
Integrated Analysis of the microRNA–mRNA Network Predicts Potential Regulators of Atrial Fibrillation in Humans


 ,
,
Abstract
:1. Introduction
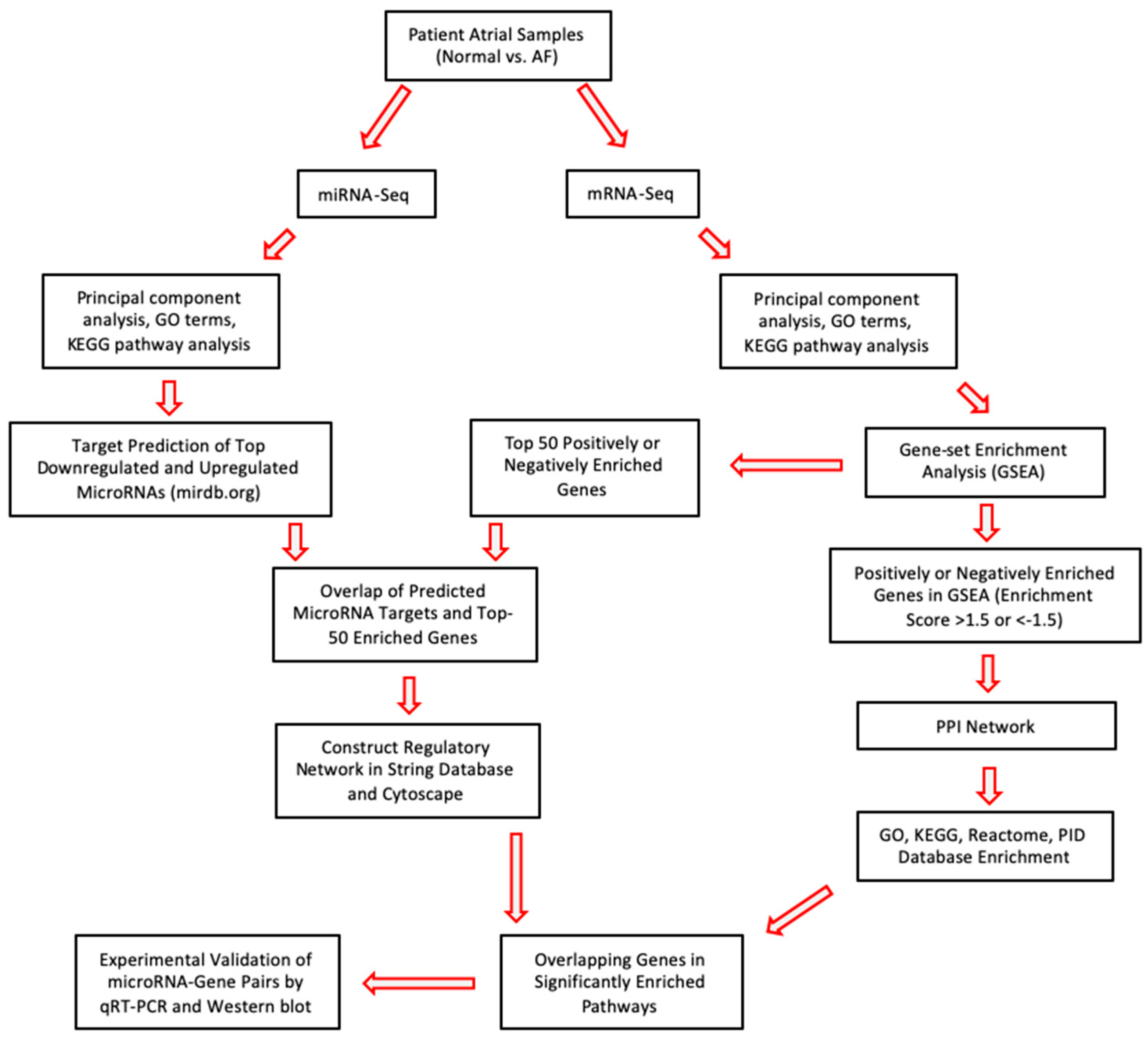
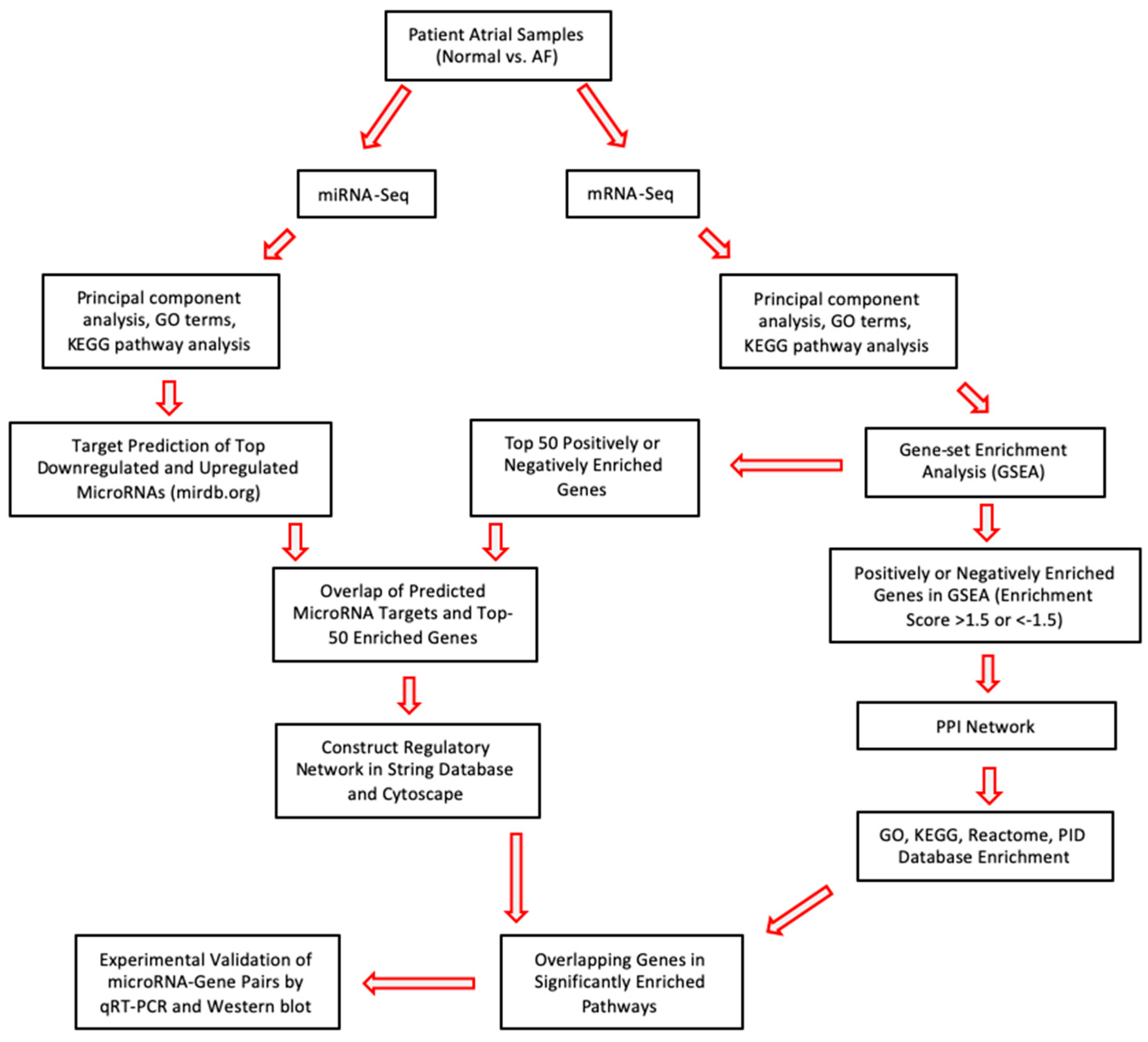
2. Results
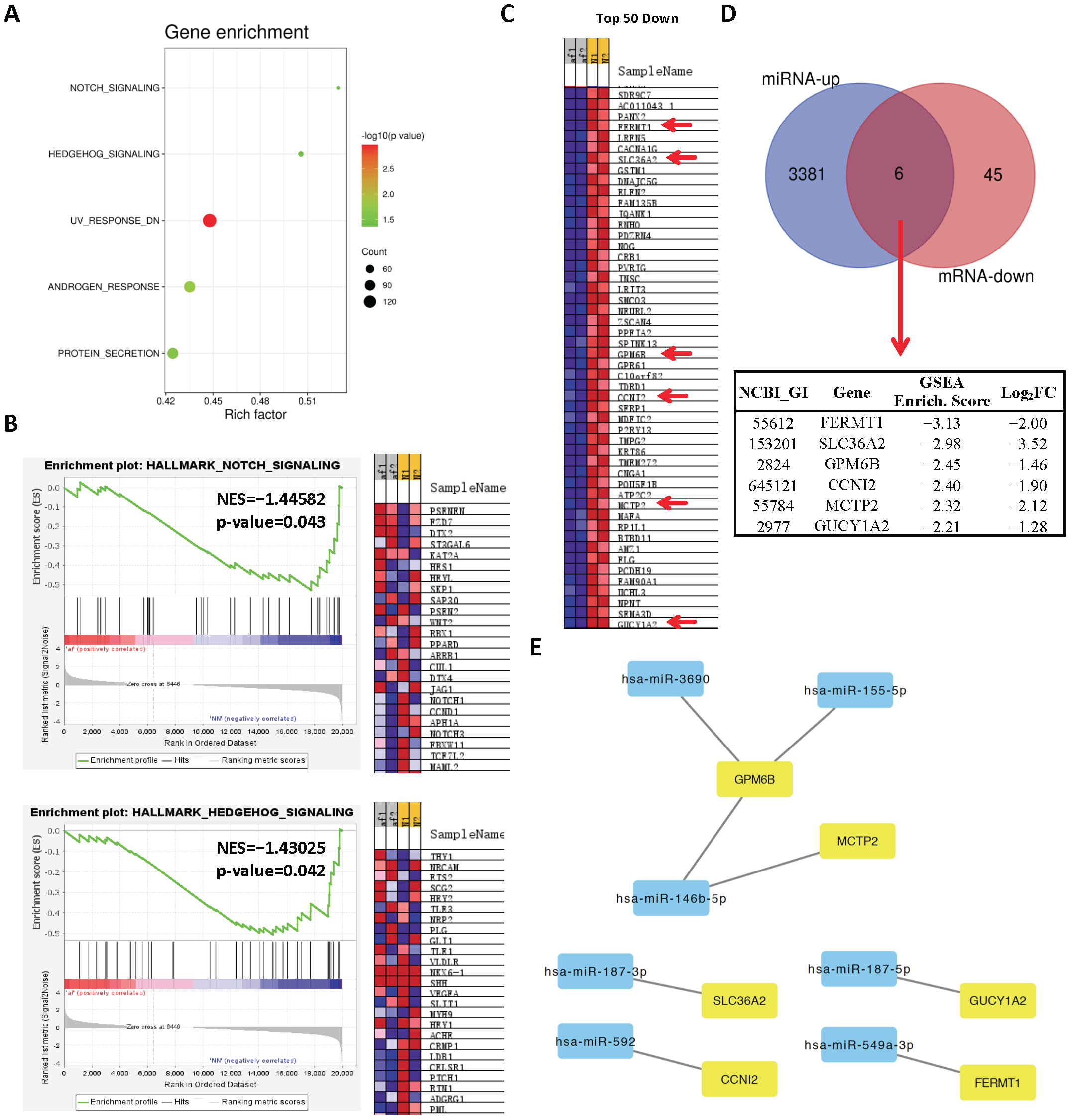
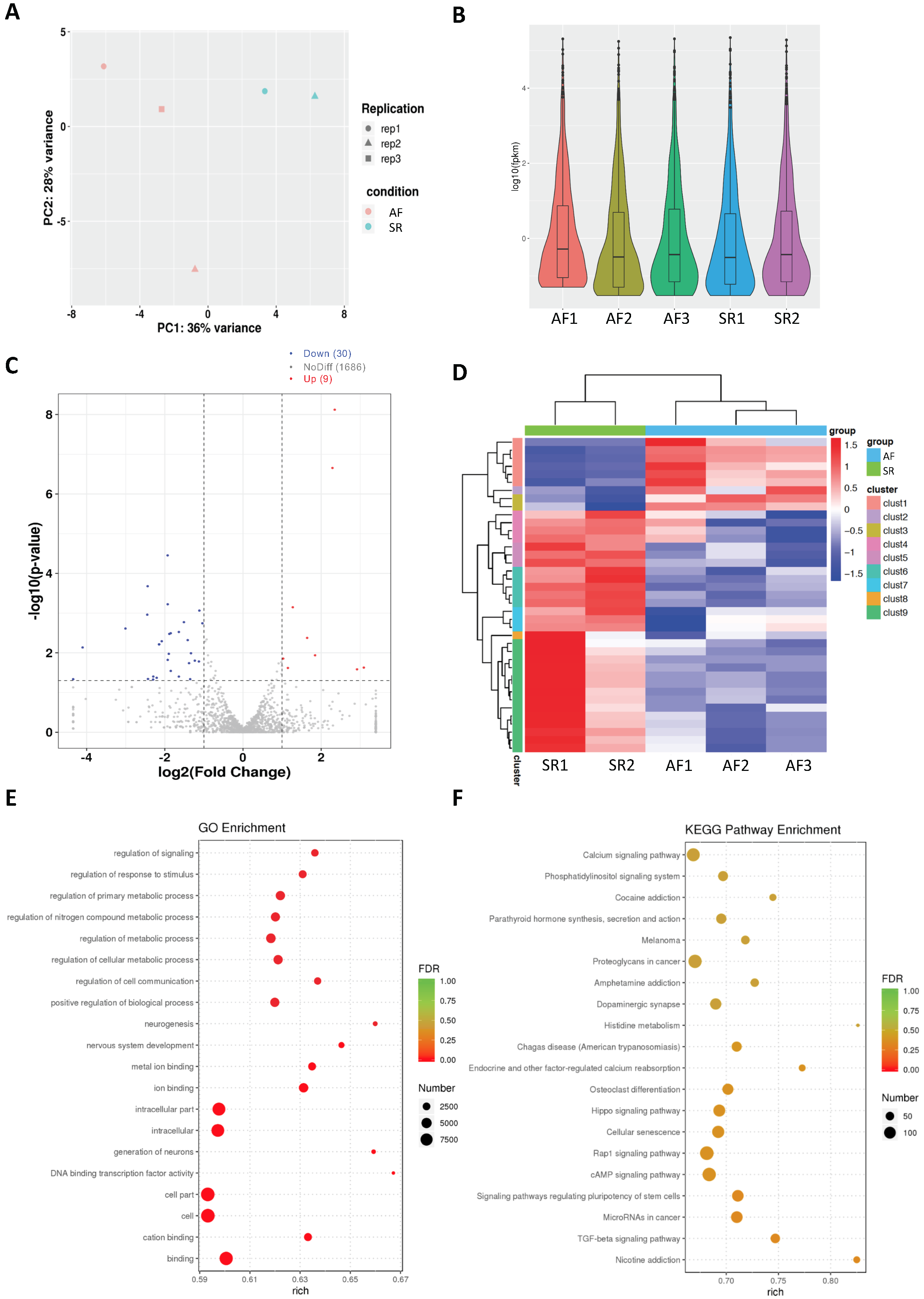
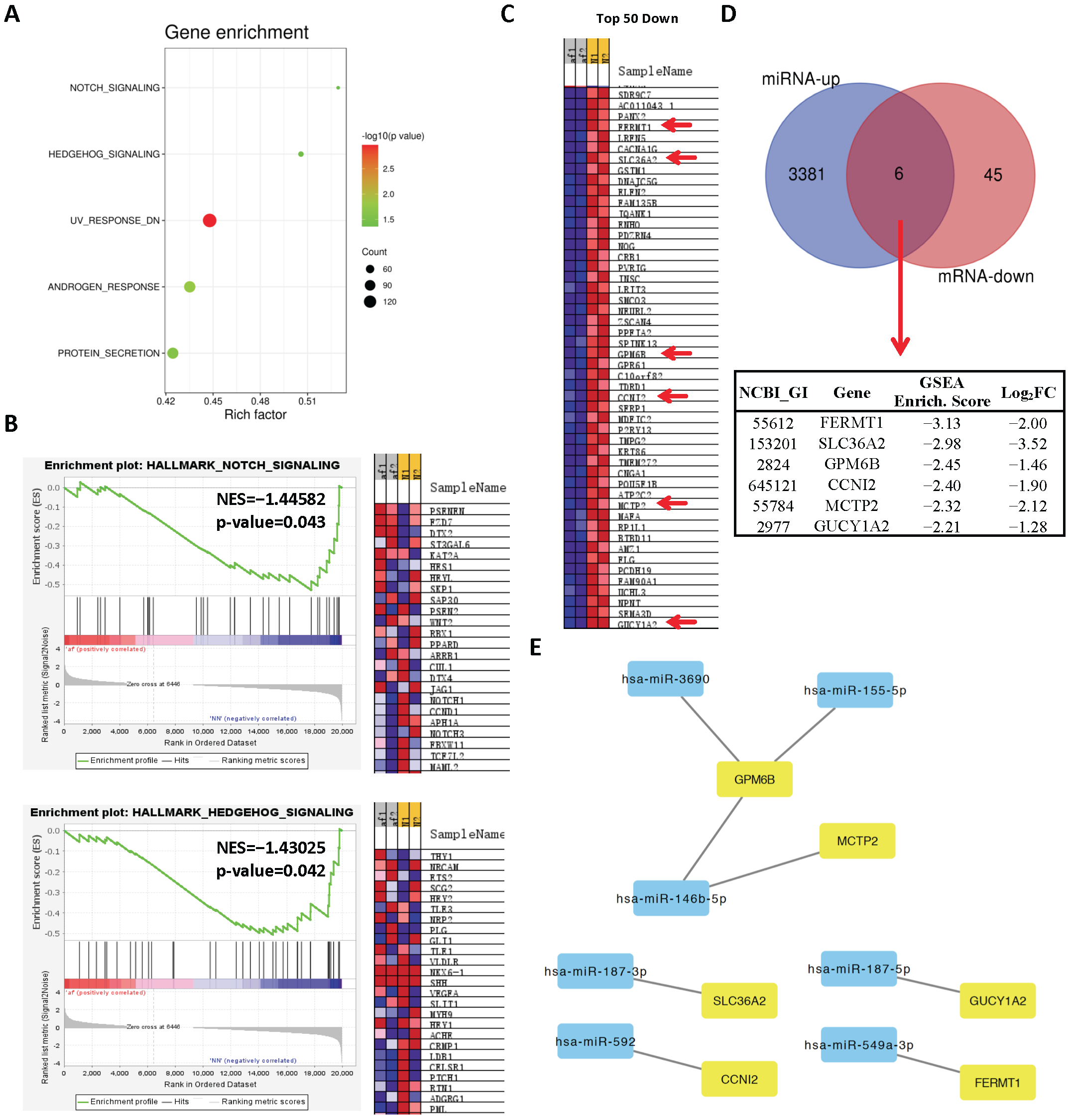
2.1. Analysis of Differentially Expressed miRs in Persistent AF
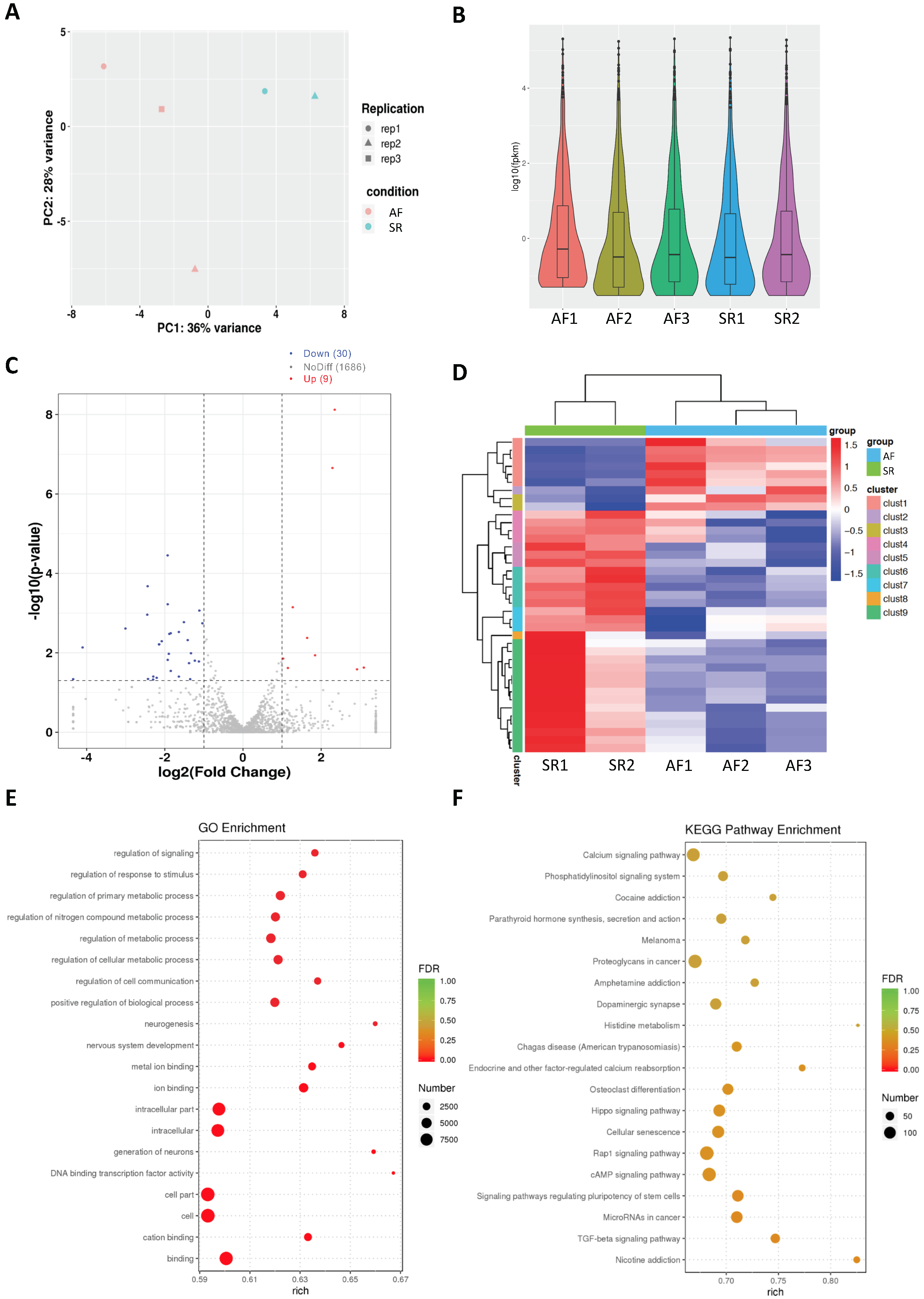
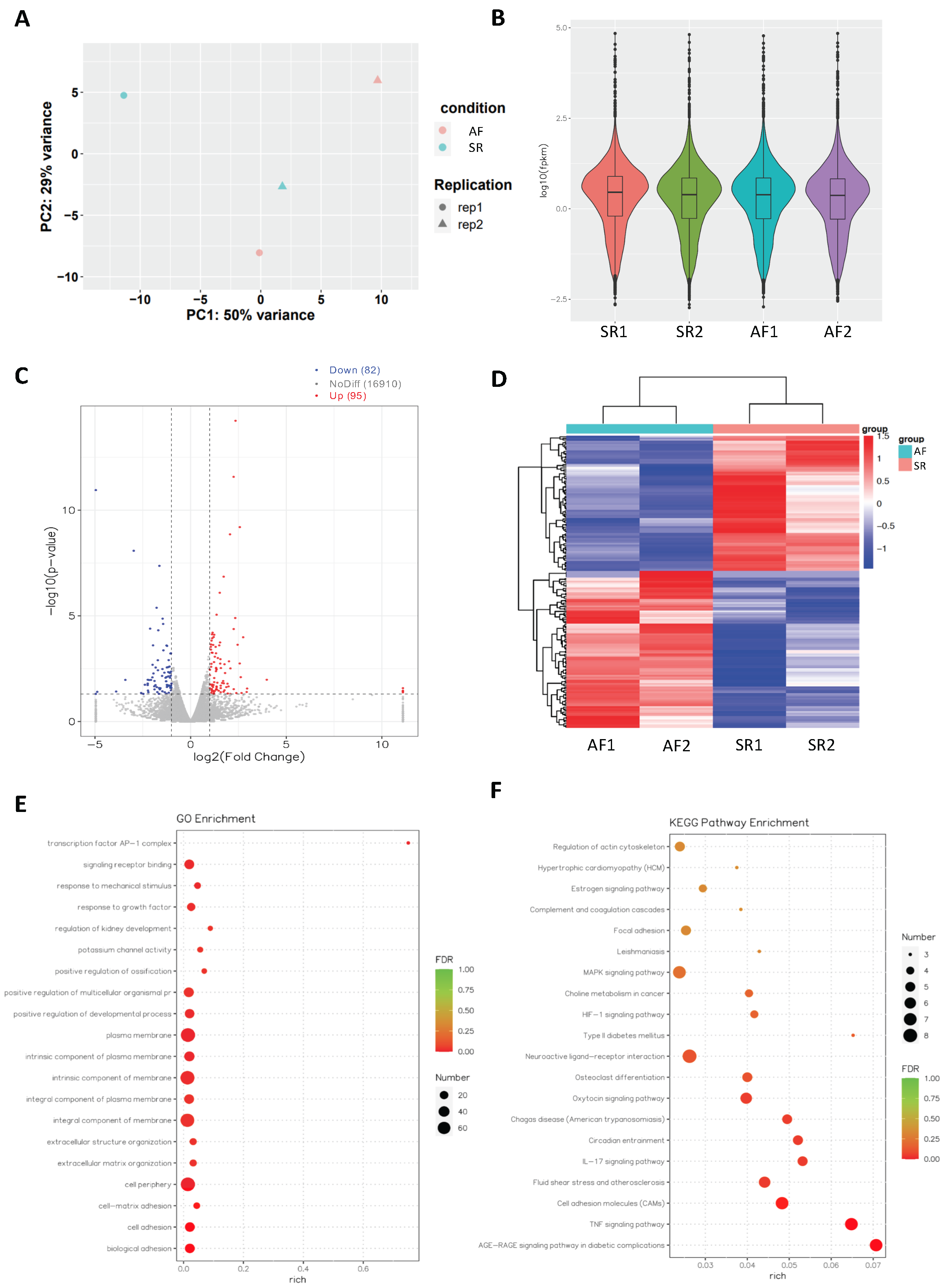
2.2. Analysis of Differentially Expressed mRNAs in Persistent AF
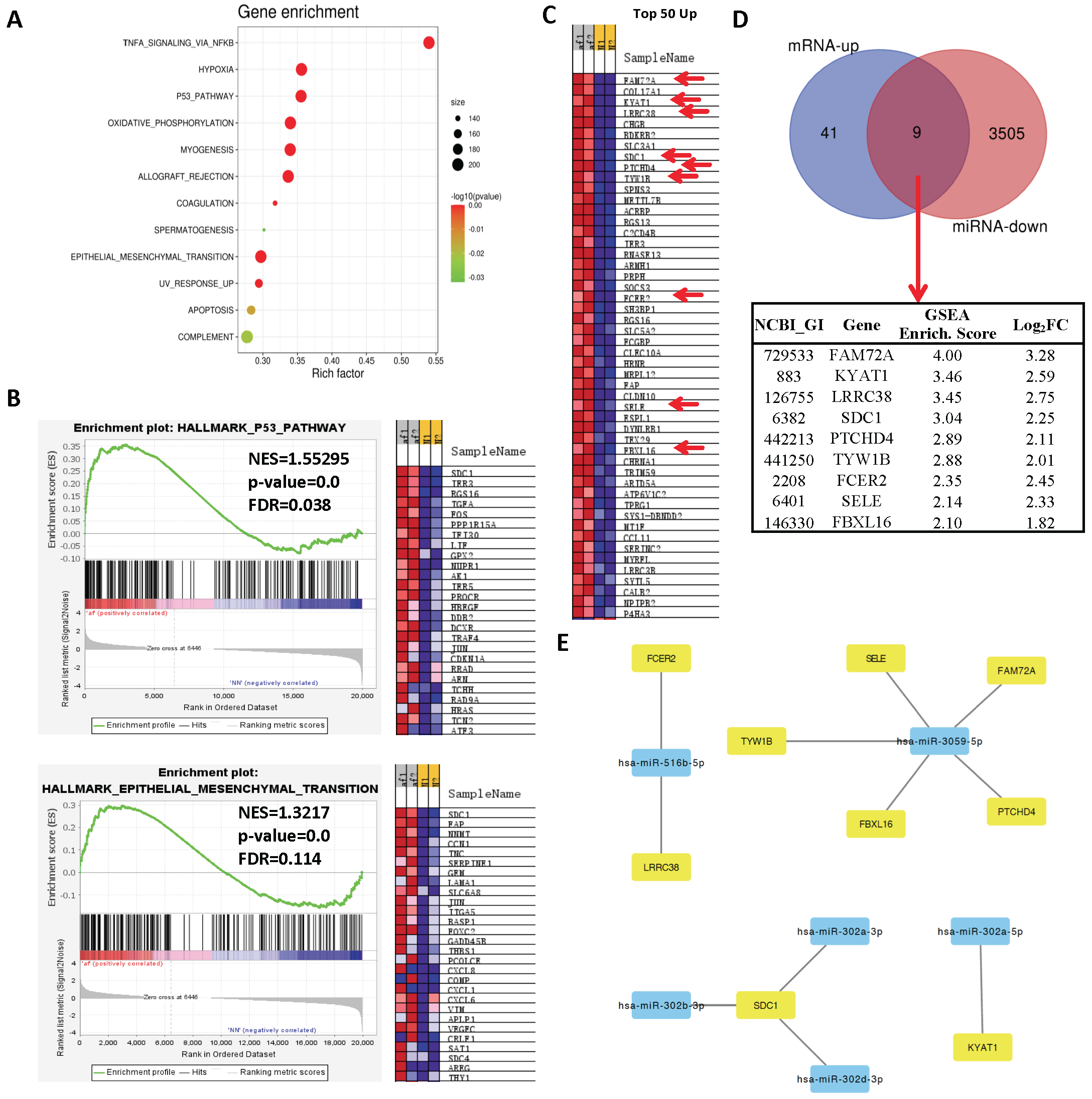
2.3. Network Analysis Downregulated miRs and Upregulated Genes and Selection of Candidate Pairs
2.4. Network Analysis of Upregulated miRs and Downregulated Genes and Selection of Candidate Pairs
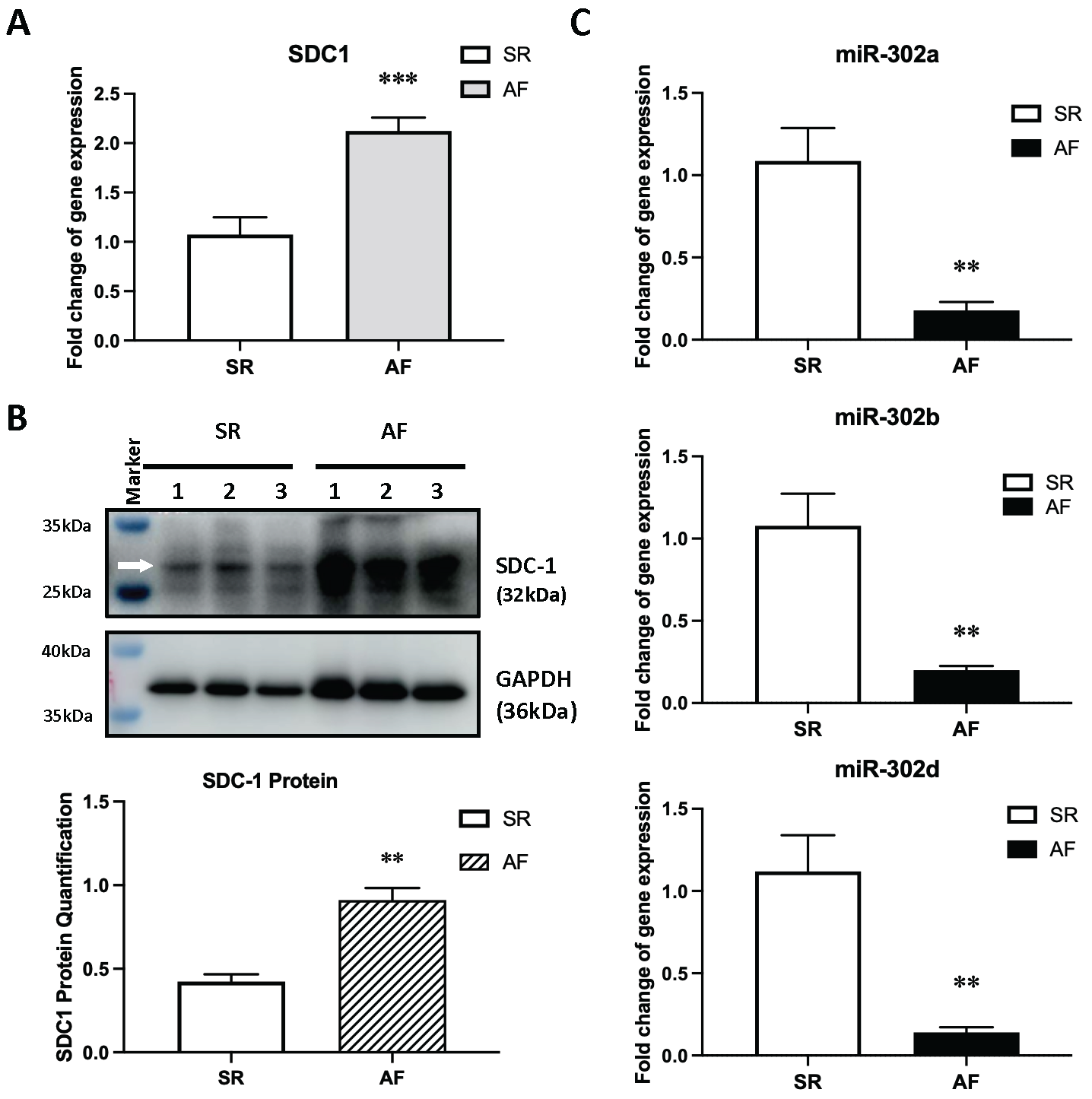
2.5. Selecting a Candidate Gene and Experimentally Validating Its Expression
2.6. MicroRNA-302b-3p Regulates SDC1 Expression In Vitro and May Involve in Atrial Fibrosis
3. Discussion
4. Materials and Methods
4.1. Collection of Human Atrial Samples and Ethical Approval Statement
4.2. Preparation of RNA Samples and mRNA Sequencing
4.3. mRNA-Seq Data Analysis
4.4. Preparation of RNA Samples and microRNA Sequencing
4.5. MicroRNA Sequencing Data Analysis
4.6. Cell Culture and Transfection
4.7. Gene Expression Analysis by qRT-PCR
4.8. Protein Expression Analysis with Western Blot
4.9. Luciferase Assay
4.10. Immunohistochemistry and Picrosirius Red Staining
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wyndham, C.R. Atrial fibrillation: The most common arrhythmia. Tex. Heart Inst. J. 2000, 27, 257–267. [Google Scholar] [PubMed]
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: Implications for management. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Dong, J.; Ma, C. Is Atrial Fibrillation a Preventable Disease? J. Am. Coll. Cardiol. 2017, 69, 1968–1982. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H., Jr.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, D.; Calvert, M.; Deeks, J.J.; Griffith, M.; Kirchhof, P.; Lip, G.Y.; Mehta, S.; Slinn, G.; Stanbury, M.; Steeds, R.P.; et al. A review of rate control in atrial fibrillation, and the rationale and protocol for the RATE-AF trial. BMJ Open 2017, 7, e015099. [Google Scholar] [CrossRef]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. J. Cardiothorac. Surg. 2016, 50, e1–e88. [Google Scholar] [CrossRef]
- Hijazi, Z.; Oldgren, J.; Lindback, J.; Alexander, J.H.; Connolly, S.J.; Eikelboom, J.W.; Ezekowitz, M.D.; Held, C.; Hylek, E.M.; Lopes, R.D.; et al. A biomarker-based risk score to predict death in patients with atrial fibrillation: The ABC (age, biomarkers, clinical history) death risk score. Eur. Heart J. 2018, 39, 477–485. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Yin, X.; Gona, P.; Larson, M.G.; Beiser, A.S.; McManus, D.D.; Newton-Cheh, C.; Lubitz, S.A.; Magnani, J.W.; Ellinor, P.T.; et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: A cohort study. Lancet 2015, 386, 154–162. [Google Scholar] [CrossRef]
- Kirchhof, P. The future of atrial fibrillation management: Integrated care and stratified therapy. Lancet 2017, 390, 1873–1887. [Google Scholar] [CrossRef]
- Chiang, C.E.; Wang, K.L.; Lip, G.Y. Stroke prevention in atrial fibrillation: An Asian perspective. Thromb. Haemost. 2014, 111, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef]
- Schmitt, N.; Grunnet, M.; Olesen, S.P. Cardiac potassium channel subtypes: New roles in repolarization and arrhythmia. Physiol. Rev. 2014, 94, 609–653. [Google Scholar] [CrossRef]
- Nattel, S.; Harada, M. Atrial remodeling and atrial fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef]
- de Vos, C.B.; Pisters, R.; Nieuwlaat, R.; Prins, M.H.; Tieleman, R.G.; Coelen, R.J.; van den Heijkant, A.C.; Allessie, M.A.; Crijns, H.J. Progression from paroxysmal to persistent atrial fibrillation clinical correlates and prognosis. J. Am. Coll. Cardiol. 2010, 55, 725–731. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, Y.H. MicroRNAs: Novel Regulators of the Heart. J. Thorac. Dis. 2010, 2, 43–47. [Google Scholar]
- Wang, Z.; Lu, Y.; Yang, B. MicroRNAs and atrial fibrillation: New fundamentals. Cardiovasc. Res. 2011, 89, 710–721. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Yang, D.; Wan, X.; Dennis, A.T.; Bektik, E.; Wang, Z.; Costa, M.G.S.; Fagnen, C.; Venien-Bryan, C.; Xu, X.; Gratz, D.H.; et al. MicroRNA Biophysically Modulates Cardiac Action Potential via Directly Binding to Ion Channel. Circulation 2021, 143, 1597–1613. [Google Scholar] [CrossRef]
- Han, Q.; Liu, D.; Convertino, M.; Wang, Z.; Jiang, C.; Kim, Y.H.; Luo, X.; Zhang, X.; Nackley, A.; Dokholyan, N.V.; et al. miRNA-711 Binds and Activates TRPA1 Extracellularly to Evoke Acute and Chronic Pruritus. Neuron 2018, 99, 449–463.e6. [Google Scholar] [CrossRef]
- Kwon, C.; Han, Z.; Olson, E.N.; Srivastava, D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 18986–18991. [Google Scholar] [CrossRef]
- Luo, X.; Pan, Z.; Shan, H.; Xiao, J.; Sun, X.; Wang, N.; Lin, H.; Xiao, L.; Maguy, A.; Qi, X.Y.; et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Investig. 2013, 123, 1939–1951. [Google Scholar] [CrossRef]
- Li, H.; Gao, F.; Wang, X.; Wu, J.; Lu, K.; Liu, M.; Li, R.; Ding, L.; Wang, R. Circulating microRNA-378 levels serve as a novel biomarker for assessing the severity of coronary stenosis in patients with coronary artery disease. Biosci. Rep. 2019, 39, BSR20182016. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Syndecan-1: A critical mediator in cardiac fibrosis. Hypertension 2010, 55, 233–235. [Google Scholar] [CrossRef]
- Lei, J.; Xue, S.; Wu, W.; Zhou, S.; Zhang, Y.; Yuan, G.; Wang, J. Sdc1 overexpression inhibits the p38 MAPK pathway and lessens fibrotic ventricular remodeling in MI rats. Inflammation 2013, 36, 603–615. [Google Scholar] [CrossRef]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wang, T.; Wang, B. Identification of microRNA-mRNA interactions in atrial fibrillation using microarray expression profiles and bioinformatics analysis. Mol. Med. Rep. 2016, 13, 4535–4540. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Zhang, M.; Voigt, N.; Alsina, K.M.; Jakob, H.; Martin, J.F.; Dobrev, D.; Wehrens, X.H.T.; Li, N. Identification of microRNA-mRNA dysregulations in paroxysmal atrial fibrillation. Int. J. Cardiol. 2015, 184, 190–197. [Google Scholar] [CrossRef]
- Jiang, S.; Guo, C.; Zhang, W.; Che, W.; Zhang, J.; Zhuang, S.; Wang, Y.; Zhang, Y.; Liu, B. The Integrative Regulatory Network of circRNA, microRNA, and mRNA in Atrial Fibrillation. Front. Genet. 2019, 10, 526. [Google Scholar] [CrossRef]
- Ruan, Z.B.; Wang, F.; Yu, Q.P.; Chen, G.C.; Zhu, L. Integrative analysis of the circRNA-miRNA regulatory network in atrial fibrillation. Sci. Rep. 2020, 10, 20451. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, Z.; Guo, Z.; Zheng, M.; Li, K.; Yang, X. Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. Biomed. Pharmacother. 2020, 121, 109634. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.K.; Arnett, D.K.; Assimes, T.L.; Basson, C.T.; Chakravarti, A.; Ellinor, P.T.; Engler, M.B.; Goldmuntz, E.; Herrington, D.M.; Hershberger, R.E.; et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: Update: A scientific statement from the American Heart Association. Circulation 2013, 128, 2813–2851. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Eckel, R.H.; McPherson, R. Triglycerides and heart disease: Still a hypothesis? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Corlin, L.; Liu, C.; Lin, H.; Leone, D.; Yang, Q.; Ngo, D.; Levy, D.; Cupples, L.A.; Gerszten, R.E.; Larson, M.G.; et al. Proteomic Signatures of Lifestyle Risk Factors for Cardiovascular Disease: A Cross-Sectional Analysis of the Plasma Proteome in the Framingham Heart Study. J. Am. Heart Assoc. 2021, 10, e018020. [Google Scholar] [CrossRef]
- Yoshida, M.; Takano, Y.; Sasaoka, T.; Izumi, T.; Kimura, A. E-selectin polymorphism associated with myocardial infarction causes enhanced leukocyte-endothelial interactions under flow conditions. Arterioscler Thromb. Vasc. Biol. 2003, 23, 783–788. [Google Scholar] [CrossRef]
- Liao, B.; Chen, K.; Xiong, W.; Chen, R.; Mai, A.; Xu, Z.; Dong, S. Relationship of SELE A561C and G98T Variants with the Susceptibility to CAD. Medicine 2016, 95, e1255. [Google Scholar] [CrossRef]
- Sandoval-Pinto, E.; Padilla-Gutierrez, J.R.; Valdes-Alvarado, E.; Garcia-Gonzalez, I.J.; Valdez-Haro, A.; Munoz-Valle, J.F.; Flores-Salinas, H.E.; Rivas, F.; Valle, Y. Assessment of the E-selectin rs5361 (561A>C) polymorphism and soluble protein concentration in acute coronary syndrome: Association with circulating levels. Mediat. Inflamm. 2014, 2014, 158367. [Google Scholar] [CrossRef]
- Li, Y.; Wei, Y.S.; Wang, M.; Zhang, P.A.; Jiang, X.J.; Huang, C.X. Association between the Ser128Arg variant of the E-selectin and risk of coronary artery disease in the central China. Int. J. Cardiol. 2005, 103, 33–36. [Google Scholar] [CrossRef]
- Freestone, B.; Chong, A.Y.; Nuttall, S.; Blann, A.D.; Lip, G.Y. Soluble E-selectin, von Willebrand factor, soluble thrombomodulin, and total body nitrate/nitrite product as indices of endothelial damage/dysfunction in paroxysmal, persistent, and permanent atrial fibrillation. Chest 2007, 132, 1253–1258. [Google Scholar] [CrossRef]
- Freestone, B.; Chong, A.Y.; Nuttall, S.; Lip, G.Y. Impaired flow mediated dilatation as evidence of endothelial dysfunction in chronic atrial fibrillation: Relationship to plasma von Willebrand factor and soluble E-selectin levels. Thromb. Res. 2008, 122, 85–90. [Google Scholar] [CrossRef]
- Kwon, M.J.; Jang, B.; Yi, J.Y.; Han, I.O.; Oh, E.S. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012, 586, 2207–2211. [Google Scholar] [CrossRef] [Green Version]
- Couchman, J.R. Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114. [Google Scholar] [CrossRef]
- Akl, M.R.; Nagpal, P.; Ayoub, N.M.; Prabhu, S.A.; Gliksman, M.; Tai, B.; Hatipoglu, A.; Goy, A.; Suh, K.S. Molecular and clinical profiles of syndecan-1 in solid and hematological cancer for prognosis and precision medicine. Oncotarget 2015, 6, 28693–28715. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Y.; Xie, Y.; Shen, J.; Xiang, M. Emerging roles of proteoglycans in cardiac remodeling. Int. J. Cardiol. 2019, 278, 192–198. [Google Scholar] [CrossRef]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef]
- Teng, Y.H.; Aquino, R.S.; Park, P.W. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef]
- Palaiologou, M.; Delladetsima, I.; Tiniakos, D. CD138 (syndecan-1) expression in health and disease. Histol. Histopathol. 2014, 29, 177–189. [Google Scholar] [CrossRef]
- Bertrand, J.; Bollmann, M. Soluble syndecans: Biomarkers for diseases and therapeutic options. Br. J. Pharmacol. 2019, 176, 67–81. [Google Scholar] [CrossRef]
- Chechi, K.; Voisine, P.; Mathieu, P.; Laplante, M.; Bonnet, S.; Picard, F.; Joubert, P.; Richard, D. Functional characterization of the Ucp1-associated oxidative phenotype of human epicardial adipose tissue. Sci. Rep. 2017, 7, 15566. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, H.; Chang, P.; Zhao, H.; Xia, Y.; Zhang, L.; Guo, X.; Huang, C.; Yan, F.; Hu, L.; et al. Glycoprotein M6B Interacts with TbetaRI to Activate TGF-beta-Smad2/3 Signaling and Promote Smooth Muscle Cell Differentiation. Stem. Cells 2019, 37, 190–201. [Google Scholar] [CrossRef]
- Park, J.H.; Pak, H.N.; Lee, S.; Park, H.K.; Seo, J.W.; Chang, B.C. The clinical significance of the atrial subendocardial smooth muscle layer and cardiac myofibroblasts in human atrial tissue with valvular atrial fibrillation. Cardiovasc. Pathol. 2013, 22, 58–64. [Google Scholar] [CrossRef]
- Shin, O.H.; Han, W.; Wang, Y.; Sudhof, T.C. Evolutionarily conserved multiple C2 domain proteins with two transmembrane regions (MCTPs) and unusual Ca2+ binding properties. J. Biol. Chem. 2005, 280, 1641–1651. [Google Scholar] [CrossRef]
- Lalani, S.R.; Ware, S.M.; Wang, X.; Zapata, G.; Tian, Q.; Franco, L.M.; Jiang, Z.; Bucasas, K.; Scott, D.A.; Campeau, P.M.; et al. MCTP2 is a dosage-sensitive gene required for cardiac outflow tract development. Hum. Mol. Genet. 2013, 22, 4339–4348. [Google Scholar] [CrossRef]
- Parker, L.E.; Landstrom, A.P. Genetic Etiology of Left-Sided Obstructive Heart Lesions: A Story in Development. J. Am. Heart Assoc. 2021, 10, e019006. [Google Scholar] [CrossRef]
- Verweij, N.; van de Vegte, Y.J.; van der Harst, P. Genetic study links components of the autonomous nervous system to heart-rate profile during exercise. Nat. Commun. 2018, 9, 898. [Google Scholar] [CrossRef]
- van de Vegte, Y.J.; Tegegne, B.S.; Verweij, N.; Snieder, H.; van der Harst, P. Genetics and the heart rate response to exercise. Cell Mol. Life Sci. 2019, 76, 2391–2409. [Google Scholar] [CrossRef]
- Leineweber, K.; Moosmang, S.; Paulson, D. Genetics of NO Deficiency. Am. J. Cardiol. 2017, 120, S80–S88. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, Q.; Ma, X.; Bai, S.; Chen, P.; Zhao, Y.; Bai, C.; Liu, Y.; Liu, K.; Xin, M.; et al. MicroRNA-146b-5p promotes atrial fibrosis in atrial fibrillation by repressing TIMP4. J. Cell Mol. Med. 2021, 25, 10543–10553. [Google Scholar] [CrossRef]
- Schellings, M.W.; Vanhoutte, D.; van Almen, G.C.; Swinnen, M.; Leenders, J.J.; Kubben, N.; van Leeuwen, R.E.; Hofstra, L.; Heymans, S.; Pinto, Y.M. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 2010, 55, 249–256. [Google Scholar] [CrossRef]
- Tromp, J.; van der Pol, A.; Klip, I.T.; de Boer, R.A.; Jaarsma, T.; van Gilst, W.H.; Voors, A.A.; van Veldhuisen, D.J.; van der Meer, P. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ. Heart Fail. 2014, 7, 457–462. [Google Scholar] [CrossRef]
- Morita, H.; Komuro, I. Heart Failure as an Aging-Related Phenotype. Int. Heart J. 2018, 59, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Men, H.; Cai, H.; Cheng, Q.; Zhou, W.; Wang, X.; Huang, S.; Zheng, Y.; Cai, L. The regulatory roles of p53 in cardiovascular health and disease. Cell Mol. Life Sci. 2021, 78, 2001–2018. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Krainock, M.; Toubat, O.; Danopoulos, S.; Beckham, A.; Warburton, D.; Kim, R. Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease. J. Clin. Med. 2016, 5, 27. [Google Scholar] [CrossRef]
- Jesel, L.; Abbas, M.; Park, S.H.; Matsushita, K.; Kindo, M.; Hasan, H.; Auger, C.; Sato, C.; Ohlmann, P.; Mazzucotelli, J.P.; et al. Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression. J. Clin. Med. 2019, 9, 36. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef]
- George, B.; Rivera Rolon, M.D.M.; Mohit, S.; Stevenson, H.L. Epithelial to mesenchymal transition in endomyocardial biopsies from orthotopic heart transplant recipients. BMJ Case Rep. 2019, 12, e229175. [Google Scholar] [CrossRef]
- Guo, T.; Yu, W.; Lv, S.; Zhang, C.; Tian, Y. MiR-302a inhibits the tumorigenicity of ovarian cancer cells by suppression of SDC1. Int. J. Clin. Exp. Pathol. 2015, 8, 4869–4880. [Google Scholar]
- Zhang, J.; Cao, Z.; Ding, X.; Wei, X.; Zhang, X.; Hou, J.; Ouyang, J. The lncRNA XIST regulates the tumorigenicity of renal cell carcinoma cells via the miR-302c/SDC1 axis. Int. J. Clin. Exp. Pathol. 2017, 10, 7481–7491. [Google Scholar]
- Sattayaprasert, P.; Vasireddi, S.K.; Bektik, E.; Jeon, O.; Hajjiri, M.; Mackall, J.A.; Moravec, C.S.; Alsberg, E.; Fu, J.; Laurita, K.R. Human Cardiac Mesenchymal Stem Cells Remodel in Disease and Can Regulate Arrhythmia Substrates. Circ. Arrhythm. Electrophysiol. 2020, 13, e008740. [Google Scholar] [CrossRef]
- Miftode, R.S.; Serban, I.L.; Timpau, A.S.; Miftode, I.L.; Ion, A.; Buburuz, A.M.; Costache, A.D.; Costache, I.-I. Syndecan-1: A Review on Its Role in Heart Failure and Chronic Liver Disease Patients’ Assessment. Cardiol. Res. Pract. 2019, 2019, 4750580. [Google Scholar] [CrossRef]
- Zhang, H.; Tian, L.; Shen, M.; Tu, C.; Wu, H.; Gu, M.; Paik, D.T.; Wu, J.C. Generation of Quiescent Cardiac Fibroblasts from Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ. Res. 2019, 125, 552–566. [Google Scholar] [CrossRef]
- Bektik, E.; Cowan, D.B.; Wang, D.Z. Long Non-Coding RNAs in Atrial Fibrillation: Pluripotent Stem Cell-Derived Cardiomyocytes as a Model System. Int. J. Mol. Sci. 2020, 21, 5424. [Google Scholar] [CrossRef]
- Blazeski, A.; Zhu, R.; Hunter, D.W.; Weinberg, S.H.; Boheler, K.R.; Zambidis, E.T.; Tung, L. Electrophysiological and contractile function of cardiomyocytes derived from human embryonic stem cells. Prog. Biophys. Mol. Biol. 2012, 110, 178–195. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Han, J.; Li, Y.; Xie, C.; Xie, L.; Shi, J.; Zhang, J.; Yang, B.; Chen, D.; et al. Integrated analysis of microRNA and mRNA expression profiles in the left atrium of patients with nonvalvular paroxysmal atrial fibrillation: Role of miR-146b-5p in atrial fibrosis. Heart Rhythm 2015, 12, 1018–1026. [Google Scholar] [CrossRef]
- Wang, J.; Ye, Q.; Bai, S.; Chen, P.; Zhao, Y.; Ma, X.; Bai, C.; Liu, Y.; Xin, M.; Zeng, C.; et al. Inhibiting microRNA-155 attenuates atrial fibrillation by targeting CACNA1C. J. Mol. Cell Cardiol. 2021, 155, 58–65. [Google Scholar] [CrossRef]
- Cao, Y.; Cui, L. Identifying the key microRNAs implicated in atrial fibrillation. Anatol. J. Cardiol. 2021, 25, 429–436. [Google Scholar] [CrossRef]
- Tran, K.V.; Majka, J.; Sanghai, S.; Sardana, M.; Lessard, D.; Milstone, Z.; Tanriverdi, K.; Freedman, J.E.; Fitzgibbons, T.P.; McManus, D. Micro-RNAs Are Related to Epicardial Adipose Tissue in Participants with Atrial Fibrillation: Data from the MiRhythm Study. Front. Cardiovasc. Med. 2019, 6, 115. [Google Scholar] [CrossRef]
- Freestone, B.; Lip, G.Y.; Chong, A.Y.; Nadar, S.; Lee, K.W.; Blann, A.D. Circulating endothelial cells in atrial fibrillation with and without acute cardiovascular disease. Thromb. Haemost. 2005, 94, 702–706. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef]
- Henrich Lobo, R. Enzymatic Immunohistochemistry. Methods Mol. Biol. 2022, 2422, 125–129. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Age | Gender | Diagnosis | History of AF | Surgery | Site of Sampling |
|---|---|---|---|---|---|---|
| Normal | 60 | Female | Congenital heart disease, atrial septal defect | No AF | Atrial septal repair | Right atrium |
| Normal | 66 | Male | Tricuspid vegetations | No AF | Excision of vegetations | Right atrium |
| AF | 64 | Female | Rheumatic heart disease, mitral stenosis | Persistent AF | Mitral valve replacement | Right atrium |
| AF | 60 | Male | Rheumatic heart disease, mitral stenosis | Persistent AF | Mitral valve replacement | Right atrium |
| AF | 57 | Female | Rheumatic heart disease, mitral stenosis | Persistent AF | Mitral valve replacement | Right atrium |
| microRNA ID | log2(FoldChange) | p-Value |
|---|---|---|
| hsa-miR-302b-3p * | −1.927993099 | 3.52446 × 10−5 |
| hsa-miR-3059-5p * | −2.439099768 | 0.000210321 |
| hsa-miR-302d-3p * | −1.925669264 | 0.000598812 |
| hsa-miR-378d * | −1.1161761 | 0.000863964 |
| hsa-miR-516a-5p * | −2.449280109 | 0.001097802 |
| hsa-miR-302a-5p * | −1.511297201 | 0.001691902 |
| hsa-miR-378i * | −1.038634253 | 0.00180456 |
| hsa-miR-518c-3p * | −3.007366398 | 0.002448495 |
| hsa-miR-516b-5p * | −1.640951948 | 0.002989037 |
| hsa-miR-5708 * | −1.848531157 | 0.003210251 |
| hsa-miR-302a-3p * | −1.884247241 | 0.003340718 |
| hsa-miR-378e | −1.41425312 | 0.004785214 |
| hsa-miR-302c-3p | −2.078986595 | 0.005110454 |
| hsa-miR-517a-3p | −2.146095895 | 0.006125071 |
| hsa-miR-517b-3p | −2.146095895 | 0.006125071 |
| hsa-miR-526b-5p | −4.103564723 | 0.00733209 |
| hsa-miR-371a-3p | −1.330621285 | 0.01025266 |
| hsa-miR-520g-3p | −1.89787358 | 0.01062743 |
| hsa-miR-585-3p | −1.925975099 | 0.01506384 |
| hsa-miR-372-3p | −1.232670652 | 0.015762249 |
| hsa-miR-4662a-5p | −1.128104683 | 0.016532088 |
| hsa-miR-378h | −1.380015786 | 0.018242992 |
| hsa-miR-520c-3p | −1.84779246 | 0.028579751 |
| hsa-miR-523-3p | −2.295223982 | 0.039597527 |
| hsa-miR-520a-3p | −1.640624273 | 0.03967688 |
| hsa-miR-519a-5p | −2.207467024 | 0.04264619 |
| hsa-miR-1323 | −2.430852745 | 0.045529514 |
| hsa-miR-520f-3p | −1.351910815 | 0.046170691 |
| hsa-miR-520b-3p | −2.29949029 | 0.047158202 |
| hsa-miR-1323 | −2.430852745 | 0.045529514 |
| hsa-miR-146b-5p * | 2.347289373 | 7.56934 × 10−9 |
| hsa-miR-146b-3p * | 2.289327335 | 2.21466 × 10−7 |
| hsa-miR-155-5p * | 1.274620071 | 0.00071305 |
| hsa-miR-3690 * | 1.641664997 | 0.004215924 |
| hsa-miR-187-5p * | 1.844437031 | 0.011552148 |
| hsa-miR-187-3p * | 1.028719714 | 0.013972754 |
| hsa-miR-592 * | 3.091036819 | 0.023513587 |
| hsa-miR-212-3p * | 1.148408853 | 0.024049925 |
| hsa-miR-549a-3p * | 2.91790048 | 0.026012753 |
| Gene Name | NCBI_GI | Rank among Top 50 Upregulated Genes | miRNA ID | Rank among Downregulated miRs | miRDB Target Score (Out of 100) |
|---|---|---|---|---|---|
| FAM72A | 729533 | 1 | hsa-miR-3059-5p | 2 | 67 |
| KYAT1 | 883 | 3 | hsa-miR-302a-5p | 6 | 51 |
| LRRC38 | 126755 | 4 | hsa-miR-516b-5p | 9 | 50 |
| SDC1 | 6382 | 8 | hsa-miR-302b-3p | 1 | 96 |
| hsa-miR-302d-3p | 3 | 96 | |||
| hsa-miR-302a-3p | 11 | 96 | |||
| PTCHD4 | 442213 | 9 | hsa-miR-3059-5p | 2 | 93 |
| TYW1B | 441250 | 10 | hsa-miR-3059-5p | 2 | 77 |
| FCER2 | 2208 | 21 | hsa-miR-516b-5p | 9 | 82 |
| SELE | 6401 | 31 | hsa-miR-3059-5p | 2 | 65 |
| FBXL16 | 146330 | 35 | has-miR-3059-5p | 2 | 63 |
| Gene Name | NCBI_GI | Rank among Top 50 Downregulated mRNAs | miRNA ID | Rank among Upregulated miRs | miRDB Target Score (Out of 100) |
|---|---|---|---|---|---|
| FERMT1 | 55612 | 4 | hsa-miR-549a-3p | 9 | 67 |
| SLC36A2 | 153201 | 7 | hsa-miR-187-3p | 6 | 57 |
| GPM6B | 2824 | 25 | hsa-miR-146b-5p | 1 | 86 |
| hsa-miR-155-5p | 3 | 82 | |||
| hsa-miR-3690 | 4 | 80 | |||
| CCNI2 | 645121 | 29 | hsa-miR-592 | 7 | 57 |
| MCTP2 | 55784 | 39 | hsa-miR-146b-5p | 1 | 78 |
| GUCY1A2 | 2977 | 50 | hsa-miR-187-5p | 5 | 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Bektik, E.; Sakon, P.; Wang, X.; Huang, S.; Meng, X.; Chen, M.; Han, W.; Chen, J.; Wang, Y.; et al. Integrated Analysis of the microRNA–mRNA Network Predicts Potential Regulators of Atrial Fibrillation in Humans. Cells 2022, 11, 2629. https://doi.org/10.3390/cells11172629
Wang R, Bektik E, Sakon P, Wang X, Huang S, Meng X, Chen M, Han W, Chen J, Wang Y, et al. Integrated Analysis of the microRNA–mRNA Network Predicts Potential Regulators of Atrial Fibrillation in Humans. Cells. 2022; 11(17):2629. https://doi.org/10.3390/cells11172629
Chicago/Turabian StyleWang, Rong, Emre Bektik, Phraew Sakon, Xiaowei Wang, Shanying Huang, Xiangbin Meng, Mo Chen, Wenqiang Han, Jie Chen, Yanhong Wang, and et al. 2022. "Integrated Analysis of the microRNA–mRNA Network Predicts Potential Regulators of Atrial Fibrillation in Humans" Cells 11, no. 17: 2629. https://doi.org/10.3390/cells11172629
APA StyleWang, R., Bektik, E., Sakon, P., Wang, X., Huang, S., Meng, X., Chen, M., Han, W., Chen, J., Wang, Y., & Zhong, J. (2022). Integrated Analysis of the microRNA–mRNA Network Predicts Potential Regulators of Atrial Fibrillation in Humans. Cells, 11(17), 2629. https://doi.org/10.3390/cells11172629