
Neutral Sphingomyelinase 2 Inhibition Limits Hepatic Steatosis and Inflammation

 , , ,
, , ,  , , , , , , , ,
, , , , , , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Immunohistochemical (IHC) Staining
2.3. Hematoxylin and Eosin (H&E) Staining and Steatosis Scoring
2.4. Quantification of Triglyceride Levels in the Liver Tissue
2.5. RNA Sequencing and Data Analysis
2.6. Bioinformatics Analyses
2.7. aSMase/nSMase2 Activity Assays
2.8. HepG2 Cell Model of Steatosis
2.9. MTT Assay
2.10. Flow Cytometry
2.11. BODIPY Lipid Staining and Quantification
2.12. Confocal Microscopy
2.13. Real-Time RT-qPCR
2.14. Western Blotting
2.15. Enzyme-Linked Immunosorbent Assay (ELISA)
2.16. Statistical Analyses
3. Results
3.1. Chronic HFD Feeding Induces NAFLD Phenotype in Mice
3.2. Transcriptomic/Bioinformatic Analyses Reveal Smpd3 (nSMase2) as a Potential Candidate Gene Associated with Liver Steatosis in HFD Mice
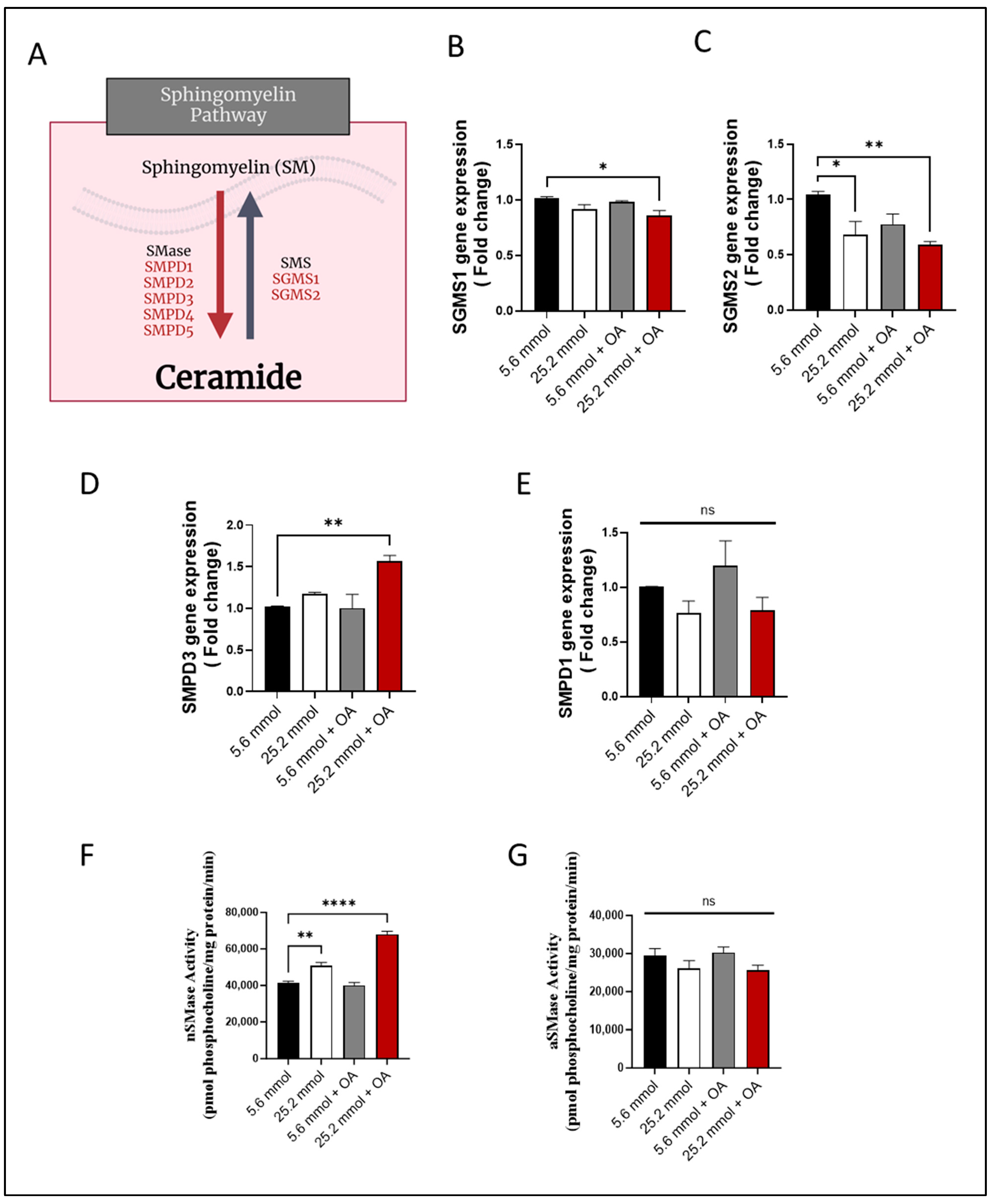
3.3. Characterization of HepG2 Cell Model of Steatosis and Role of SM Pathway
3.4. Effect of nSMase2 Inhibition in HepG2 Cell Model of Steatosis
3.5. nSMase2 Inhibition Reduces Inflammation, Oxidative Stress, and Apoptosis in HepG2 Cells
3.6. nSMase2 Deficiency Improves Steatosis-Associated Pathological Changes in HepG2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stefan, N.; Cusi, K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol. 2022, 10, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr. Diabetes Rep. 2010, 10, 306–315. [Google Scholar] [CrossRef]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Ahmad, Z.; Snider, A.J.; Thomas, R.; Kochumon, S.; Melhem, M.; Sindhu, S.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; et al. Ceramide kinase regulates tnf-α-induced immune responses in human monocytic cells. Sci. Rep. 2021, 11, 8259. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in non-alcoholic fatty liver disease and hepatocellular carcinoma: Ceramide turnover. Int. J. Mol. Sci. 2019, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef]
- Goñi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Eccleston, H.B.; Andringa, K.K.; Betancourt, A.M.; King, A.L.; Mantena, S.K.; Swain, T.M.; Tinsley, H.N.; Nolte, R.N.; Nagy, T.R.; Abrams, G.A.; et al. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid. Redox Signal. 2011, 15, 447–459. [Google Scholar] [CrossRef]
- de Meijer, V.E.; Le, H.D.; Meisel, J.A.; Akhavan Sharif, M.R.; Pan, A.; Nosé, V.; Puder, M. Dietary fat intake promotes the development of hepatic steatosis independently from excess caloric consumption in a murine model. Metabolism 2010, 59, 1092–1105. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Al-Mass, A.; Atizado, V.; Al-Hubail, A.; Al-Ghimlas, F.; Al-Arouj, M.; Bennakhi, A.; Dermime, S.; Behbehani, K. Elevated expression of the toll like receptors 2 and 4 in obese individuals: Its significance for obesity-induced inflammation. J. Inflamm. 2012, 9, 48. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: An integrated protocol. Bio Protoc. 2019, 9, e4610. [Google Scholar] [CrossRef]
- Yen, K.; Le, T.T.; Bansal, A.; Narasimhan, S.D.; Cheng, J.X.; Tissenbaum, H.A. A comparative study of fat storage quantitation in nematode caenorhabditis elegans using label and label-free methods. PLoS ONE 2010, 5, e12810. [Google Scholar] [CrossRef]
- Liu, Y.; Han, R.; Zhou, L.; Luo, M.; Zeng, L.; Zhao, X.; Ma, Y.; Zhou, Z.; Sun, L. Comparative performance of the genolab m and novaseq 6000 sequencing platforms for transcriptome and lncrna analysis. BMC Genom. 2021, 22, 829. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. Fqc dashboard: Integrates fastqc results into a web-based, interactive, and extensible fastq quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq--a python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Voronova, A.; Coyne, E.; Al Madhoun, A.; Fair, J.V.; Bosiljcic, N.; St-Louis, C.; Li, G.; Thurig, S.; Wallace, V.A.; Wiper-Bergeron, N.; et al. Hedgehog signaling regulates myod expression and activity. J. Biol. Chem. 2013, 288, 4389–4404. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Maher, A.; Nunez-Toldra, R.; Carrio, N.; Ferres-Padro, E.; Ali, H.; Montori, S.; Al Madhoun, A. The effect of commercially available endodontic cements and biomaterials on osteogenic differentiation of dental pulp pluripotent-like stem cells. Dent. J. 2018, 6, 48. [Google Scholar] [CrossRef]
- Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin induces inflammation and insulin resistance via the nf-κb and stat3 signaling pathways in hepatocytes. J. Diabetes Res. 2019, 2019, 4021623. [Google Scholar] [CrossRef]
- Akhter, N.; Wilson, A.; Thomas, R.; Al-Rashed, F.; Kochumon, S.; Al-Roub, A.; Arefanian, H.; Al-Madhoun, A.; Al-Mulla, F.; Ahmad, R.; et al. Ros/tnf-α crosstalk triggers the expression of il-8 and mcp-1 in human monocytic thp-1 cells via the nf-κb and erk1/2 mediated signaling. Int. J. Mol. Sci. 2021, 22, 10519. [Google Scholar] [CrossRef]
- Khavandgar, Z.; Murshed, M. Sphingolipid metabolism and its role in the skeletal tissues. Cell Mol. Life Sci. 2015, 72, 959–969. [Google Scholar] [CrossRef]
- Chatterjee, S. Neutral sphingomyelinase: Past, present and future. Chem. Phys. Lipids 1999, 102, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, S.; Sun, J.; Han, P.; Xu, B.; Li, X.; Zhong, Y.; Xu, Z.; Zhang, P.; Mi, P.; et al. m6A modification-tuned sphingolipid metabolism regulates postnatal liver development in male mice. Nat. Metab. 2023, 5, 842–860. [Google Scholar] [CrossRef] [PubMed]
- Wandrer, F.; Liebig, S.; Marhenke, S.; Vogel, A.; John, K.; Manns, M.P.; Teufel, A.; Itzel, T.; Longerich, T.; Maier, O.; et al. Tnf-receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in nafld mice. Cell Death Dis. 2020, 11, 212. [Google Scholar] [CrossRef]
- Potoupni, V.; Georgiadou, M.; Chatzigriva, E.; Polychronidou, G.; Markou, E.; Zapantis Gakis, C.; Filimidou, I.; Karagianni, M.; Anastasilakis, D.; Evripidou, K.; et al. Circulating tumor necrosis factor-α levels in non-alcoholic fatty liver disease: A systematic review and a meta-analysis. J. Gastroenterol. Hepatol. 2021, 36, 3002–3014. [Google Scholar] [CrossRef]
- Biswas, P.; Datta, C.; Rathi, P.; Bhattacharjee, A. Fatty acids and their lipid mediators in the induction of cellular apoptosis in cancer cells. Prostaglandins Other Lipid Mediat. 2022, 160, 106637. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. Cyp2e1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome proliferator-activated receptors (ppars) and oxidative stress in physiological conditions and in cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral sphingomyelinase 2 regulates inflammatory responses in monocytes/macrophages induced by tnf-α. Sci. Rep. 2020, 10, 16802. [Google Scholar] [CrossRef]
- Li, C.; Guo, S.; Pang, W.; Zhao, Z. Crosstalk between acid sphingomyelinase and inflammasome signaling and their emerging roles in tissue injury and fibrosis. Front. Cell Dev. Biol. 2019, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.G.; Way, G.W.; Zhou, H. Bile acids and sphingolipids in non-alcoholic fatty liver disease. Chin. Med. J. 2022, 135, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Torta, F.; Lin, M.K.; Olona, A.; Bagnati, M.; Moreno-Moral, A.; Ko, J.H.; Ji, S.; Burla, B.; Wenk, M.R.; et al. Immunolipidomics reveals a globoside network during the resolution of pro-inflammatory response in human macrophages. Front. Immunol. 2022, 13, 926220. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.D.; Wang, J.W. Ceramide de novo synthesis in non-alcoholic fatty liver disease: Pathogenic mechanisms and therapeutic perspectives. Biochem. Pharmacol. 2022, 202, 115157. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Bickel, P.E.; Scherer, P.E. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat. Rev. Drug Discov. 2016, 15, 639–660. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Lee, H.Y.; Jurczak, M.J.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.A.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid hormone receptor-α gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 2012, 153, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Sachwani, G.R.; Jaehne, A.K.; Jayaprakash, N.; Kuzich, M.; Onkoba, V.; Blyden, D.; Rivers, E.P. The association between blood glucose levels and matrix-metalloproteinase-9 in early severe sepsis and septic shock. J. Inflamm. 2016, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Shinke, T.; Otake, H.; Sugiyama, D.; Takaya, T.; Takahashi, H.; Terashita, D.; Uzu, K.; Tahara, N.; Kashiwagi, D.; et al. Effects of daily glucose fluctuations on the healing response to everolimus-eluting stent implantation as assessed using continuous glucose monitoring and optical coherence tomography. Cardiovasc. Diabetol. 2016, 15, 79. [Google Scholar] [CrossRef][Green Version]
- Al-Rashed, F.; Sindhu, S.; Arefanian, H.; Al Madhoun, A.; Kochumon, S.; Thomas, R.; Al-Kandari, S.; Alghaith, A.; Jacob, T.; Al-Mulla, F.; et al. Repetitive intermittent hyperglycemia drives the m1 polarization and inflammatory responses in thp-1 macrophages through the mechanism involving the tlr4-irf5 pathway. Cells 2020, 9, 1892. [Google Scholar] [CrossRef]
- Ishii, Y.; Tsuchiya, A.; Natsui, K.; Koseki, Y.; Takeda, N.; Tomiyoshi, K.; Yamazaki, F.; Yoshida, Y.; Shimbo, T.; Tamai, K.; et al. Synthesized hmgb1 peptide prevents the progression of inflammation, steatosis, fibrosis, and tumor occurrence in a nonalcoholic steatohepatitis mouse model. Hepatol. Res. 2022, 52, 985–997. [Google Scholar] [CrossRef]
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C. Pathogenesis and therapeutic strategies related to non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2022, 23, 7841. [Google Scholar] [CrossRef]
- Meng, L.C.; Zheng, J.Y.; Qiu, Y.H.; Zheng, L.; Liu, Y.Q.; Miao, X.L.; Lu, X.Y. Salvianolic acid b ameliorates non-alcoholic fatty liver disease by inhibiting hepatic lipid accumulation and nlrp3 inflammasome in ob/ob mice. Int. Immunopharmacol. 2022, 111, 109099. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, T.; Wang, T.; Liu, X.; Hamada, K.; Sun, D.; Sun, Y.; Yang, Y.; Wang, J.; Takahashi, S.; et al. Crosstalk between cyp2e1 and ppar. Acta Pharm. Sin. B 2022, 12, 2224–2238. [Google Scholar] [CrossRef]
- Li, Z.; Chiang, Y.P.; He, M.; Worgall, T.S.; Zhou, H.; Jiang, X.C. Liver sphingomyelin synthase 1 deficiency causes steatosis, steatohepatitis, fibrosis, and tumorigenesis: An effect of glucosylceramide accumulation. iScience 2021, 24, 103449. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective tnf-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Ahn, H.K.; Lee, E.J.; Kim, S.J.; Kim, Y.R.; Park, J.W.; Park, W.J. Hepatic inflammatory cytokine production can be regulated by modulating sphingomyelinase and ceramide synthase 6. Int. J. Mol. Med. 2017, 39, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol. Res. 2012, 42, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M.; Karakashian, A.; Rutkute, K. Role of neutral sphingomyelinases in aging and inflammation. Subcell. Biochem. 2008, 49, 469–486. [Google Scholar] [PubMed]
- Jin, J.; Zhang, X.; Lu, Z.; Perry, D.M.; Li, Y.; Russo, S.B.; Cowart, L.A.; Hannun, Y.A.; Huang, Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E853–E867. [Google Scholar] [CrossRef] [PubMed]
- Tibbles, L.A.; Woodgett, J.R. The stress-activated protein kinase pathways. Cell Mol. Life Sci. 1999, 55, 1230–1254. [Google Scholar] [CrossRef]
- Moran, E.; Ding, L.; Wang, Z.; Cheng, R.; Chen, Q.; Moore, R.; Takahashi, Y.; Ma, J.X. Protective and antioxidant effects of pparα in the ischemic retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4568–4576. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Q.; Li, H.; Lan, X.; Kan, M.; Lin, J.; Wang, J.; Zhang, Z.; Ming, S.; Li, Z.; et al. The anti-aging effect of velvet antler polypeptide is dependent on modulation of the gut microbiota and regulation of the pparα/apoe4 pathway. J. Integr. Neurosci. 2021, 20, 573–583. [Google Scholar] [PubMed]
- Kim, M.J.; Kim, D.H.; Bang, E.; Noh, S.G.; Chun, P.; Yokozawa, T.; Moon, H.R.; Chung, H.Y. Pparα agonist, mhy3200, alleviates renal inflammation during aging via regulating ros/akt/foxo1 signaling. Molecules 2021, 26, 3197. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver pparα is crucial for whole-body fatty acid homeostasis and is protective against nafld. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Rashed, F.; Arefanian, H.; Madhoun, A.A.; Bahman, F.; Sindhu, S.; AlSaeed, H.; Jacob, T.; Thomas, R.; Al-Roub, A.; Alzaid, F.; et al. Neutral Sphingomyelinase 2 Inhibition Limits Hepatic Steatosis and Inflammation. Cells 2024, 13, 463. https://doi.org/10.3390/cells13050463
Al-Rashed F, Arefanian H, Madhoun AA, Bahman F, Sindhu S, AlSaeed H, Jacob T, Thomas R, Al-Roub A, Alzaid F, et al. Neutral Sphingomyelinase 2 Inhibition Limits Hepatic Steatosis and Inflammation. Cells. 2024; 13(5):463. https://doi.org/10.3390/cells13050463
Chicago/Turabian StyleAl-Rashed, Fatema, Hossein Arefanian, Ashraf Al Madhoun, Fatemah Bahman, Sardar Sindhu, Halemah AlSaeed, Texy Jacob, Reeby Thomas, Areej Al-Roub, Fawaz Alzaid, and et al. 2024. "Neutral Sphingomyelinase 2 Inhibition Limits Hepatic Steatosis and Inflammation" Cells 13, no. 5: 463. https://doi.org/10.3390/cells13050463
APA StyleAl-Rashed, F., Arefanian, H., Madhoun, A. A., Bahman, F., Sindhu, S., AlSaeed, H., Jacob, T., Thomas, R., Al-Roub, A., Alzaid, F., Malik, M. Z., Nizam, R., Thanaraj, T. A., Al-Mulla, F., Hannun, Y. A., & Ahmad, R. (2024). Neutral Sphingomyelinase 2 Inhibition Limits Hepatic Steatosis and Inflammation. Cells, 13(5), 463. https://doi.org/10.3390/cells13050463