

Tofogliflozin Delays Portal Hypertension and Hepatic Fibrosis by Inhibiting Sinusoidal Capillarization in Cirrhotic Rats

 , , , and
, , , and 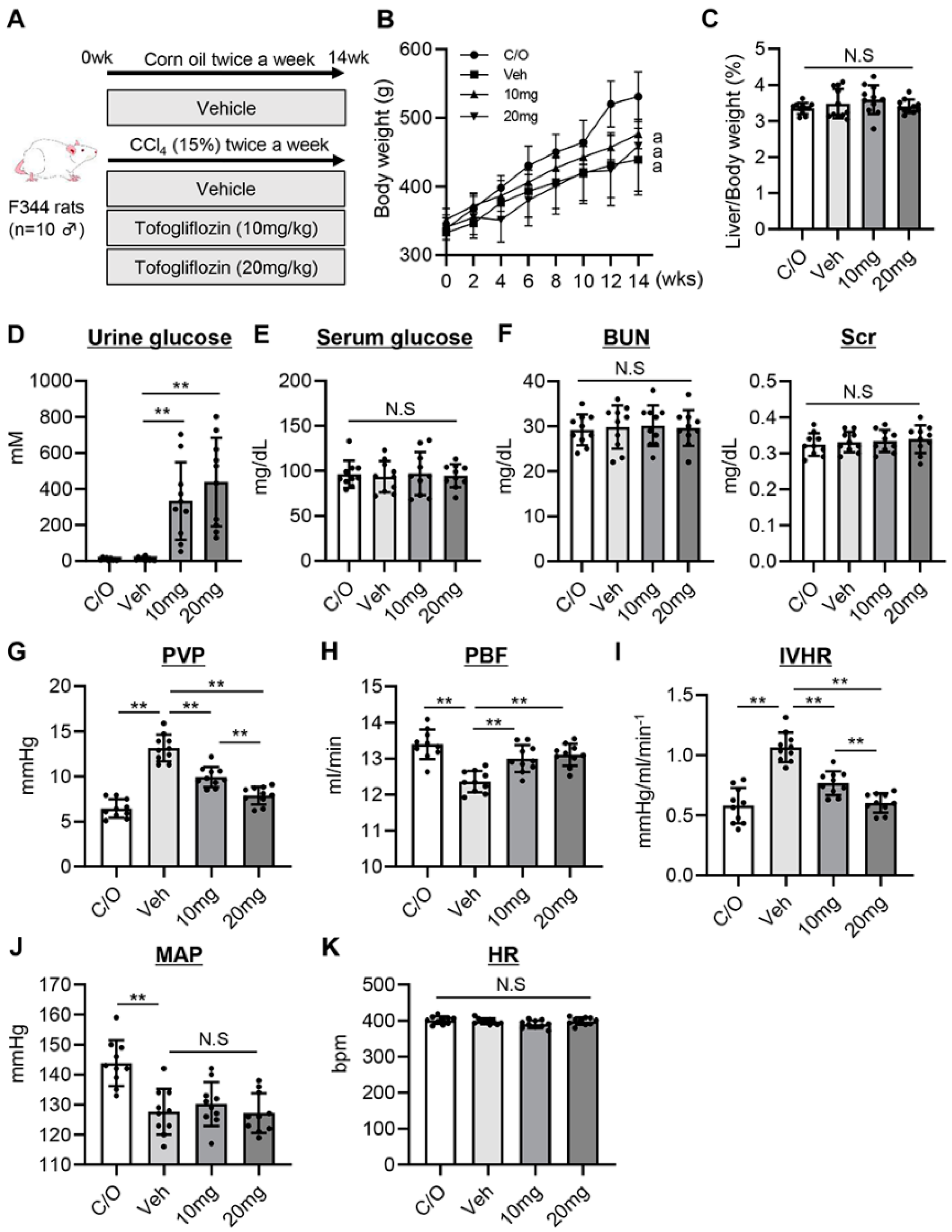{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Compounds
2.2. In Vivo Experimental Protocol
2.3. In Vivo Hemodynamic Evaluation
2.4. Cell Culture
2.5. Statistical Analyzes
3. Results
3.1. Tofogliflozin Prevents Portal Hypertension Development in CCl4-Treated Rats
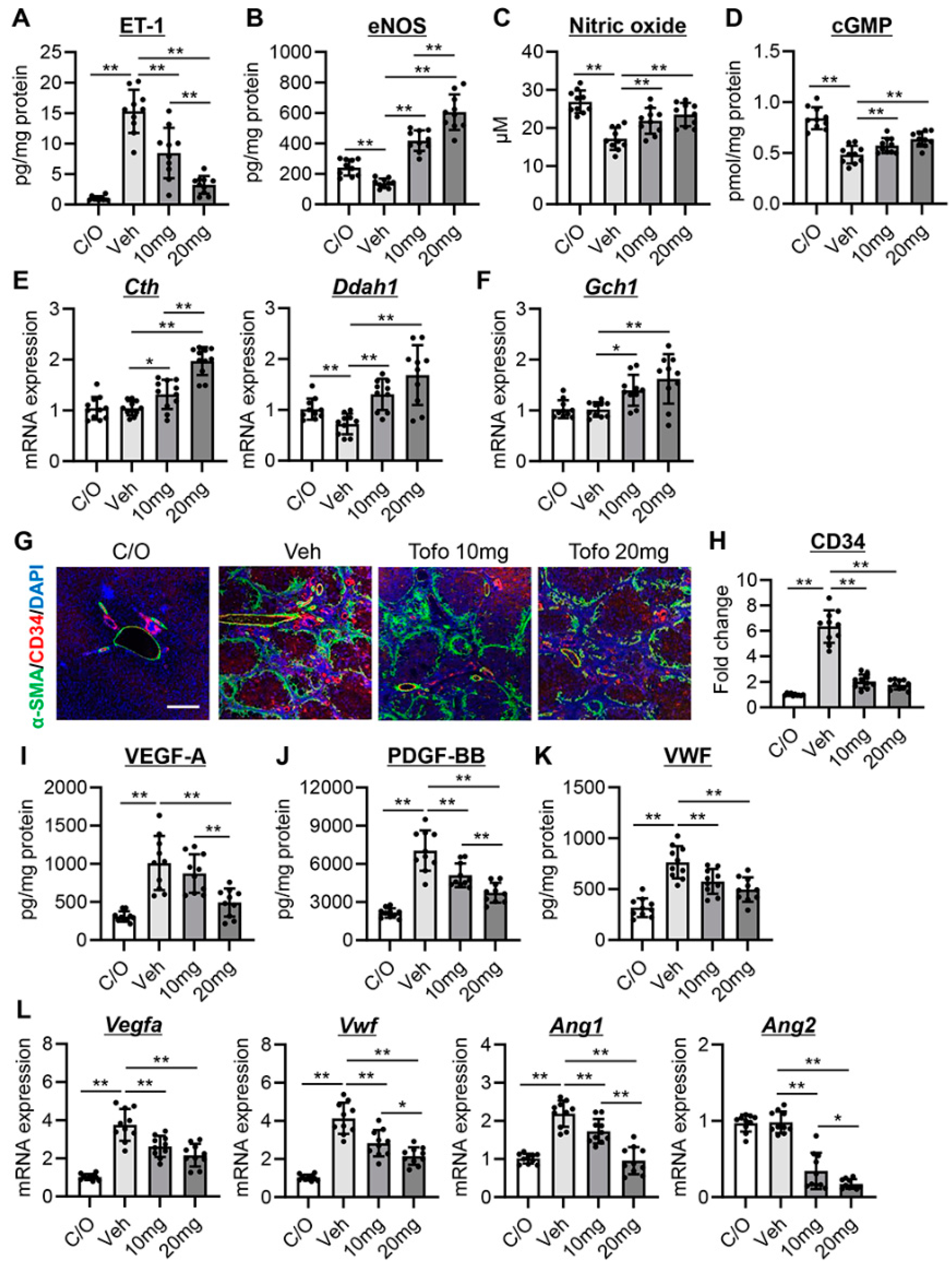
3.2. Tofogliflozin Attenuates Intrahepatic Vasoconstriction and Vascular Resistance in CCl4-Treated Rats
3.3. Tofogliflozin Inhibits Sinusoidal Capillarization and Remodeling in CCl4-Treated Rats
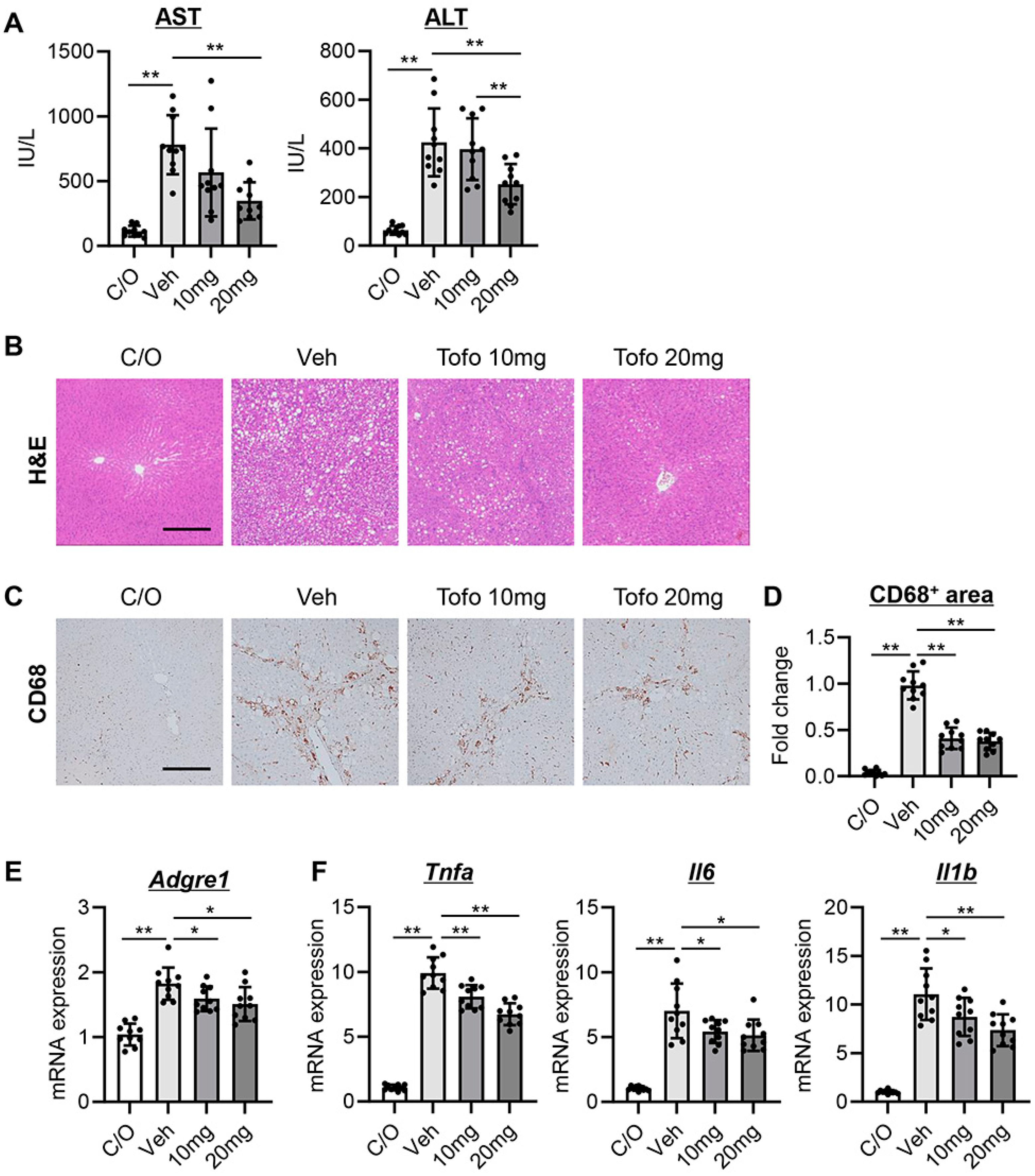
3.4. Tofogliflozin Suppresses Hepatic Necroinflammation and Macrophage Infiltration in CCl4-Treated Rats
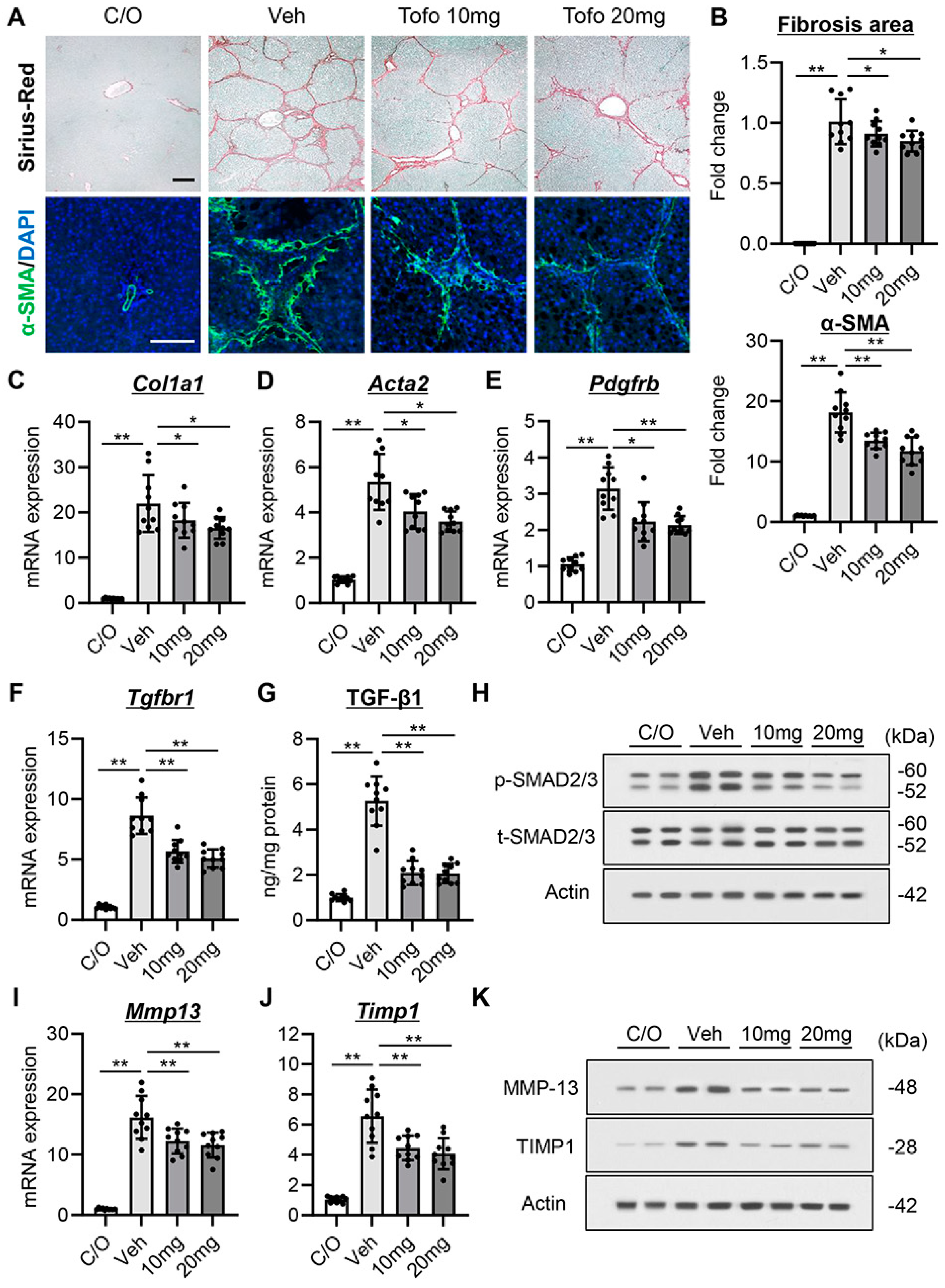
3.5. Tofogliflozin Exerts Antifibrotic Effects in CCl4-Treated Rats
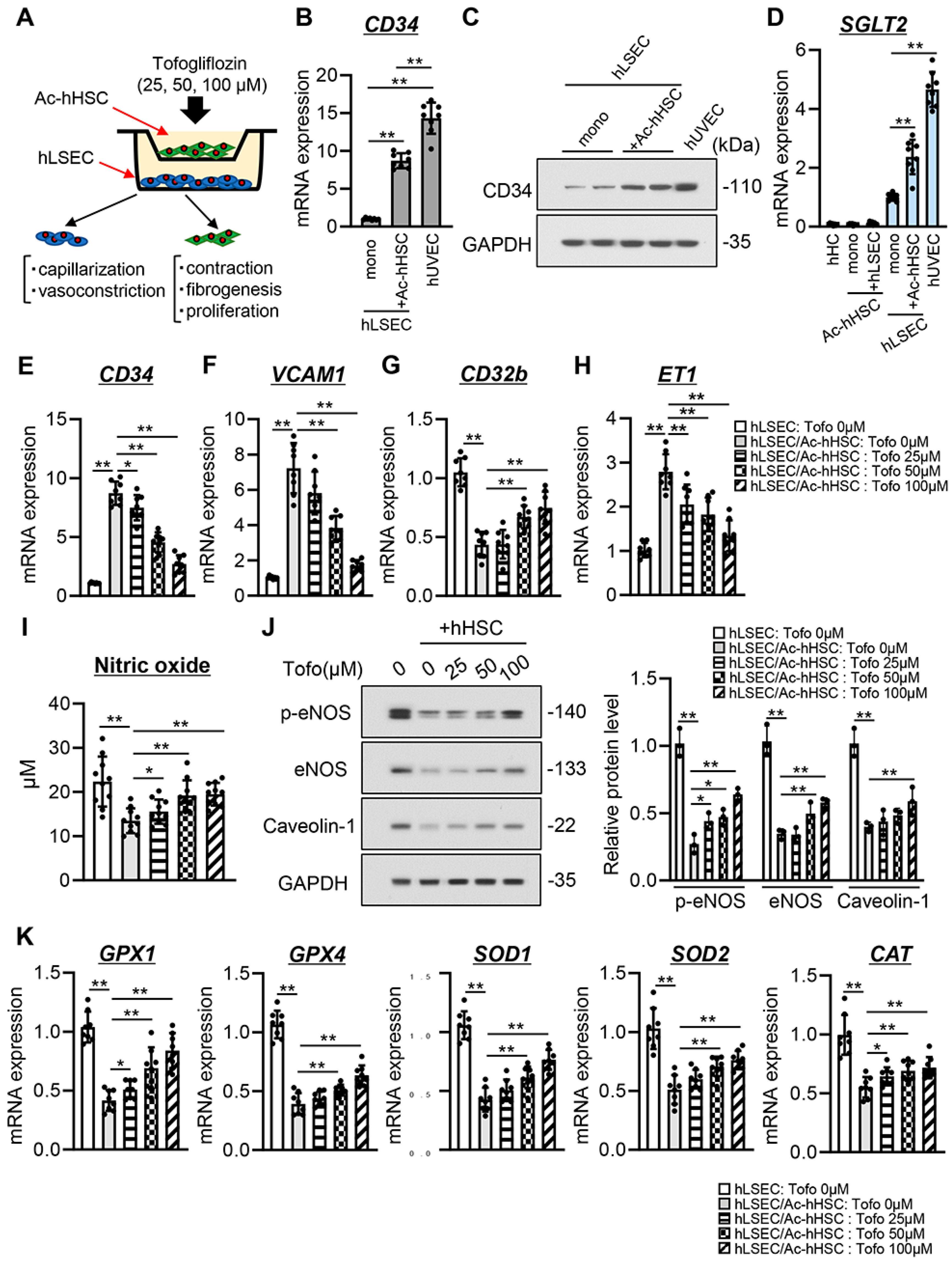
3.6. Tofogliflozin Suppresses Ac-hHSC-Stimulated Capillarization and Vasoconstrictive Activity in hLSECs
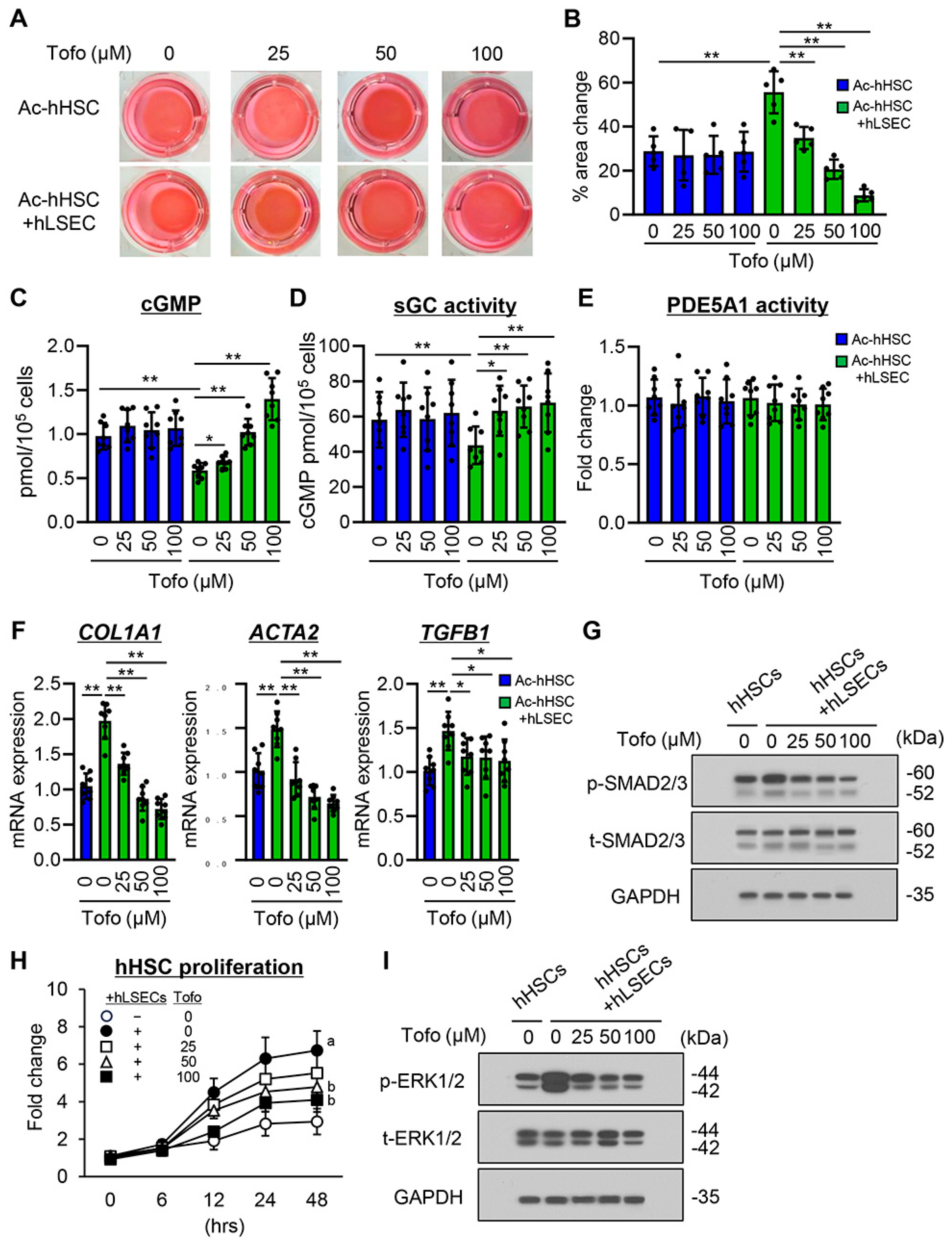
3.7. Effects of Tofogliflozin on hLSEC-Stimulated Contractive, Profibrogenic, and Proliferative Activities of Ac-hHSCs
4. Discussion
5. Supplementary Materials and Methods
5.1. Biochemical Analysis
5.2. Transforming Growth Factor-β1 (TGF-β1) Level
5.3. Vascular Endothelial Growth Factor (VEGF)-A, Platelet-Derived Growth Factor (PDGF)-BB, and Von Willebrand Factor (VWF) Levels
5.4. Histological and Immunohistochemical Analyses
5.5. TMNK-1 Cocultured with LX-2
5.6. The Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
5.7. Western Blotting Assays
5.8. ET-1, eNOS, NO, and Cyclic Guanosine Monophosphate (cGMP) Levels
5.9. Collagen Gel Contraction Assay
5.10. Soluble Guanylyl Cyclase (sGC) and Phosphodiesterase 5A1 (PDE5A1) Activity Assay
5.11. Cell Proliferation Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef]
- Engelmann, C.; Clària, J.; Szabo, G.; Bosch, J.; Bernardi, M. Pathophysiology of decompensated cirrhosis: Portal hypertension, circulatory dysfunction, inflammation, metabolism and mitochondrial dysfunction. J. Hepatol. 2021, 75 (Suppl. S1), S49–S66. [Google Scholar] [CrossRef]
- McConnell, M.; Iwakiri, Y. Biology of portal hypertension. Hepatol. Int. 2018, 12, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Trebicka, J. Portal hypertension in cirrhosis: Pathophysiological mechanisms and therapy. JHEP Rep. 2021, 3, 100316. [Google Scholar] [CrossRef] [PubMed]
- Thabut, D.; Shah, V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? J. Hepatol. 2010, 53, 976–980. [Google Scholar] [CrossRef]
- Kitto, L.J.; Henderson, N.C. Hepatic Stellate Cell Regulation of Liver Regeneration and Repair. Hepatol. Commun. 2021, 5, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Intercellular crosstalk of liver sinusoidal endothelial cells in liver fibrosis, cirrhosis and hepatocellular carcinoma. Dig. Liver Dis. 2022, 54, 598–613. [Google Scholar] [CrossRef] [PubMed]
- de Franchis, R.; Bosch, J.; Garcia-Tsao, G.; Reiberger, T.; Ripoll, C.; Baveno VII Faculty. Baveno VII—Renewing consensus in portal hypertension. J. Hepatol. 2022, 76, 959–974. [Google Scholar] [CrossRef]
- Idris, I.; Donnelly, R. Sodium-glucose co-transporter-2 inhibitors: An emerging new class of oral antidiabetic drug. Diabetes Obes. Metab. 2009, 11, 79–88. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Targher, G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: A systematic review. Lancet Gastroenterol. Hepatol. 2022, 7, 367–378. [Google Scholar] [CrossRef]
- Arai, T.; Atsukawa, M.; Tsubota, A.; Mikami, S.; Haruki, U.; Yoshikata, K.; Ono, H.; Kawano, T.; Yoshida, Y.; Tanabe, T.; et al. Antifibrotic effect and long-term outcome of SGLT2 inhibitors in patients with NAFLD complicated by diabetes mellitus. Hepatol. Commun. 2022, 6, 3073–3082. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Montalvo-Gordon, I.; Chi-Cervera, L.A.; García-Tsao, G. Sodium-Glucose Cotransporter 2 Inhibitors Ameliorate Ascites and Peripheral Edema in Patients with Cirrhosis and Diabetes. Hepatology 2020, 72, 1880–1882. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Wang, L.X.; Dong, S.S.; Hong, Y.; Zhou, X.H.; Zheng, W.W.; Zheng, C. Phlorizin Exerts Direct Protective Effects on Palmitic Acid (PA)-Induced Endothelial Dysfunction by Activating the PI3K/AKT/eNOS Signaling Pathway and Increasing the Levels of Nitric Oxide (NO). Med. Sci. Monit. Basic. Res. 2018, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.; Zhou, Y.; Fu, L.; Ding, H.; Zhou, Y.; Yin, Z.; Yang, R.; Liu, Z.; Zhou, S. Dapagliflozin impedes endothelial cell senescence by activating the SIRT1 signaling pathway in type 2 diabetes. Heliyon 2023, 9, e19152. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud Refaie, M.M.; Bayoumi, A.M.; Mokhemer, S.A.; Shehata, S.; Abd El-Hameed, N.M. Role of hypoxia inducible factor/vascular endothelial growth factor/endothelial nitric oxide synthase signaling pathway in mediating the cardioprotective effect of dapagliflozin in cyclophosphamide-induced cardiotoxicity. Hum. Exp. Toxicol. 2023, 42, 9603271231193392. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, Z.; Chang, X.; Li, Z.; Wu, F.; He, J.; Cao, T.; Wang, K.; Shi, N.; Zhou, H.; et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 2022, 52, 102288. [Google Scholar] [CrossRef]
- Suzuki, M.; Honda, K.; Fukazawa, M.; Ozawa, K.; Hagita, H.; Kawai, T.; Takeda, M.; Yata, T.; Kawai, M.; Fukuzawa, T.; et al. Tofogliflozin, a potent and highly specific sodium/glucose cotransporter 2 inhibitor, improves glycemic control in diabetic rats and mice. J. Pharmacol. Exp. Ther. 2012, 341, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, R.P.; Mehta, G.; Balasubramaniyan, V.; Mohamed Fel, Z.; Davies, N.; Sharma, V.; Iwakiri, Y.; Jalan, R. Hepatic dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis and is a target for therapy in portal hypertension. J. Hepatol. 2015, 62, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Furuta, K.; Islam, S.; Caporarello, N.; Kostallari, E.; Dielis, K.; Tschumperlin, D.J.; Hirsova, P.; Ibrahim, S.H. Liver sinusoidal endothelial cell expressed vascular cell adhesion molecule 1 promotes liver fibrosis. Front. Immunol. 2022, 13, 983255. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Fernández, M.; Bosch, J.; García-Pagán, J.C. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology 2008, 47, 1248–1256. [Google Scholar] [CrossRef]
- Nishimura, N.; Kitade, M.; Noguchi, R.; Namisaki, T.; Moriya, K.; Takeda, K.; Okura, Y.; Aihara, Y.; Douhara, A.; Kawaratani, H.; et al. Ipragliflozin, a sodium–glucose cotransporter 2 inhibitor, ameliorates the development of liver fibrosis in diabetic Otsuka Long–Evans Tokushima fatty rats. J. Gastroenterol. 2016, 51, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wu, B.; He, Y.; Qiu, R.; Yang, T.; Wang, S.; Lei, Y.; Li, H.; Zheng, F. Canagliflozin ameliorates the development of NAFLD by preventing NLRP3-mediated pyroptosis through FGF21-ERK1/2 pathway. Hepatol. Commun. 2023, 7, e0045. [Google Scholar] [CrossRef]
- Lombardi, R.; Mantovani, A.; Cespiati, A.; Francione, P.; Maffi, G.; Del Zanna, E.; Maffeis, C.; Colecchia, A.; Passigato, N.; Ferrarese, A.; et al. Evolution of liver fibrosis in diabetic patients with NAFLD in a follow-up study: Hepatoprotective effects of sodium-glucose co-transporter-2 inhibitors. Dig. Liver Dis. 2023. [Google Scholar] [CrossRef]
- Taheri, H.; Malek, M.; Ismail-Beigi, F.; Zamani, F.; Sohrabi, M.; Reza Babaei, M.; Khamseh, M.E. Effect of Empagliflozin on Liver Steatosis and Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease Without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv. Ther. 2020, 37, 4697–4708. [Google Scholar] [CrossRef]
- Saffo, S.; Garcia-Tsao, G.; Taddei, T. SGLT2 inhibitors in patients with cirrhosis and diabetes mellitus: A tertiary center cohort study and insights about a potential therapeutic target for portal hypertension. J. Diabetes 2021, 13, 265–269. [Google Scholar] [CrossRef]
- Muskiet, M.H.A.; van Bommel, E.J.; van Raalte, D.H. Antihypertensive effects of SGLT2 inhibitors in type 2 diabetes. Lancet Diabetes Endocrinol. 2016, 4, 188–189. [Google Scholar] [CrossRef]
- Kario, K.; Okada, K.; Kato, M.; Nishizawa, M.; Yoshida, T.; Asano, T.; Uchiyama, K.; Niijima, Y.; Katsuya, T.; Urata, H.; et al. Twenty-Four-Hour Blood Pressure-Lowering Effect of a Sodium-Glucose Cotransporter 2 Inhibitor in Patients with Diabetes and Uncontrolled Nocturnal Hypertension: Results from the Randomized, Placebo-Controlled SACRA Study. Circulation 2019, 139, 2089–2097. [Google Scholar] [CrossRef]
- El-Daly, M.; Pulakazhi Venu, V.K.; Saifeddine, M.; Mihara, K.; Kang, S.; Fedak, P.W.M.; Alston, L.A.; Hirota, S.A.; Ding, H.; Triggle, C.R.; et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: Prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vascul. Pharmacol. 2018, 109, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Pegoli, W.; Clemens, M.G. Endothelin-1 induces direct constriction of hepatic sinusoids. Am. J. Physiol. 1994, 266, 624–632. [Google Scholar] [CrossRef]
- Bocca, C.; Novo, E.; Miglietta, A.; Parola, M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasan, D. Endothelin-1 in portal hypertension: The intricate role of hepatic stellate cells. Exp. Biol. Med. 2020, 245, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Ohara, N.; Futagawa, S.; Watanabe, S.; Fukasawa, M.; Takamori, S. Clinical investigation of endothelin-1 and nitric oxide in patients with portal hypertension focusing on plasma levels and immunohistological staining of liver tissues. Hepatol. Res. 2001, 21, 40–54. [Google Scholar] [CrossRef]
- Wereszczynka-Siemiatkowska, U.; Swidnicka-Siergiejko, A.; Siemiatkowski, A.; Bondyra, Z.; Wasielica-Berger, J.; Mroczko, B.; Janica, J.; Dabrowski, A. Endothelin 1 and transforming growth factor-β1 correlate with liver function and portal pressure in cirrhotic patients. Cytokine 2015, 76, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Chung, J.J. Reduced Nitric Oxide Production by Endothelial Cells in Cirrhotic Rat Liver: Endothelial Dysfunction in Portal Hypertension. Gastroenterology 1998, 114, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Osorio, H.; Coronel, I.; Arellano, A.; Pacheco, U.; Bautista, R.; Franco, M.; Escalante, B. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid. Med. Cell Longev. 2012, 2012, 542042. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, W.; Wan, S.; Chen, Y.; Fu, M.; Wang, Z.; Xiong, F.; Zhang, Y. Canagliflozin ameliorates high glucose-induced apoptosis in NRK-52E cells via inhibiting oxidative stress and activating AMPK/mTOR-mediated autophagy. Mol. Biol. Rep. 2023, 50, 10325–10337. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Van Beuge, M.M.; Granzow, M.; Beljaars, L.; Schierwagen, R.; Kilic, S.; Heidari, I.; Huss, S.; Sauerbruch, T.; Poelstra, K.; et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J. Hepatol. 2012, 57, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Failli, P.; DeFRANCO, R.M.; Caligiuri, A.; Gentilini, A.; Romanelli, R.G.; Marra, F.; Batignani, G.; Guerra, C.T.; Laffi, G.; Gentilini, P.; et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology 2000, 119, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Langer, D.A.; Das, A.; Semela, D.; Kang-Decker, N.; Hendrickson, H.; Bronk, S.F.; Katusic, Z.S.; Gores, G.J.; Shah, V.H. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology 2008, 47, 1983–1993. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef]
- Tateya, S.; Rizzo, N.O.; Handa, P.; Cheng, A.M.; Morgan-Stevenson, V.; Daum, G.; Clowes, A.W.; Morton, G.J.; Schwartz, M.W.; Kim, F. Endothelial NO/cGMP/VASP signaling attenuates kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 2011, 60, 2792–2801. [Google Scholar] [CrossRef]
- Abdollahi, E.; Keyhanfar, F.; Delbandi, A.A.; Falak, R.; Hajimiresmaiel, S.J.; Shafiei, M. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-κB activation in human endothelial cells and differentiated macrophages. Eur. J. Pharmacol. 2022, 918, 174715. [Google Scholar] [CrossRef]
- Noah, A.A.; El-Mezayen, N.S.; El-Ganainy, S.O.; Darwish, I.E.; Afify, E.A. Reversal of fibrosis and portal hypertension by Empagliflozin treatment of CCl4-induced liver fibrosis: Emphasis on gal-1/NRP-1/TGF-β and gal-1/NRP-1/VEGFR2 pathways. Eur. J. Pharmacol. 2023, 959, 176066. [Google Scholar] [CrossRef]
- Kreisel, W.; Lazaro, A.; Trebicka, J.; Grosse Perdekamp, M.; Schmitt-Graeff, A.; Deibert, P. Cyclic GMP in Liver Cirrhosis-Role in Pathophysiology of Portal Hypertension and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 10372. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asada, S.; Kaji, K.; Nishimura, N.; Koizumi, A.; Matsuda, T.; Tanaka, M.; Yorioka, N.; Sato, S.; Kitagawa, K.; Namisaki, T.; et al. Tofogliflozin Delays Portal Hypertension and Hepatic Fibrosis by Inhibiting Sinusoidal Capillarization in Cirrhotic Rats. Cells 2024, 13, 538. https://doi.org/10.3390/cells13060538
Asada S, Kaji K, Nishimura N, Koizumi A, Matsuda T, Tanaka M, Yorioka N, Sato S, Kitagawa K, Namisaki T, et al. Tofogliflozin Delays Portal Hypertension and Hepatic Fibrosis by Inhibiting Sinusoidal Capillarization in Cirrhotic Rats. Cells. 2024; 13(6):538. https://doi.org/10.3390/cells13060538
Chicago/Turabian StyleAsada, Shohei, Kosuke Kaji, Norihisa Nishimura, Aritoshi Koizumi, Takuya Matsuda, Misako Tanaka, Nobuyuki Yorioka, Shinya Sato, Koh Kitagawa, Tadashi Namisaki, and et al. 2024. "Tofogliflozin Delays Portal Hypertension and Hepatic Fibrosis by Inhibiting Sinusoidal Capillarization in Cirrhotic Rats" Cells 13, no. 6: 538. https://doi.org/10.3390/cells13060538
APA StyleAsada, S., Kaji, K., Nishimura, N., Koizumi, A., Matsuda, T., Tanaka, M., Yorioka, N., Sato, S., Kitagawa, K., Namisaki, T., Akahane, T., & Yoshiji, H. (2024). Tofogliflozin Delays Portal Hypertension and Hepatic Fibrosis by Inhibiting Sinusoidal Capillarization in Cirrhotic Rats. Cells, 13(6), 538. https://doi.org/10.3390/cells13060538