.png)
Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Primary Ciliary Dyskinesia Diagnostic Evaluation
2.3. In Silico Studies of the Candidate Variant
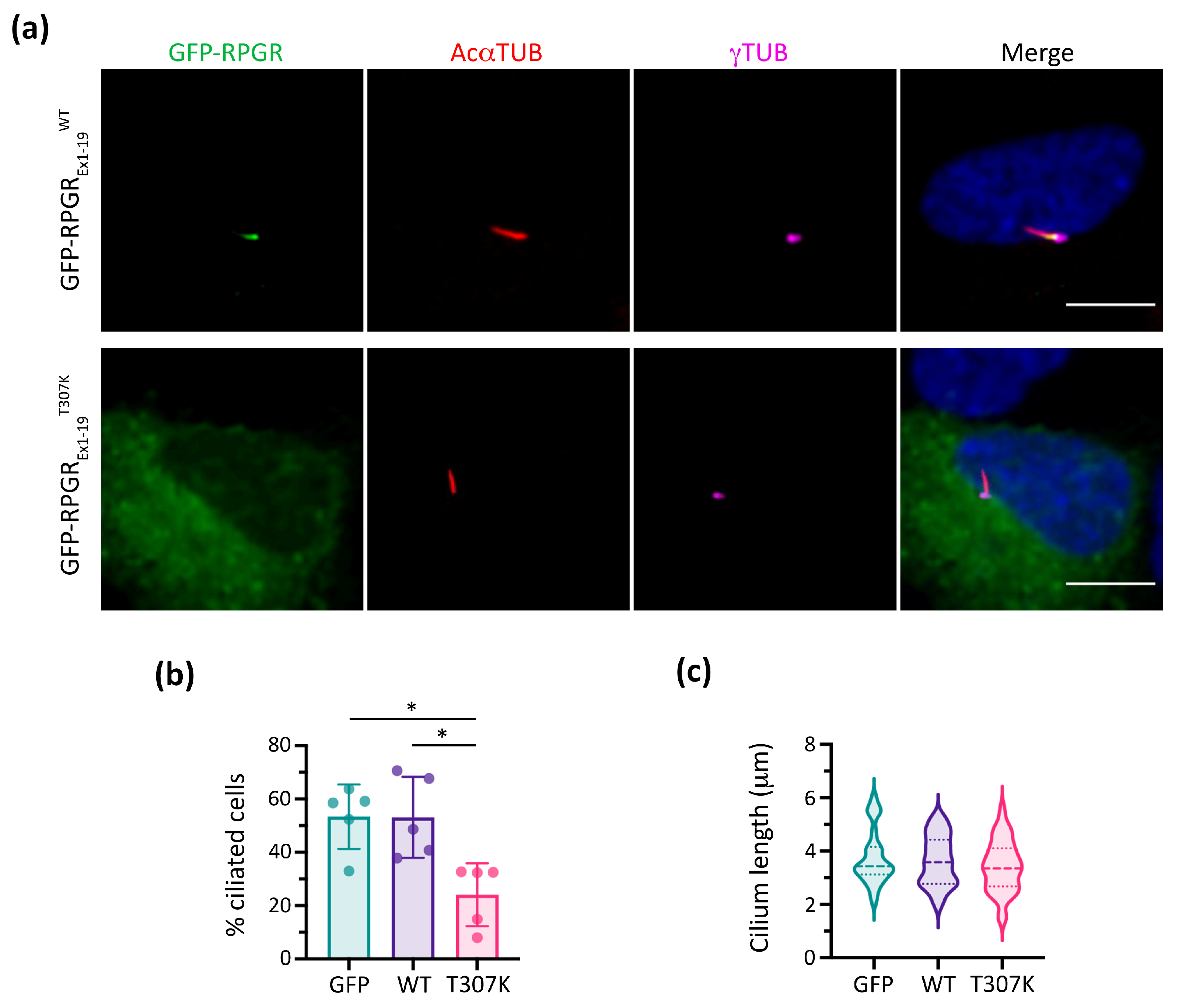
2.4. In Vitro Functional Studies
2.5. Immunofluorescence Analyses of RPGR and CEP290 in Nasal Brushing Samples
2.6. Statistics
3. Results
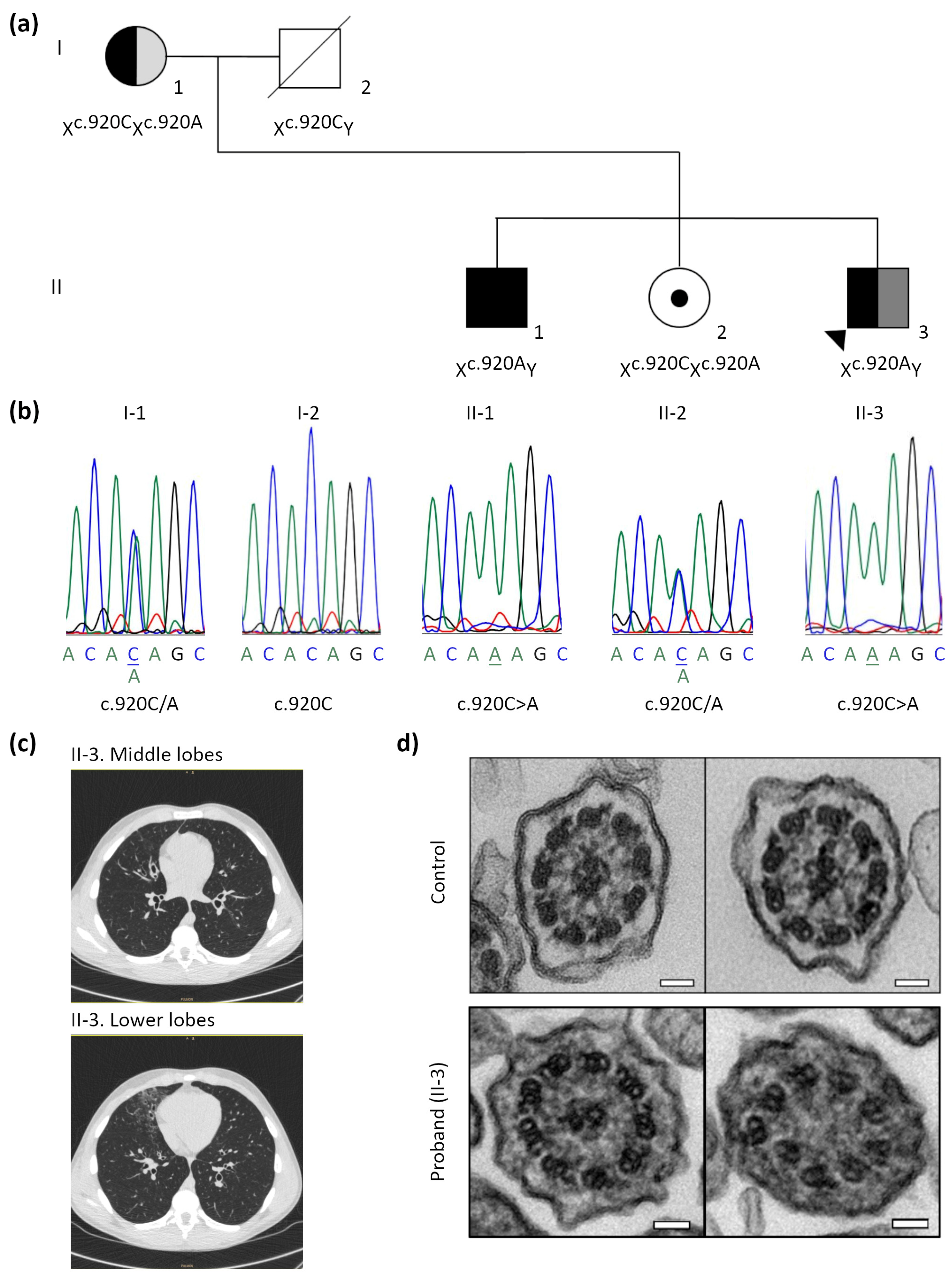
3.1. PCD Clinical Manifestation and Diagnosis
3.2. Identification of the Pathogenic RPGR Variant
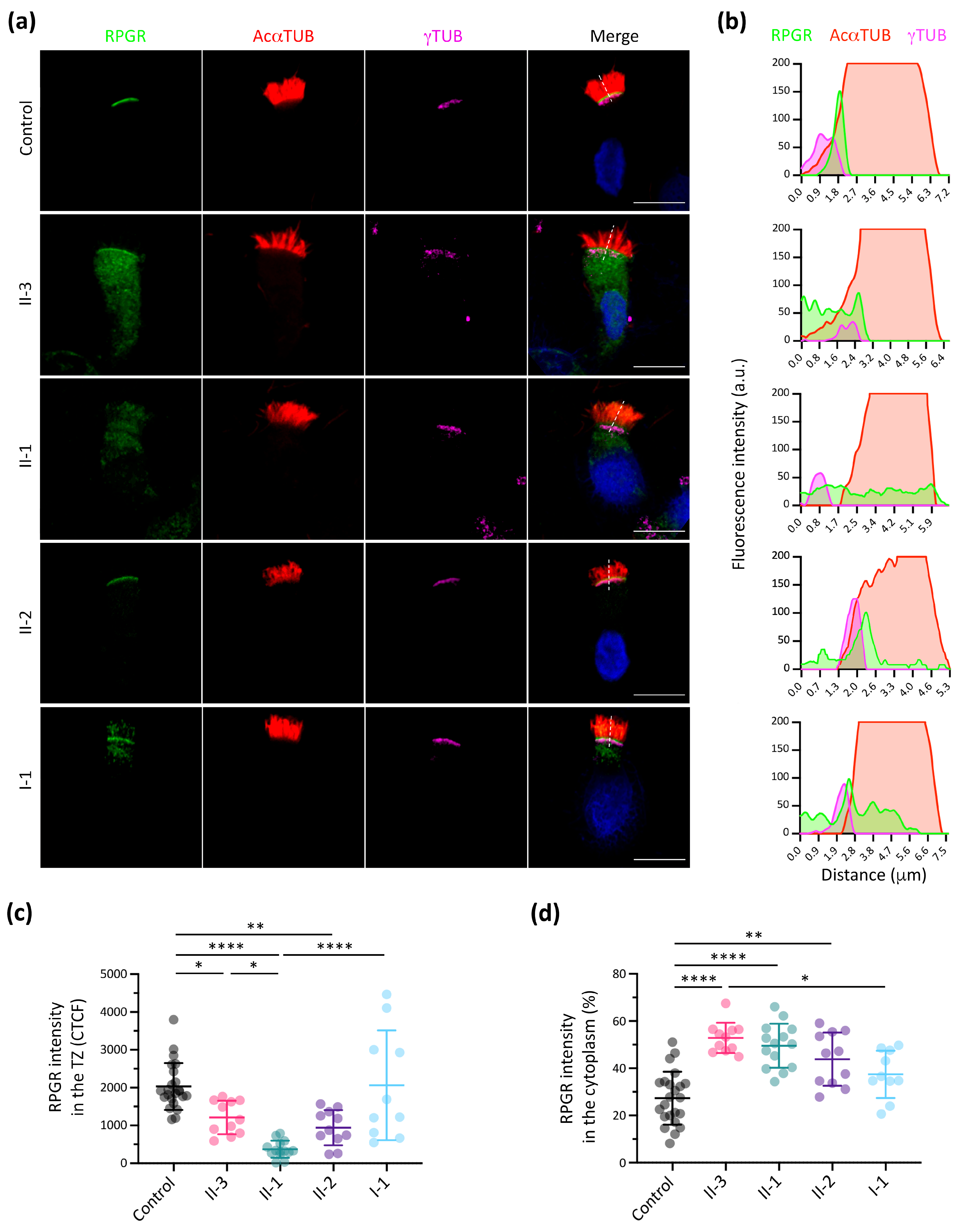
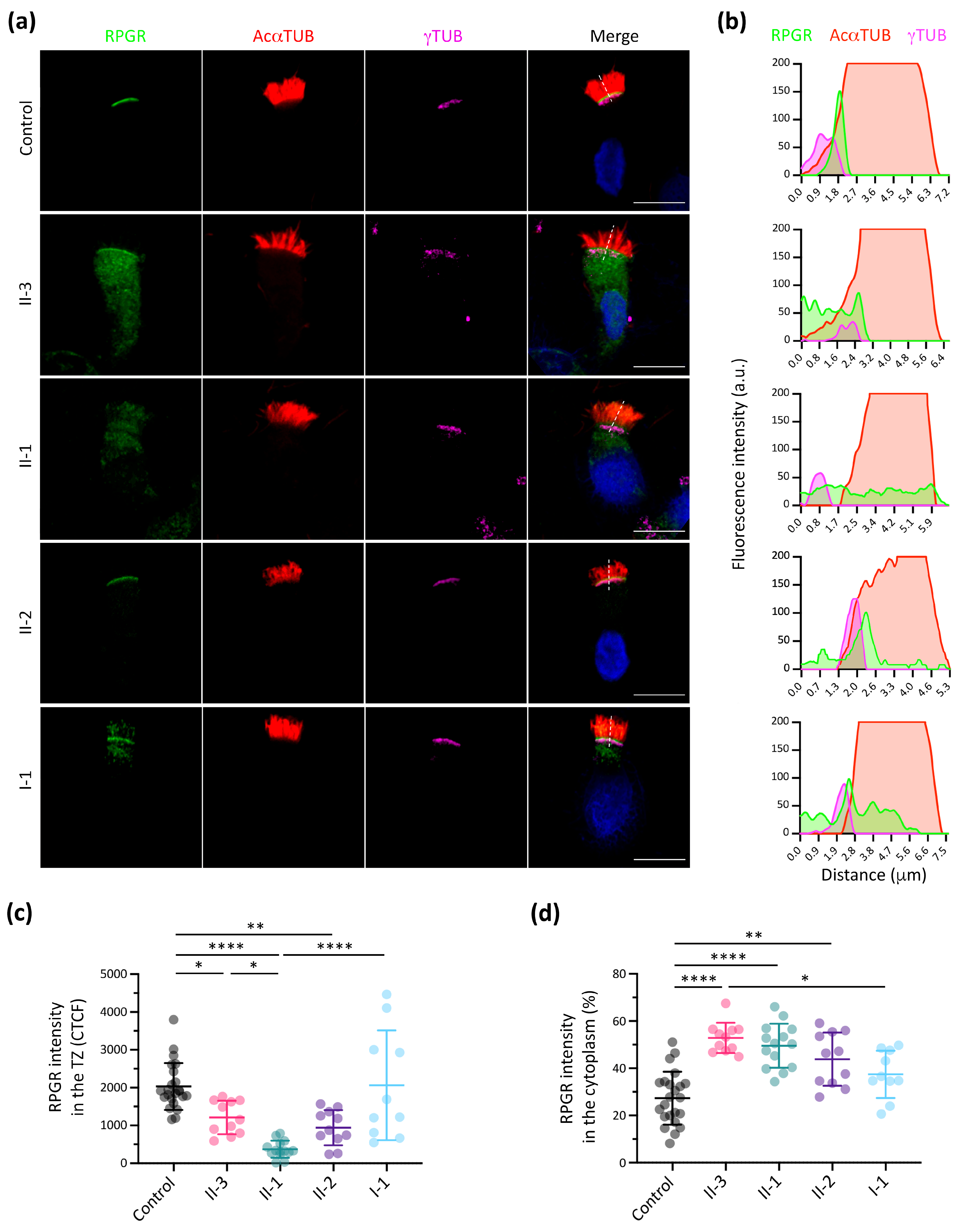
3.3. RPGR Immunofluorescence Analysis in Respiratory Ciliated Cells
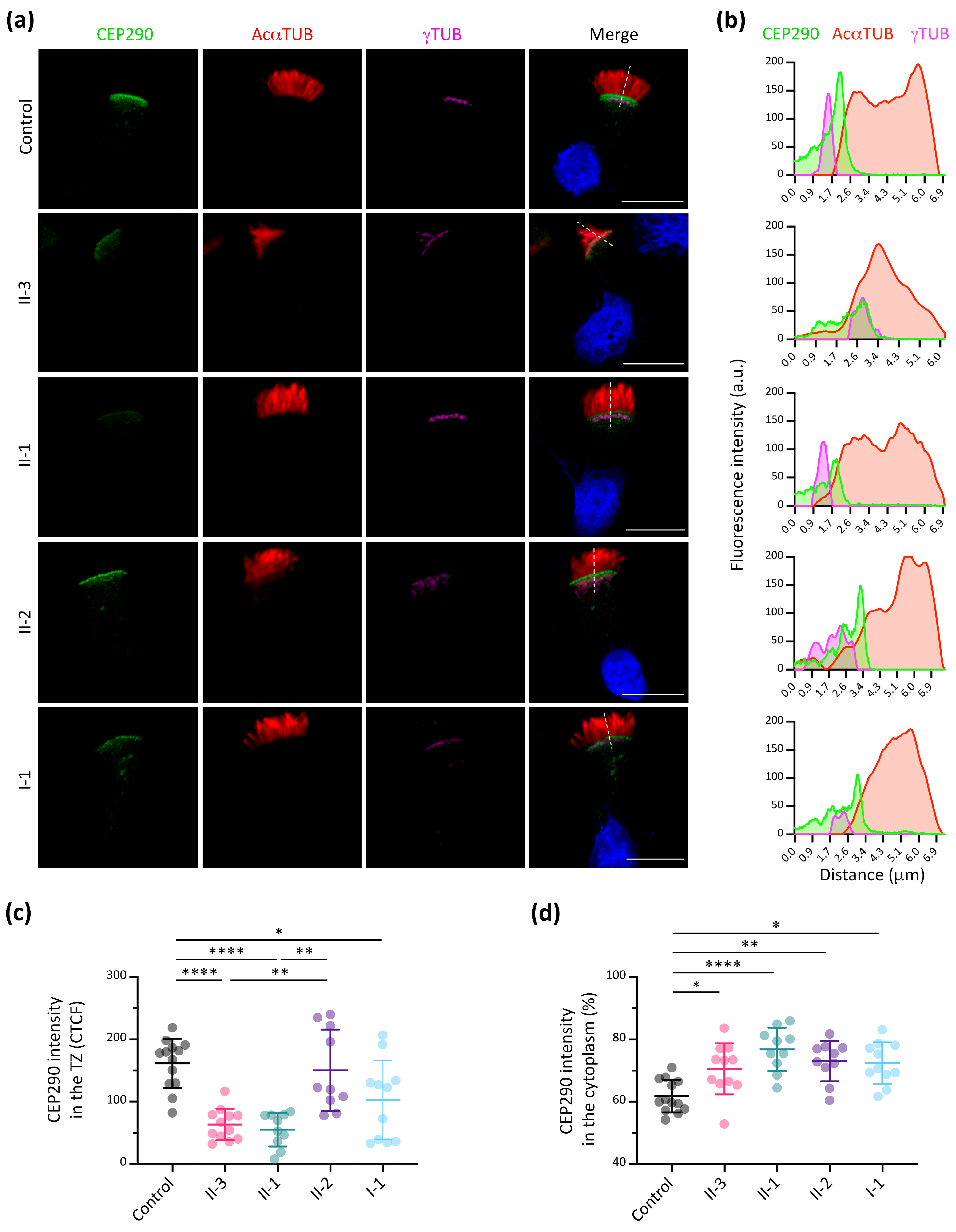
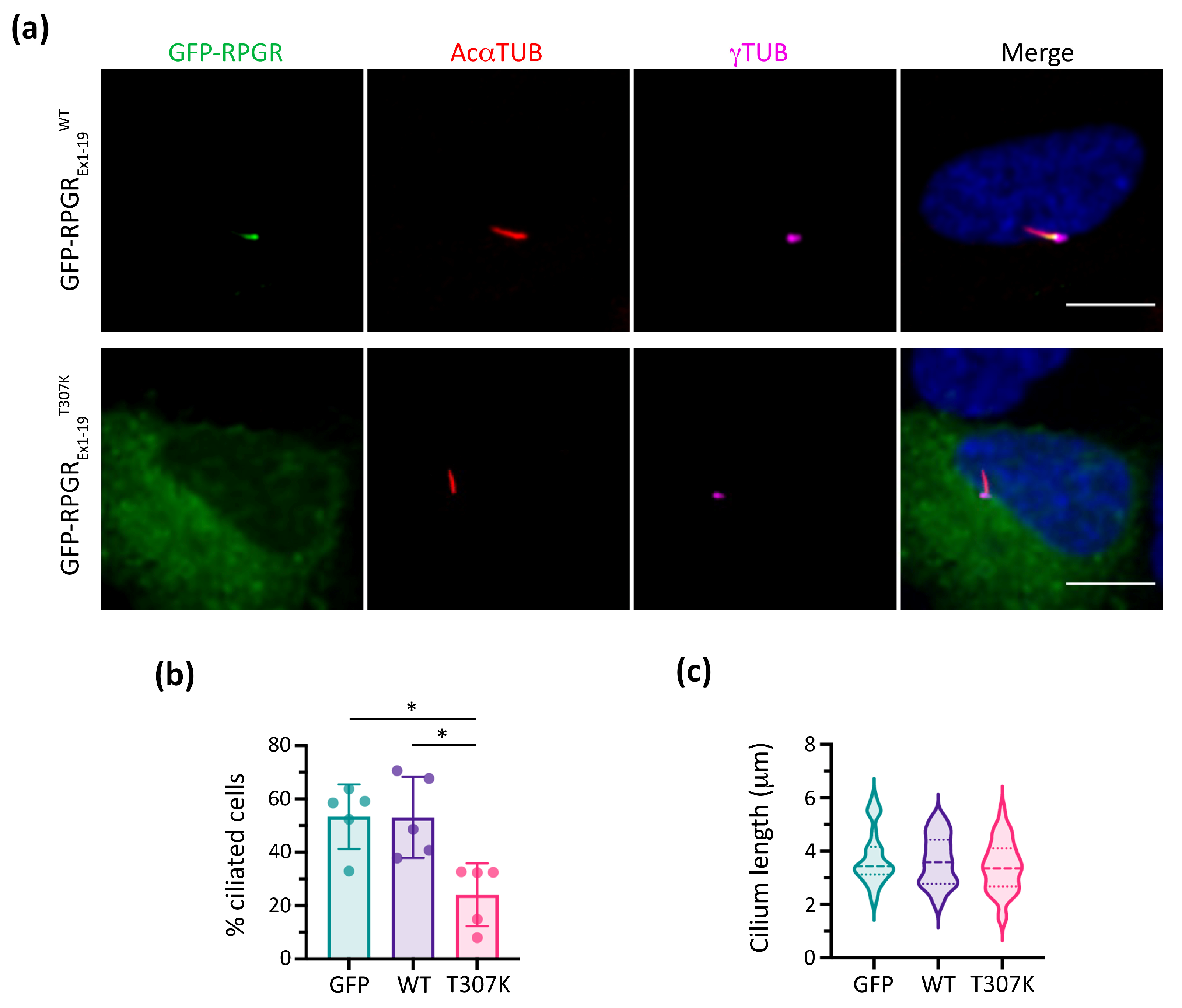
3.4. Additive Effect of Modifier Genes on the PCD Phenotype
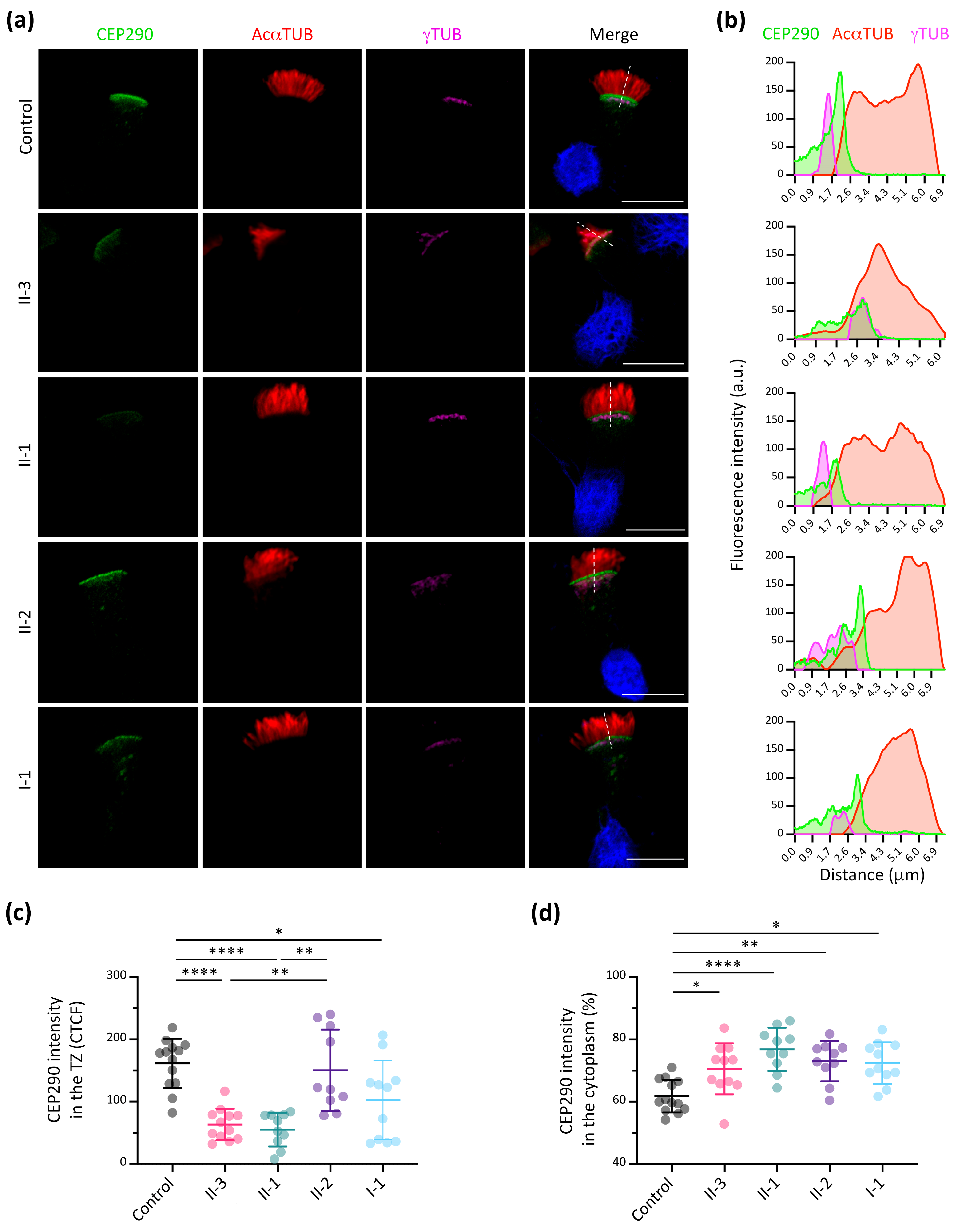
3.5. CEP290 Immunofluorescence Analysis in Respiratory Ciliated Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile Ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 77. [Google Scholar] [CrossRef]
- Legendre, M.; Zaragosi, L.E.; Mitchison, H.M. Motile Cilia and Airway Disease. Semin. Cell Dev. Biol. 2021, 110, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.; Seifert, B.; Truty, R.; Zariwala, M.; Ameel, K.; Zhao, Y.; Nykamp, K.; Gaston, B. The Global Prevalence and Ethnic Heterogeneity of Primary Ciliary Dyskinesia Gene Variants: A Genetic Database Analysis. Lancet Respir. Med. 2022, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Reula, A.; Lucas, J.; Moreno-Galdó, A.; Romero, T.; Milara, X.; Carda, C.; Mata-Roig, M.; Escribano, A.; Dasi, F.; Armengot-Carceller, M. New Insights in Primary Ciliary Dyskinesia. Expert Opin. Orphan Drugs 2017, 5, 537–548. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Davis, S.D.; Ferkol, T.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Wolf, W.; et al. Laterality Defects Other than Situs Inversus Totalis in Primary Ciliary Dyskinesia: Insights into Situs Ambiguus and Heterotaxy. Chest 2014, 146, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. X-Linked Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2018, 1085, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Meindl, A.; Dry, K.; Herrmann, K.; Manson, F.; Ciccodicola, A.; Edgar, A.; Carvalho, M.R.S.; Achatz, H.; Hellebrand, H.; Lennon, A.; et al. A Gene (RPGR) with Homology to the RCC1 Guanine Nucleotide Exchange Factor Is Mutated in X–Linked Retinitis Pigmentosa (RP3). Nat. Genet. 1996, 13, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Giacalone, J.C.; Searby, C.; Stone, E.M.; Tucker, B.A.; Sheffield, V.C. Disruption of RPGR Protein Interaction Network Is the Common Feature of RPGR Missense Variations That Cause XLRP. Proc. Natl. Acad. Sci. USA 2019, 116, 1353–1360. [Google Scholar] [CrossRef]
- Boulanger-Scemama, E.; Mohand-Saïd, S.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.P.; Letexier, M.; et al. Phenotype Analysis of Retinal Dystrophies in Light of the Underlying Genetic Defects: Application to Cone and Cone-Rod Dystrophies. Int. J. Mol. Sci. 2019, 20, 4854. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Loyer, M.; Hand, C.K.; Al Mahdi, H.; Dembinska, O.; Beneish, R.; Racine, J.; Rouleau, G.A. Novel RPGR Mutations with Distinct Retinitis Pigmentosa Phenotypes in French-Canadian Families. Am. J. Ophthalmol. 2003, 136, 678–687. [Google Scholar] [CrossRef]
- Breuer, D.K.; Yashar, B.M.; Filippova, E.; Hiriyanna, S.; Lyons, R.H.; Mears, A.J.; Asaye, B.; Acar, C.; Vervoort, R.; Wright, A.F.; et al. A Comprehensive Mutation Analysis of RP2 and RPGR in a North American Cohort of Families with X-Linked Retinitis Pigmentosa. Am. J. Hum. Genet. 2002, 70, 1545–1554. [Google Scholar] [CrossRef]
- van Dorp, D.B.; Wright, A.F.; Carothers, A.D.; Bleeker-Wagemakers, E.M. A Family with RP3 Type of X-Linked Retinitis Pigmentosa: An Association with Ciliary Abnormalities. Hum. Genet. 1992, 88, 331–334. [Google Scholar] [CrossRef]
- Bukowy-Bieryłło, Z.; Ziȩtkiewicz, E.; Loges, N.T.; Wittmer, M.; Geremek, M.; Olbrich, H.; Fliegauf, M.; Voelkel, K.; Rutkiewicz, E.; Rutland, J.; et al. RPGR Mutations Might Cause Reduced Orientation of Respiratory Cilia. Pediatr. Pulmonol. 2013, 48, 352–363. [Google Scholar] [CrossRef]
- Moore, A.; Escudier, E.; Roger, G.; Tamalet, A.; Pelosse, B.; Marlin, S.; Clément, A.; Geremek, M.; Delaisi, B.; Bridoux, A.M.; et al. RPGR Is Mutated in Patients with a Complex X Linked Phenotype Combining Primary Ciliary Dyskinesia and Retinitis Pigmentosa. J. Med. Genet. 2006, 43, 326–333. [Google Scholar] [CrossRef]
- Iannaccone, A.; Breuer, D.K.; Wang, X.; Kuo, S.; Normando, E.; Filippova, E.; Baldi, A.; Hiriyanna, S.; MacDonald, C.; Baldi, F.; et al. Clinical and Immunohistochemical Evidence for an X Linked Retinitis Pigmentosa Syndrome with Recurrent Infections and Hearing Loss in Association with an RPGR Mutation. J. Med. Genet. 2003, 40, e118. [Google Scholar] [CrossRef]
- Han, R.C.; Taylor, L.J.; Martinez-Fernandez de la Camara, C.; Henderson, R.H.; Thompson, D.A.; Cehajic-Kapetanovic, J.; MacLaren, R.E. Is RPGR-Related Retinal Dystrophy Associated with Systemic Disease? A Case Series. Ophthalmic Genet. 2023, 44, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Kolkova, Z.; Durdik, P.; Holubekova, V.; Durdikova, A.; Jesenak, M.; Banovcin, P. Identification of a Novel RPGR Mutation Associated with Retinitis Pigmentosa and Primary Ciliary Dyskinesia in a Slovak Family: A Case Report. Front. Pediatr. 2024, 12, 1339664. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, A.; Namkoong, H.; Iwami, E.; Tsutsumi, A.; Nakajima, T.; Shinoda, H.; Katada, Y.; Iimura, J.; Suzuki, H.; Kosaki, K.; et al. X -Linked Inheritance of Primary Ciliary Dyskinesia and Retinitis Pigmentosa Due to RPGR Variant: A Case Report and Literature Review. Respirol. Case Rep. 2023, 11, e01240. [Google Scholar] [CrossRef] [PubMed]
- McCray, G.; Griffin, P.; Martinello, P.; De Iongh, R.; Ruddle, J.; Robinson, P. Altered Airway Ciliary Orientation in Patients with X-Linked Retinitis Pigmentosa. Thorax 2019, 74, 914–916. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-Linked Retinopathies: Physiological Insights, Pathogenic Mechanisms, Phenotypic Features and Novel Therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR Isoforms in Photoreceptor Connecting Cilia and the Transitional Zone of Motile Cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef]
- Vössing, C.; Atigbire, P.; Eilers, J.; Markus, F.; Stieger, K.; Song, F.; Neidhardt, J. The Major Ciliary Isoforms of Rpgr Build Different Interaction Complexes with Inpp5e and Rpgrip1l. Int. J. Mol. Sci. 2021, 22, 3583. [Google Scholar] [CrossRef]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its Role in Photoreceptor Physiology, Human Disease, and Future Therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef]
- Patnaik, S.R.; Raghupathy, R.K.; Zhang, X.; Mansfield, D.; Shu, X. The Role of RPGR and Its Interacting Proteins in Ciliopathies. J. Ophthalmol. 2015, 2015, 414781. [Google Scholar] [CrossRef]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society Guidelines for the Diagnosis of Primary Ciliary Dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef]
- Behan, L.; Dimitrov, B.D.; Kuehni, C.E.; Hogg, C.; Carroll, M.; Evans, H.J.; Goutaki, M.; Harris, A.; Packham, S.; Walker, W.T.; et al. PICADAR: A Diagnostic Predictive Tool for Primary Ciliary Dyskinesia. Eur. Respir. J. 2016, 47, 1103–1112. [Google Scholar] [CrossRef]
- Beydon, N.; Kouis, P.; Marthin, J.K.; Latzin, P.; Colas, M.; Davis, S.D.; Haarman, E.; Harris, A.L.; Hogg, C.; Kilbride, E.; et al. Nasal nitric oxide measurement in children for the diagnosis of primary ciliary dyskinesia: European Respiratory Society technical standard. Eur. Respir. J. 2023, 61, 2202031. [Google Scholar] [CrossRef] [PubMed]
- Kempeneers, C.; Seaton, C.; Garcia Espinosa, B.; Chilvers, M.A. Ciliary functional analysis: Beating a path towards standardization. Pediatr. Pulmonol. 2019, 54, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Baz-Redón, N.; Rovira-Amigo, S.; Fernández-Cancio, M.; Castillo-Corullón, S.; Cols, M.; Caballero-Rabasco, M.A.; Asensio, Ó.; de Vicente, C.M.; del Mar Martínez-Colls, M.M.; Torrent-Vernetta, A.; et al. Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3603. [Google Scholar] [CrossRef] [PubMed]
- Baz-Redón, N.; Rovira-Amigo, S.; Paramonov, I.; Castillo-Corullón, S.; Cols Roig, M.; Antolín, M.; García Arumí, E.; Torrent-Vernetta, A.; de Mir Messa, I.; Gartner, S.; et al. Implementation of a Gene Panel for Genetic Diagnosis of Primary Ciliary Dyskinesia. Arch. Bronconeumol. 2021, 57, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Schröder, J.; Penington, J.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.; Papenfuss, A. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Cols, N.; Marfany, G.; Atrian, S.; Gonzàlez-Duarte, R. Effect of site-directed mutagenesis on conserved positions of Drosophila alcohol dehydrogenase. FEBS Lett. 1993, 319, 90–94. [Google Scholar] [CrossRef]
- Knowles, M.R.; Leigh, M.W.; Carson, J.L.; Davis, S.D.; Dell, S.D.; Ferkol, T.W.; Olivier, K.N.; Sagel, S.D.; Rosenfeld, M.; Burns, K.A.; et al. Mutations of DNAH11 in Primary Ciliary Dyskinesia Patients with Normal Ciliary Ultrastructure. Thorax 2012, 67, 441. [Google Scholar] [CrossRef]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations in RSPH1 Cause Primary Ciliary Dyskinesia with a Unique Clinical and Ciliary Phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef]
- Vertii, A.; Hung, H.F.; Hehnly, H.; Doxsey, S. Human Basal Body Basics. Cilia 2016, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F. Basal Bodies Platforms for Building Cilia. Curr. Top. Dev. Biol. 2008, 85, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S.; Branham, K.; Wheaton, D.K.; Jones, K.D.; Avery, C.E.; Cadena, E.D.; Heckenlively, J.R.; Birch, D.G. Molecular Findings in Families with an Initial Diagnose of Autosomal Dominant Retinitis Pigmentosa (AdRP). Adv. Exp. Med. Biol. 2018, 1074, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pang, N.; Zhang, Y.; Chen, H.; Peng, Y.; Fu, J.; Wei, Q. CEP290 Is Essential for the Initiation of Ciliary Transition Zone Assembly. PLoS Biol. 2020, 18, e3001034. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.N.; Zhang, W.; Li, L.; Ronquillo, C.; Baehr, W.; Khanna, H. Ciliopathy-Associated Protein CEP290 Modifies the Severity of Retinal Degeneration Due to Loss of RPGR. Hum. Mol. Genet. 2016, 25, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a Gene with Many Faces: Mutation Overview and Presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.A.; May-simera, H.L.; Veleri, S.; Gotoh, N.; Choi, B.Y.; Murga-zamalloa, C.; McIntyre, J.C.; Marek, J.; Lopez, I.; Hackett, A.N.; et al. Combining Cep290 and Mkks Ciliopathy Alleles in Mice Rescues Sensory Defects and Restores Ciliogenesis. J. Cinical Investig. 2012, 122, 1233–1245. [Google Scholar] [CrossRef]
- Helou, J.; Otto, E.A.; Attanasio, M.; Allen, S.J.; Parisi, M.A.; Glass, I.; Utsch, B.; Hashmi, S.; Fazzi, E.; Omran, H.; et al. Mutation Analysis of NPHP6/CEP290 in Patients with Joubert Syndrome and Senior-Løken Syndrome. J. Med. Genet. 2007, 44, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Papon, J.F.; Perrault, I.; Coste, A.; Louis, B.; Gérard, X.; Hanein, S.; Fares-Taie, L.; Gerber, S.; Defoort-Dhellemmes, S.; Vojtek, A.M.; et al. Abnormal Respiratory Cilia in Non-Syndromic Leber Congenital Amaurosis with CEP290 Mutations. J. Med. Genet. 2010, 47, 829–834. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | I-1 | I-2 | II-1 | II-2 | II-3 (Proband) |
|---|---|---|---|---|---|
| Clinical manifestation | RP + asthma | Healthy | RP | RP carrier | RP + PCD respiratory symptoms |
| PICADAR score | 3 | NA | 2 | 1 | 6 |
| nNO (nL/min) | 314.2 | NA | 328.2 | 533.8 | 107.8 |
| HSVM 1 | 8.46 Hz, 45.7% DC (D and S) | NA | 11.68 Hz, 23% DC (D and S) | 11.74 Hz, 26% DC (D and S) | 8.67 Hz, 95.4% DC (D, S and I) |
| IF 2 | All markers present | NA | All markers present | All markers present | All markers present |
| TEM | 33% MT disorganisation with IDA present | NA | 16% MT disorganisation with IDA present | 17% MT disorganisation with IDA present | 25% MT disorganisation with IDA present |
| Genetics | RPGR:c.920C>A het. | RPGR:c.920C>A hem. | RPGR:c.920C>A het. | RPGR:c.920C>A hem. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baz-Redón, N.; Sánchez-Bellver, L.; Fernández-Cancio, M.; Rovira-Amigo, S.; Burgoyne, T.; Ranjit, R.; Aquino, V.; Toro-Barrios, N.; Carmona, R.; Polverino, E.; et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells 2024, 13, 524. https://doi.org/10.3390/cells13060524
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, Carmona R, Polverino E, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024; 13(6):524. https://doi.org/10.3390/cells13060524
Chicago/Turabian StyleBaz-Redón, Noelia, Laura Sánchez-Bellver, Mónica Fernández-Cancio, Sandra Rovira-Amigo, Thomas Burgoyne, Rai Ranjit, Virginia Aquino, Noemí Toro-Barrios, Rosario Carmona, Eva Polverino, and et al. 2024. "Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene" Cells 13, no. 6: 524. https://doi.org/10.3390/cells13060524
APA StyleBaz-Redón, N., Sánchez-Bellver, L., Fernández-Cancio, M., Rovira-Amigo, S., Burgoyne, T., Ranjit, R., Aquino, V., Toro-Barrios, N., Carmona, R., Polverino, E., Cols, M., Moreno-Galdó, A., Camats-Tarruella, N., & Marfany, G. (2024). Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells, 13(6), 524. https://doi.org/10.3390/cells13060524