G.; Li, Z.; Talaba, N.; Genthe, W.; Brix, M.B.; Pham, B.; Wienhold, M.D.; Sandok, G.; Hernan, R.; Wynn, J.; et al. Rescuing lung development through embryonic inhibition of histone acetylation. Sci. Transl. Med. 2024, 16, eadc8930. [Google Scholar] [CrossRef] [PubMed]
- Friedmacher, F.; Rolle, U.; Puri, P. Genetically Modified Mouse Models of Congenital Diaphragmatic Hernia: Opportunities and Limitations for Studying Altered Lung Development. Front. Pediatr. 2022, 10, 867307. [Google Scholar]
- Thébaud, B.; Barlier-Mur, A.-M.; Chailley-Heu, B.; Henrion-Caude, A.; Tibboel, D.; Dinh-Xuan, A.-T.; Bourbon, J.R. Restoring Effects of Vitamin A on Surfactant Synthesis in Nitrofen-induced Congenital Diaphragmatic Hernia in Rats. Am. J. Respir. Crit. Care Med. 2001, 164, 1083–1089. [Google Scholar] [CrossRef]
- Greer, J.J.; Babiuk, R.P.; Thebaud, B. Etiology of Congenital Diaphragmatic Hernia: The Retinoid Hypothesis. Pediatr. Res. 2003, 53, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.F.G.; Clugston, R.D. The etiology of congenital diaphragmatic hernia: The retinoid hypothesis 20 years later. Pediatr. Res. 2024, 95, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Garcia Rivas, J.F.; Applin, N.H.M.; Albrechtsen, J.F.P.; Ghazanfari, A.; Doschak, M.; Clugston, R.D. Mesenchymal retinoic acid signaling is required for normal diaphragm development in mice. FASEB J. 2025, 39, e70381. [Google Scholar] [CrossRef] [PubMed]
- Merrell, A.J.; Ellis, B.J.; Fox, Z.D.; Lawson, J.A.; Weiss, J.A.; Kardon, G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat. Genet. 2015, 47, 496–504. [Google Scholar] [CrossRef]
- Cleal, L.; McHaffie, S.L.; Lee, M.; Hastie, N.; Martínez-Estrada, O.M.; Chau, Y.-Y. Resolving the heterogeneity of diaphragmatic mesenchyme: A novel mouse model of congenital diaphragmatic hernia. Dis. Model. Mech. 2021, 14, dmm046797. [Google Scholar] [CrossRef]
- Wagner, R.; Lieckfeldt, P.; Piyadasa, H.; Markel, M.; Riedel, J.; Stefanovici, C.; Peukert, N.; Patel, D.; Derraugh, G.; Min, S.A.L.; et al. Proteomic Profiling of Hypoplastic Lungs Suggests an Underlying Inflammatory Response in the Pathogenesis of Abnormal Lung Development in Congenital Diaphragmatic Hernia. Ann. Surg. 2023, 278, e411–e421. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Yu, W.; Liu, X.; Liu, C. Tandem mass tag (TMT) proteomic analysis of fetal lungs revealed differential expression of tight junction proteins in a rat model of congenital diaphragmatic hernia. Biomed. Pharmacother. 2020, 121, 109621. [Google Scholar] [CrossRef]
- Peiro, J.L.; Oria, M.; Aydin, E.; Joshi, R.; Cabanas, N.; Schmidt, R.; Schroeder, C.; Marotta, M.; Varisco, B.M. Proteomic profiling of tracheal fluid in an ovine model of congenital diaphragmatic hernia and fetal tracheal occlusion. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 315, L1028–L1041. [Google Scholar] [CrossRef]
- Bhutada, S.; Tran-Lundmark, K.; Kramer, B.; Conner, P.; Lowry, A.M.; Blackstone, E.; Frenckner, B.; Mesas-Burgos, C.; Apte, S.S. Identification of protein biomarkers associated with congenital diaphragmatic hernia in human amniotic fluid. Sci. Rep. 2023, 13, 15483. [Google Scholar] [CrossRef]
- Tachi, A.; Moriyama, Y.; Tsuda, H.; Miki, R.; Ushida, T.; Miura, M.; Ito, Y.; Imai, K.; Nakano-Kobayashi, T.; Hayakawa, M.; et al. A proteome signature of umbilical cord serum associated with congenital diaphragmatic hernia. Nagoya J. Med. Sci. 2020, 82, 345–354. [Google Scholar]
- Piersigilli, F.; Syed, M.; Lam, T.T.; Dotta, A.; Massoud, M.; Vernocchi, P.; Quagliariello, A.; Putignani, L.; Auriti, C.; Salvatori, G.; et al. An omic approach to congenital diaphragmatic hernia: A pilot study of genomic, microRNA, and metabolomic profiling. J. Perinatol. 2020, 40, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Piersigilli, F.; Syed, M.; Mondi, V.; Capolupo, I.; Campi, F.; Danhaive, O.; Bagolan, P.; Dotta, A.; Auriti, C.; Tukiet, L.; et al. Transcriptomics and Metabolomics of Congenital Diaphragmatic Hernia. Am. J. Perinatol. 2016, 33, A023. [Google Scholar] [CrossRef]
- Easton, Z.E.; Regnault, T.R.H.; Mudri, M.; Zhao, S.; Smith, S.A.; Zardini Buzatto, A.; Li, J.; Duruisseau-Kuntz, R.; Davidson, J.; Li, L.; et al. The metabolic and lipidomic profiling of the effects of tracheal occlusion in a rabbit model of congenital diaphragmatic hernia. J. Pediatr. Surg. 2023, 58, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Romero-Lopez, M.D.M.; Oria, M.; Watanabe-Chailland, M.; Varela, M.F.; Romick-Rosendale, L.; Peiro, J.L. Lung Metabolomics Profiling of Congenital Diaphragmatic Hernia in Fetal Rats. Metabolites 2021, 11, 177. [Google Scholar] [CrossRef]
- Croitor-Sava, A.; Beck, V.; Sandaite, I.; Van Huffel, S.; Dresselaers, T.; Claus, F.; Himmelreich, U.; Deprest, J. High-Resolution 1H NMR Spectroscopy Discriminates Amniotic Fluid of Fetuses with Congenital Diaphragmatic Hernia from Healthy Controls. J. Proteome Res. 2015, 14, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Boivin, F.J.; Schmidt-Ott, K.M. Transcriptional mechanisms coordinating tight junction assembly during epithelial differentiation. Ann. N. Y. Acad. Sci. 2017, 1397, 80–99. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Van Der Merwe, J.; Elowsson Rendin, L.; Larsson-Callerfelt, A.-K.; Deprest, J.; Westergren-Thorsson, G.; Toelen, J. Stretch increases alveolar type 1 cell number in fetal lungs through ROCK-Yap/Taz pathway. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L814–L826. [Google Scholar] [CrossRef]
- Shiraishi, K.; Shah, P.P.; Morley, M.P.; Loebel, C.; Santini, G.T.; Katzen, J.; Basil, M.C.; Lin, S.M.; Planer, J.D.; Cantu, E.; et al. Biophysical forces mediated by respiration maintain lung alveolar epithelial cell fate. Cell 2023, 186, 1478–1492.e15. [Google Scholar] [CrossRef]
- Nelson, C.M.; Gleghorn, J.P.; Pang, M.-F.; Jaslove, J.; Goodwin, K.; Varner, V.D.; Miller, E.; Radisky, D.C.; Stone, H.A. Microfluidic chest cavities reveal that transmural pressure controls the rate of lung development. Development 2017, 144, 4328–4335. [Google Scholar] [CrossRef]
- Keijzer, R.; Liu, J.; Deimling, J.; Tibboel, D.; Post, M. Dual-Hit Hypothesis Explains Pulmonary Hypoplasia in the Nitrofen Model of Congenital Diaphragmatic Hernia. Am. J. Pathol. 2000, 156, 1299–1306. [Google Scholar] [CrossRef]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev. Cell 2015, 34, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kahnamoui, S.; Khoshgoo, N.; Patel, D.; Wagner, R.; Keijzer, R. Yes-associated protein is dysregulated during nitrofen-induced hypoplastic lung development due to congenital diaphragmatic hernia. Pediatr. Surg. Int. 2022, 38, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Montalva, L.; Zani, A.; Keijzer, R. Basic and translational science advances in congenital diaphragmatic hernia. Semin. Perinatol. 2020, 44, 151170. [Google Scholar] [CrossRef]
- Kluth, D.; Kangah, R.; Reich, P.; Tenbrinck, R.; Tibboel, D.; Lambrecht, W. Nitrofen-induced diaphragmatic hernias in rats: An animal model. J. Pediatr. Surg. 1990, 25, 850–854. [Google Scholar] [CrossRef]
- Khoshgoo, N.; Visser, R.; Falk, L.; Day, C.A.; Ameis, D.; Iwasiow, B.M.; Zhu, F.; Öztürk, A.; Basu, S.; Pind, M.; et al. MicroRNA-200b regulates distal airway development by maintaining epithelial integrity. Sci. Rep. 2017, 7, 6382. [Google Scholar] [CrossRef]
- Mujahid, S.; Nielsen, H.C.; Volpe, M.V. MiR-221 and miR-130a Regulate Lung Airway and Vascular Development. PLoS ONE 2013, 8, e55911. [Google Scholar] [CrossRef]
- Cushing, L.; Jiang, Z.; Kuang, P.; Lü, J. The Roles of MicroRNAs and Protein Components of the MicroRNA Pathway in Lung Development and Diseases. Am. J. Respir. Cell Mol. Biol. 2015, 52, 397–408. [Google Scholar] [CrossRef]
- Pereira-Terra, P.; Deprest, J.A.; Kholdebarin, R.; Khoshgoo, N.; DeKoninck, P.; Munck, A.A.B.-D.; Wang, J.; Zhu, F.; Rottier, R.J.; Iwasiow, B.M.; et al. Unique Tracheal Fluid MicroRNA Signature Predicts Response to FETO in Patients with Congenital Diaphragmatic Hernia. Ann. Surg. 2015, 262, 1130–1140. [Google Scholar] [CrossRef]
- Khoshgoo, N.; Kholdebarin, R.; Pereira-Terra, P.; Mahood, T.H.; Falk, L.; Day, C.A.; Iwasiow, B.M.; Zhu, F.; Mulhall, D.; Fraser, C.; et al. Prenatal microRNA miR-200b Therapy Improves Nitrofen-induced Pulmonary Hypoplasia Associated with Congenital Diaphragmatic Hernia. Ann. Surg. 2019, 269, 979–987. [Google Scholar] [CrossRef]
- Amonkar, G.M.; Wagner, R.; Bankoti, K.; Shui, J.E.; Ai, X.; Lerou, P.H. Primary culture of tracheal aspirate-derived human airway basal stem cells. STAR Protoc. 2022, 3, 101390. [Google Scholar] [CrossRef]
- Bankoti, K.; Wang, W.; Amonkar, G.M.; Xiong, L.; Shui, J.E.; Zhao, C.; Van, E.; Mwase, C.; Park, J.-A.; Mou, H.; et al. Airway Basal Stem Cells in COVID-19 Exhibit a Proinflammatory Signature and Impaired Mucocililary Differentiation. Am. J. Respir. Cell Mol. Biol. 2024, 70, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, W.; Bai, Y.; Amonkar, G.; Mou, H.; Olejnik, J.; Hume, A.J.; Mühlberger, E.; Fang, Y.; Que, J.; et al. Activation of STAT3-mediated ciliated cell survival protects against severe infection by respiratory syncytial virus. J. Clin. Investig. 2024, 134, e183978. [Google Scholar] [CrossRef]
- Lally, K.P.; Bagolan, P.; Hosie, S.; Lally, P.A.; Stewart, M.; Cotten, C.M.; Van Meurs, K.P.; Alexander, G. Corticosteroids for fetuses with congenital diaphragmatic hernia: Can we show benefit? J. Pediatr. Surg. 2006, 41, 668–674. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primer 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Rana, T.M. Generation of 3D lung organoids from human induced pluripotent stem cells for modeling of lung development and viral infection. Heliyon 2023, 9, e19601. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Huang, S.X.; De Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.-H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. eLife 2015, 4, e05098. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857.e6. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Chung, M.-I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L.M. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Armendariz, A.I.; Tata, P.R. Recent advances in lung organoid development and applications in disease modeling. J. Clin. Investig. 2023, 133, e170500. [Google Scholar] [CrossRef]
- Chiu, M.C.; Li, C.; Liu, X.; Yu, Y.; Huang, J.; Wan, Z.; Xiao, D.; Chu, H.; Cai, J.-P.; Zhou, B.; et al. A bipotential organoid model of respiratory epithelium recapitulates high infectivity of SARS-CoV-2 Omicron variant. Cell Discov. 2022, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Vedaie, M.; Roberts, D.A.; Thomas, D.C.; Villacorta-Martin, C.; Alysandratos, K.-D.; Hawkins, F.; Kotton, D.N. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. 2019, 14, 3303–3332. [Google Scholar] [CrossRef]
- Katsura, H.; Sontake, V.; Tata, A.; Kobayashi, Y.; Edwards, C.E.; Heaton, B.E.; Konkimalla, A.; Asakura, T.; Mikami, Y.; Fritch, E.J.; et al. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell 2020, 27, 890–904.e8. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Weiner, A.I.; Jackson, S.R.; Zhao, G.; Quansah, K.K.; Farshchian, J.N.; Neupauer, K.M.; Littauer, E.Q.; Paris, A.J.; Liberti, D.C.; Scott Worthen, G.; et al. Mesenchyme-free expansion and transplantation of adult alveolar progenitor cells: Steps toward cell-based regenerative therapies. Npj Regen. Med. 2019, 4, 17. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Y. Isolation and culture of mouse alveolar type II cells to study type II to type I cell differentiation. STAR Protoc. 2021, 2, 100241. [Google Scholar] [CrossRef]
- Dye, B.R.; Dedhia, P.H.; Miller, A.J.; Nagy, M.S.; White, E.S.; Shea, L.D.; Spence, J.R. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. eLife 2016, 5, e19732. [Google Scholar] [CrossRef]
- Heo, H.-R.; Hong, S.-H. Generation of macrophage containing alveolar organoids derived from human pluripotent stem cells for pulmonary fibrosis modeling and drug efficacy testing. Cell Biosci. 2021, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Eenjes, E.; Van Riet, S.; Kroon, A.A.; Slats, A.M.; Khedoe, P.P.S.J.; Boerema-de Munck, A.; Buscop-van Kempen, M.; Ninaber, D.K.; Reiss, I.K.M.; Clevers, H.; et al. Disease modeling following organoid-based expansion of airway epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L775–L786. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Batie, M.R.; Fan, Q.; Varisco, B.M. Mouse lung organoid responses to reduced, increased, and cyclic stretch. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 322, L162–L173. [Google Scholar] [CrossRef]
- Kunisaki, S.M.; Jiang, G.; Biancotti, J.C.; Ho, K.K.Y.; Dye, B.R.; Liu, A.P.; Spence, J.R. Human induced pluripotent stem cell-derived lung organoids in an ex vivo model of the congenital diaphragmatic hernia fetal lung. Stem Cells Transl. Med. 2021, 10, 98–114. [Google Scholar] [CrossRef]
- Nicholas, B.; Staples, K.J.; Moese, S.; Meldrum, E.; Ward, J.; Dennison, P.; Havelock, T.; Hinks, T.S.C.; Amer, K.; Woo, E.; et al. A Novel Lung Explant Model for the Ex Vivo Study of Efficacy and Mechanisms of Anti-Influenza Drugs. J. Immunol. 2015, 194, 6144–6154. [Google Scholar] [CrossRef] [PubMed]
- Koziol-White, C.; Gebski, E.; Cao, G.; Panettieri, R.A. Precision cut lung slices: An integrated ex vivo model for studying lung physiology, pharmacology, disease pathogenesis and drug discovery. Respir. Res. 2024, 25, 231. [Google Scholar] [CrossRef]
- Xia, J.-Y.; Zeng, Y.-F.; Wu, X.-J.; Xu, F. Short-term ex vivo tissue culture models help study human lung infections: A review. Medicine 2023, 102, e32589. [Google Scholar] [CrossRef]
- Fox, Z.D.; Jiang, G.; Ho, K.K.Y.; Walker, K.A.; Liu, A.P.; Kunisaki, S.M. Fetal lung transcriptome patterns in an ex vivo compression model of diaphragmatic hernia. J. Surg. Res. 2018, 231, 411–420. [Google Scholar] [CrossRef]
- Danzer, E.; Davey, M.G.; Kreiger, P.A.; Ruchelli, E.D.; Johnson, M.P.; Adzick, N.S.; Flake, A.W.; Hedrick, H.L. Fetal tracheal occlusion for severe congenital diaphragmatic hernia in humans: A morphometric study of lung parenchyma and muscularization of pulmonary arterioles. J. Pediatr. Surg. 2008, 43, 1767–1775. [Google Scholar] [CrossRef]
- Loukogeorgakis, S.P.; Michielin, F.; Al-juffali, N.; Jimenez, J.; Shibuya, S.; Nikolic, M.Z.; Elvassore, N.; Deprest, J.; De Coppi, P. Prenatal VEGF Nano-Delivery Reverses Congenital Diaphragmatic Hernia-Associated Pulmonary Abnormalities. Am. J. Respir. Crit. Care Med. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Miyake, Y.; Tse, W.H.; Wang, J.Q.; Leon, N.D.; Mourin, M.; Patel, D.; Aptekmann, A.O.; Yamataka, A.; Keijzer, R. The effect of tracheal occlusion in congenital diaphragmatic hernia in the nitrofen rat lung explant model. Pediatr. Surg. Int. 2022, 39, 61. [Google Scholar] [CrossRef]
- Negretti, N.M.; Son, Y.; Crooke, P.; Plosa, E.J.; Benjamin, J.T.; Jetter, C.S.; Bunn, C.; Mignemi, N.; Marini, J.; Hackett, A.N.; et al. Epithelial outgrowth through mesenchymal rings drives lung alveologenesis. JCI Insight 2025, 10, e187876. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, A.C.; Ahmed, A.M.; Roberts, J.D.; Kelleher, C.M. A Novel In Vitro Model to Study Alveologenesis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 459–469. [Google Scholar] [CrossRef]
- Antounians, L.; Tzanetakis, A.; Pellerito, O.; Catania, V.D.; Sulistyo, A.; Montalva, L.; McVey, M.J.; Zani, A. The Regenerative Potential of Amniotic Fluid Stem Cell Extracellular Vesicles: Lessons Learned by Comparing Different Isolation Techniques. Sci. Rep. 2019, 9, 1837. [Google Scholar] [CrossRef]
- Khalaj, K.; Antounians, L.; Figueira, R.L.; Post, M.; Zani, A. Autophagy Is Impaired in Fetal Hypoplastic Lungs and Rescued by Administration of Amniotic Fluid Stem Cell Extracellular Vesicles. Am. J. Respir. Crit. Care Med. 2022, 206, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Khalaj, K.; Figueira, R.L.; Antounians, L.; Gandhi, S.; Wales, M.; Montalva, L.; Biouss, G.; Zani, A. Treatment with Amniotic Fluid Stem Cell Extracellular Vesicles Promotes Fetal Lung Branching and Cell Differentiation at Canalicular and Saccular Stages in Experimental Pulmonary Hypoplasia Secondary to Congenital Diaphragmatic Hernia. Stem Cells Transl. Med. 2022, 11, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Lesage, F.; Thébaud, B. Mesenchymal Stromal Cell-Derived Extracellular Vesicles for Neonatal Lung Disease: Tiny Particles, Major Promise, Rigorous Requirements for Clinical Translation. Cells 2022, 11, 1176. [Google Scholar] [CrossRef]
- Lithopoulos, M.A.; Strueby, L.; O’Reilly, M.; Zhong, S.; Möbius, M.A.; Eaton, F.; Fung, M.; Hurskainen, M.; Cyr-Depauw, C.; Suen, C.; et al. Pulmonary and Neurologic Effects of Mesenchymal Stromal Cell Extracellular Vesicles in a Multifactorial Lung Injury Model. Am. J. Respir. Crit. Care Med. 2022, 205, 1186–1201. [Google Scholar] [CrossRef]
- Valsecchi, C.; Croce, S.; Lenta, E.; Acquafredda, G.; Comoli, P.; Avanzini, M.A. New therapeutic approaches in pediatric diseases: Mesenchymal stromal cell and mesenchymal stromal cell-derived extracellular vesicles as new drugs. Pharmacol. Res. 2023, 192, 106796. [Google Scholar] [CrossRef]
- Monroe, M.N.; Zhaorigetu, S.; Gupta, V.S.; Jin, D.; Givan, K.D.; Curylo, A.L.; Olson, S.D.; Cox, C.S.; Segura, A.; Buja, L.M.; et al. Extracellular vesicles influence the pulmonary arterial extracellular matrix in congenital diaphragmatic hernia. Pediatr. Pulmonol. 2020, 55, 2402–2411. [Google Scholar] [CrossRef]
- Moskowitzova, K.; Fauza, D.O. Transamniotic stem cell therapy (TRASCET): An emerging minimally invasive strategy for intrauterine stem cell delivery. Semin. Perinatol. 2023, 47, 151728. [Google Scholar] [CrossRef] [PubMed]
- Fauza, D.O. Transamniotic stem cell therapy: A novel strategy for the prenatal management of congenital anomalies. Pediatr. Res. 2018, 83, 241–248. [Google Scholar] [CrossRef]
- Chalphin, A.V.; Lazow, S.P.; Labuz, D.F.; Tracy, S.A.; Kycia, I.; Zurakowski, D.; Fauza, D.O. Transamniotic Stem Cell Therapy for Experimental Congenital Diaphragmatic Hernia: Structural, Transcriptional, and Cell Kinetics Analyses in the Nitrofen Model. Fetal Diagn. Ther. 2021, 48, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Labuz, D.F.; Whitlock, A.E.; Kycia, I.; Zurakowski, D.; Fauza, D.O. Early functional analysis on the pulmonary hemodynamic effects of Transamniotic Stem Cell Therapy (TRASCET) in the nitrofen model of congenital diaphragmatic hernia. J. Pediatr. Surg. 2023, 58, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Dalmer, T.R.A.; Clugston, R.D. Gene ontology enrichment analysis of congenital diaphragmatic hernia-associated genes. Pediatr. Res. 2019, 85, 13–19. [Google Scholar] [CrossRef]
- Vuckovic, A.; Herber-Jonat, S.; Flemmer, A.W.; Ruehl, I.M.; Votino, C.; Segers, V.; Benachi, A.; Martinovic, J.; Nowakowska, D.; Dzieniecka, M.; et al. Increased TGF-β: A drawback of tracheal occlusion in human and experimental congenital diaphragmatic hernia? Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L311–L327. [Google Scholar] [CrossRef]

{kind=link}
| Model (Developmental Stage + Sample Type) | Omics Method | Findings | Ref. | |
|---|---|---|---|---|
| Nitrofen Rat | Canalicular; lung tissue | LS-MS/MS | >200 altered proteins, inflammatory response. | [25] |
| Pseudoglandular; lung tissue | Tandem mass tag (TMT) | 79 altered proteins. Tight junction pathways, phospholipase D pathways and HIF-1 signaling. | [26] | |
| Late gestation; lung tissue | NMR-based metabolomics | Disruption in glycolysis, nucleotide metabolism, antioxidants. | [33] | |
| Human | Canalicular and alveolar; amniotic fluid | Proteomics + ELISA validation | 218 altered proteins. GP6 signaling. MSP-RON signaling. Cardiovascular development. | [28] |
| Alveolar; umbilical cord serum | LC-MS/MS | 98 altered proteins. Complement C1q/C5. Coagulation cascades. | [29] | |
| Canalicular, saccular and alveolar; amniotic fluid and tracheal aspirates | Metabolomics | Distinct CDH metabolomic profiles. | [34] | |
| Lamb (surgical) | Saccular; tracheal fluid | Proteomics | CDH suppressed (while TO promoted), cell proliferation and AKT-related signaling cascades. | [27] |
| Rabbit (surgical) | Late saccular; lung tissue | Metabolics + pathway analysis | Enrichment in ubiquinone, tyrosine, and terpenoid-quinone biosynthesis in CDH + TO. | [32] |
| Approach | Target | Outcome | Ref. | |
|---|---|---|---|---|
| Stem cell and Regenerative Therapies | AFSC-EVs | Developmental and inflammatory pathways | Autophagy activator expression restored. Promoted branching morphogenesis. Promoted epithelial and mesenchymal maturation. Reduced inflammation. | [82,83] |
| MSC-EVs | ECM-modifying enzymes | Partial restoration of vascular structural integrity. | [87] | |
| TRASCET | Pulmonary vasculature and epithelial development | Improvement of epithelial differentiation and vascular development. | [90,91] | |
| Precision Medicine and CDH Endotyping | Transcriptomic profiling | Biomarker identification | miR-200b expression correlates with improved outcomes. Elevated GATA4 marks more severe phenotypes. | [8,93] |
| Anti-NFkB Signaling | DEX and JSH-23 | Reduction of inflammation | Benefit seen in rat model, though not in human trials (discussed in Section 3.2). | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, O.; Dinwoodie, O.M.; Wagner, R.; Ai, X. Epithelial Dysfunction in Congenital Diaphragmatic Hernia: Mechanisms, Models and Emerging Therapies. Cells 2025, 14, 687. https://doi.org/10.3390/cells14100687
Aubert O, Dinwoodie OM, Wagner R, Ai X. Epithelial Dysfunction in Congenital Diaphragmatic Hernia: Mechanisms, Models and Emerging Therapies. Cells. 2025; 14(10):687. https://doi.org/10.3390/cells14100687
Chicago/Turabian StyleAubert, Ophelia, Olivia M. Dinwoodie, Richard Wagner, and Xingbin Ai. 2025. "Epithelial Dysfunction in Congenital Diaphragmatic Hernia: Mechanisms, Models and Emerging Therapies" Cells 14, no. 10: 687. https://doi.org/10.3390/cells14100687
APA StyleAubert, O., Dinwoodie, O. M., Wagner, R., & Ai, X. (2025). Epithelial Dysfunction in Congenital Diaphragmatic Hernia: Mechanisms, Models and Emerging Therapies. Cells, 14(10), 687. https://doi.org/10.3390/cells14100687