
GLA Mutations Suppress Autophagy and Stimulate Lysosome Generation in Fabry Disease

 ,
, 
Abstract
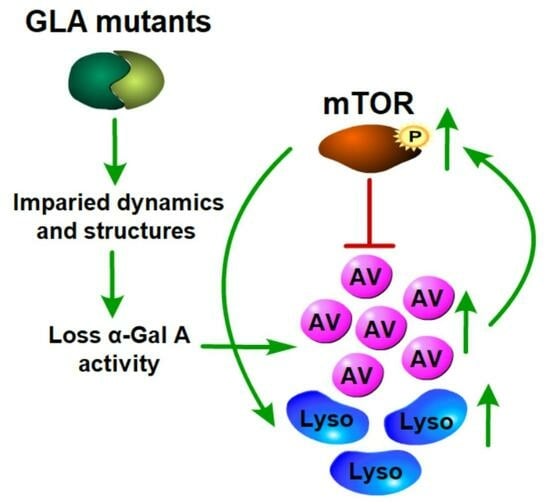
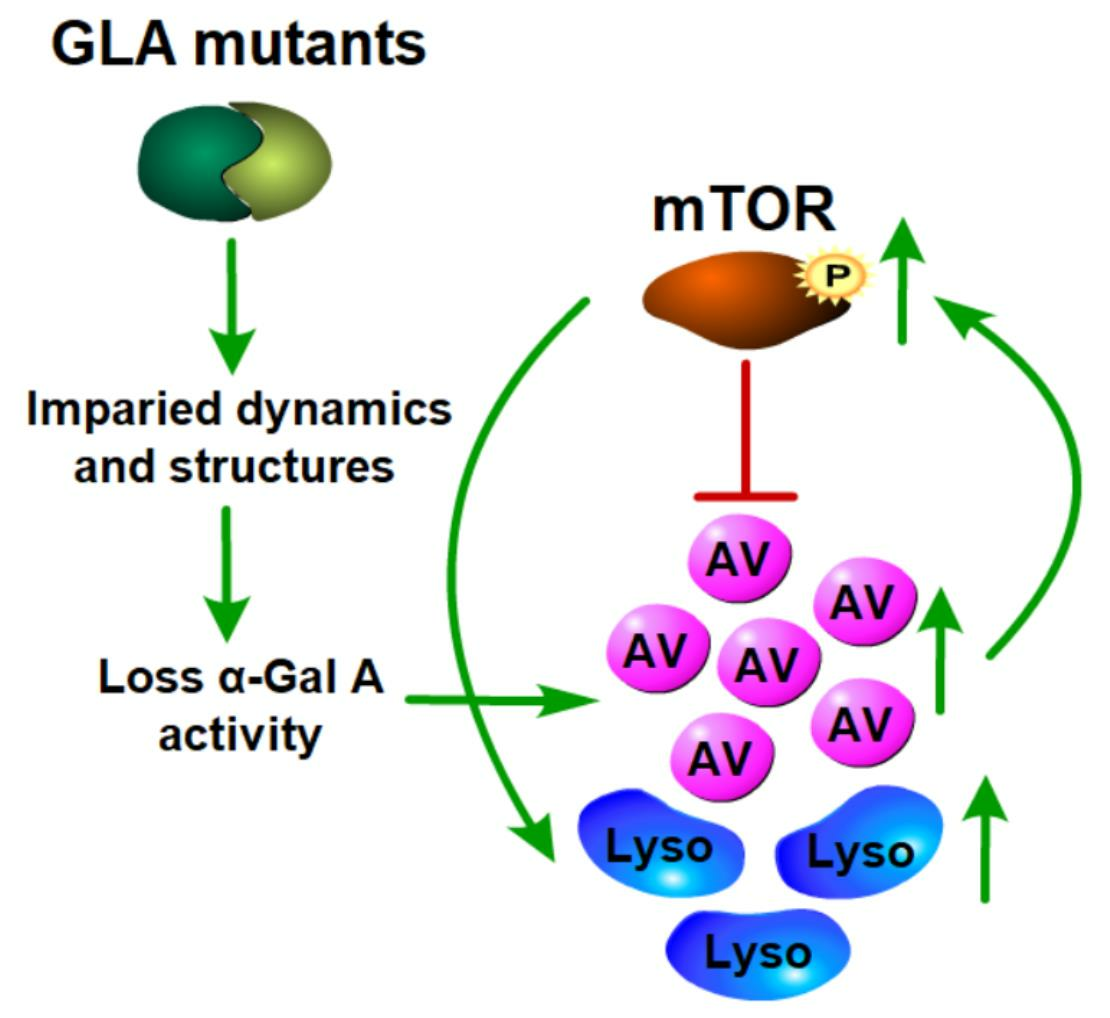{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Evolutionary Conservation and Structure Preparation
2.2. Molecular Dynamic Simulation
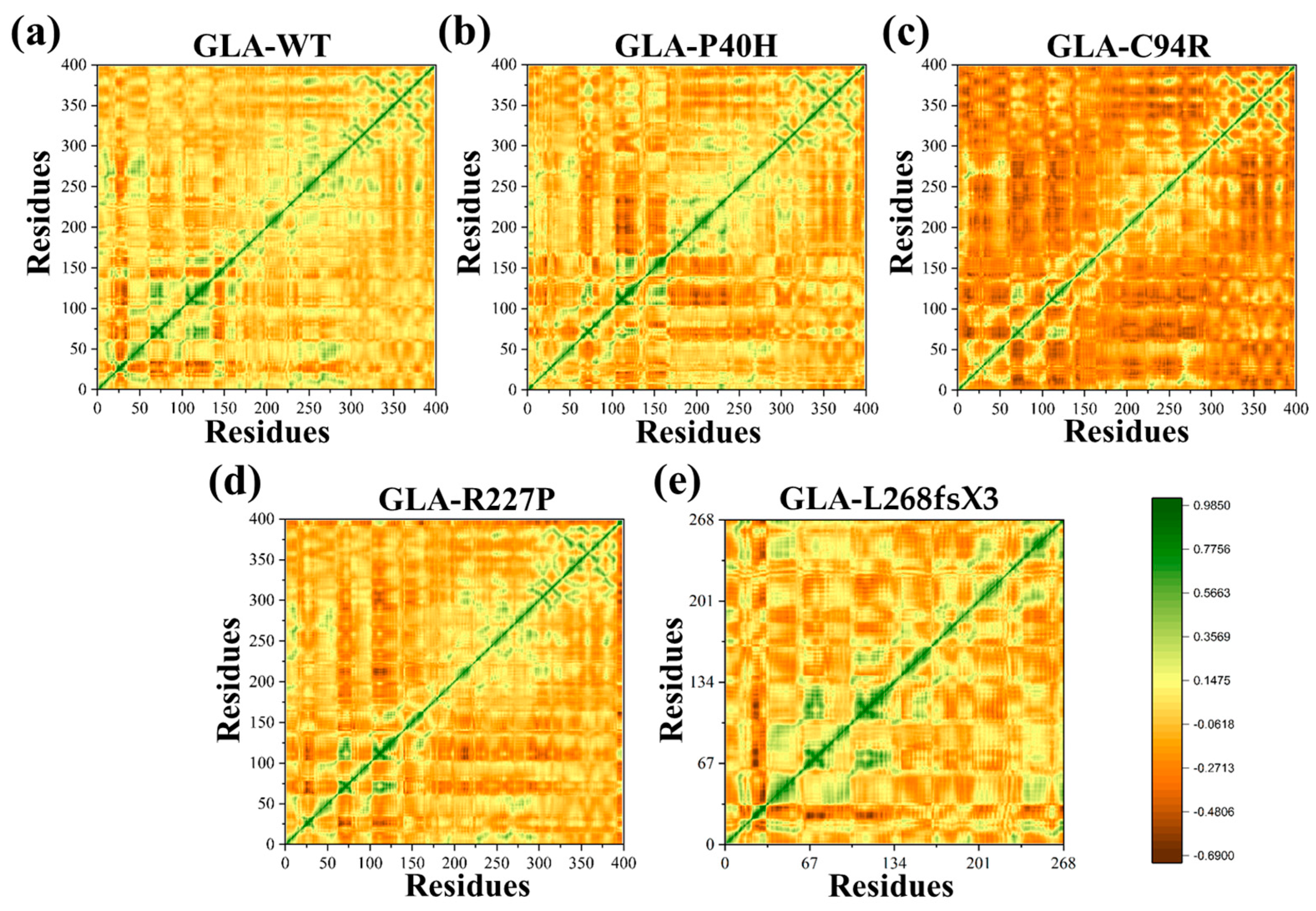
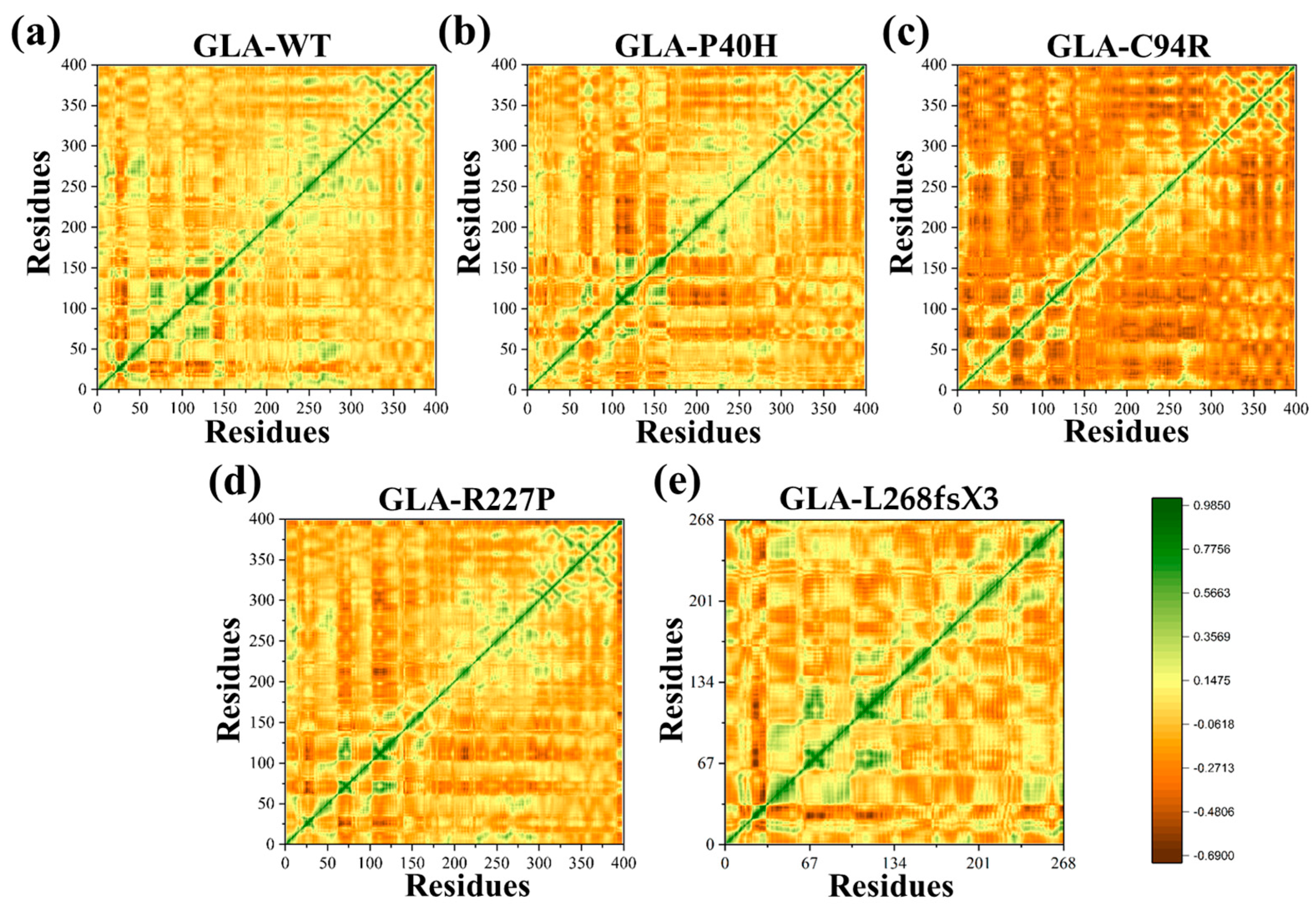
2.3. Dynamic Cross-Correlation Analysis (DCCM)
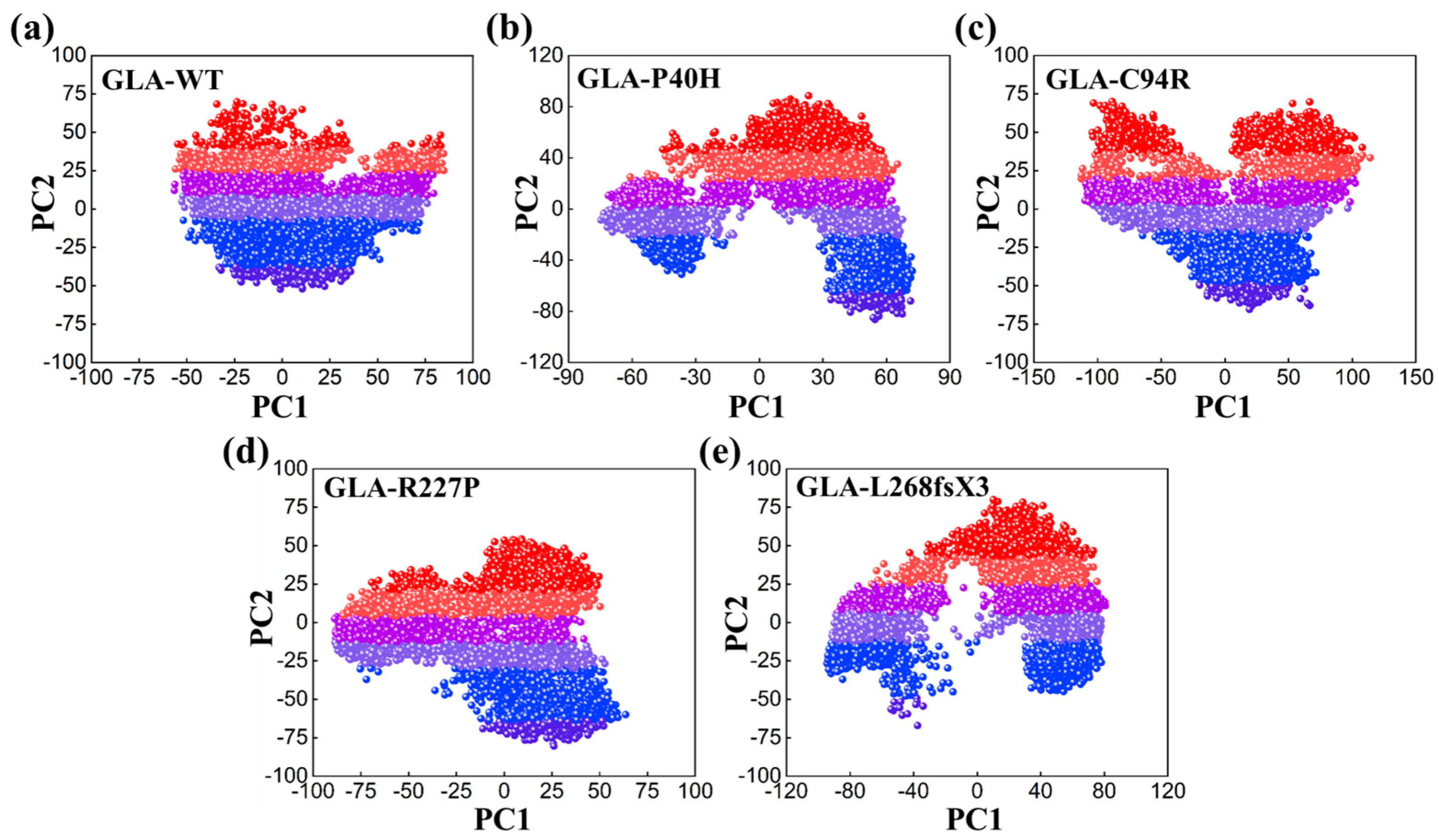
2.4. Principal Component Analysis (PCA)
2.5. Cell Culture and Ctable Cell Line Construction
2.6. GLA Enzyme Activity Assay
2.7. Western Blot Analysis
2.8. Immunofluorescence Analysis
2.9. Statistical Analysis
3. Results
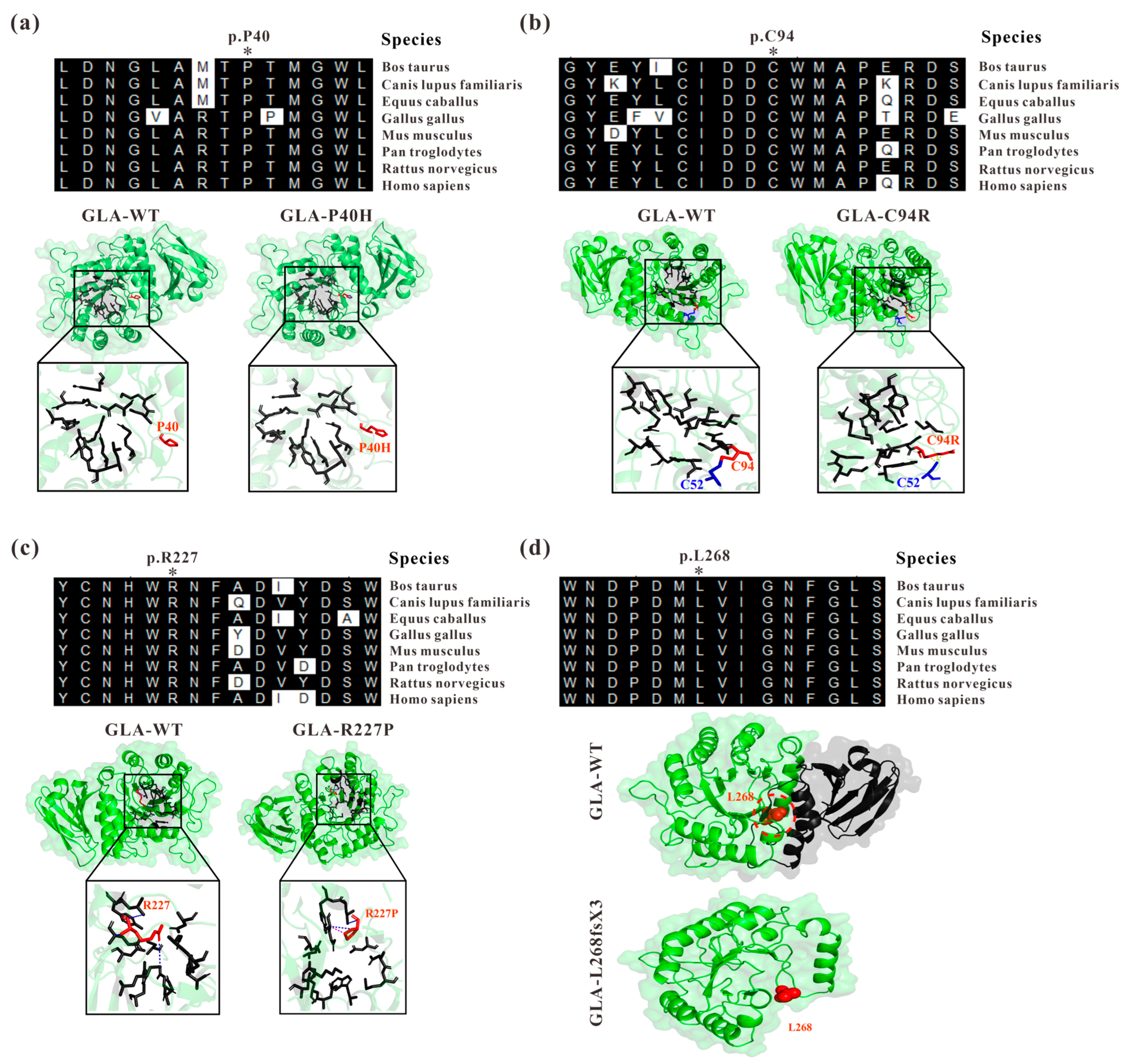
3.1. Abnormal Three-Dimensional Structure of α-Galactosidase A Caused by GLA Mutations
3.2. Structural Perturbations upon GLA Mutations
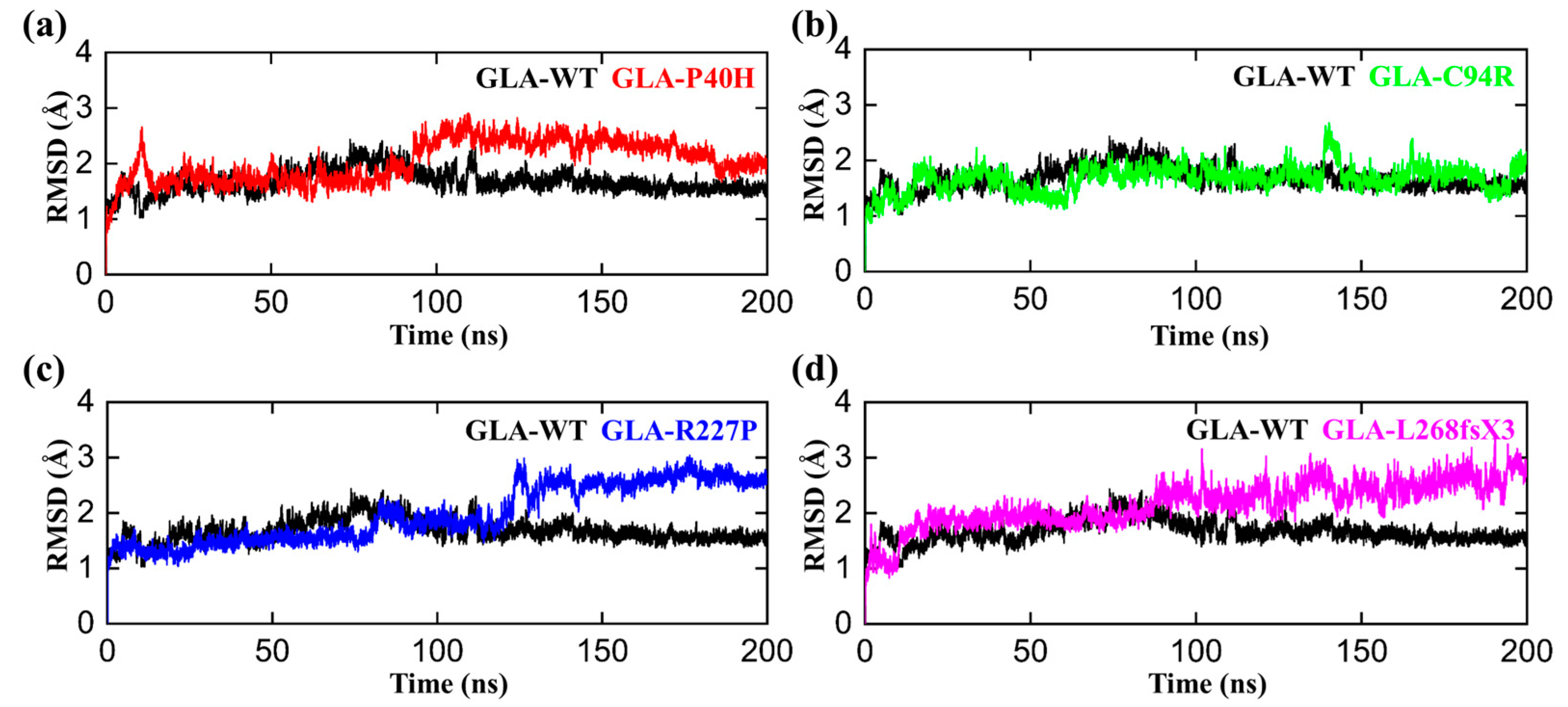
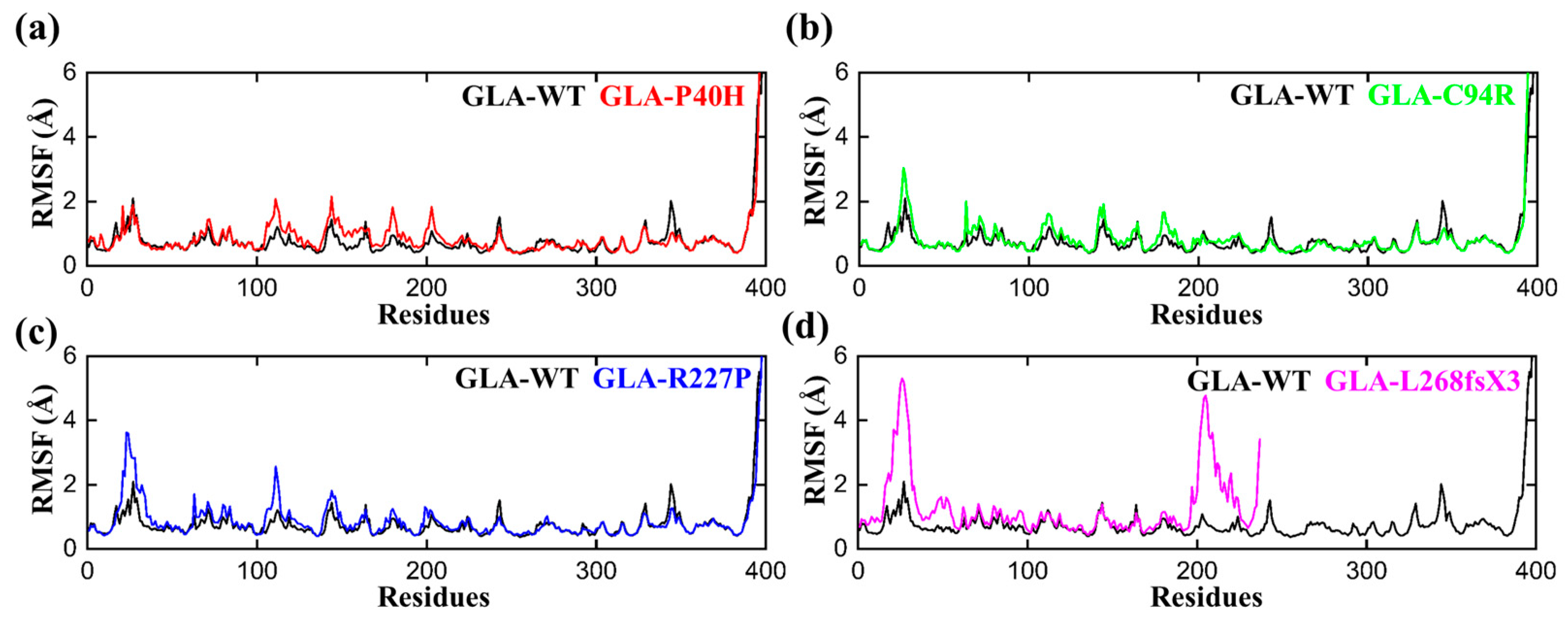
3.3. Flexibility Comparison of Wildtype and Mutant GLA Structures
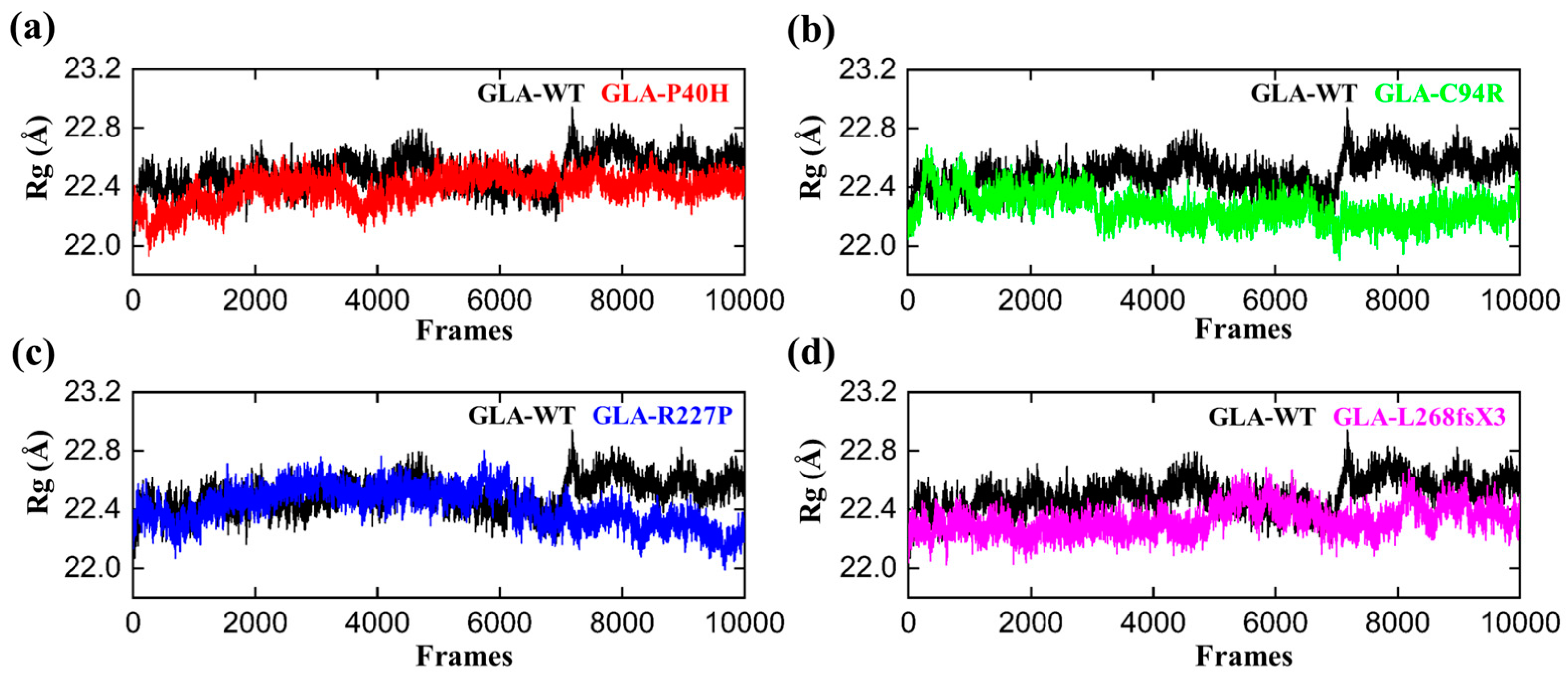
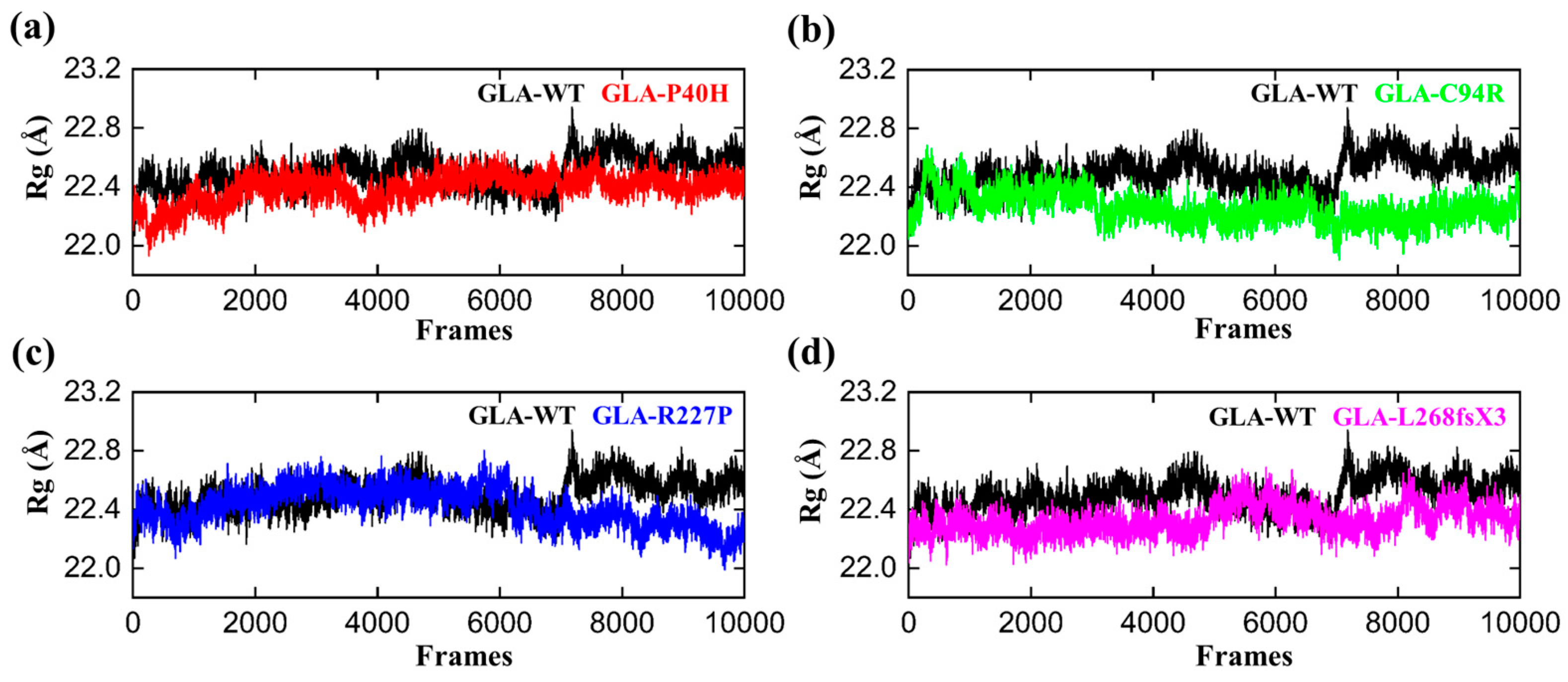
3.4. Structural Compactness of GLA upon Acquring Mutations
3.5. Functional Displacement of GLA and Mutants
3.6. Impacts on the Internal Dynamics and Structures of GLA Mutants
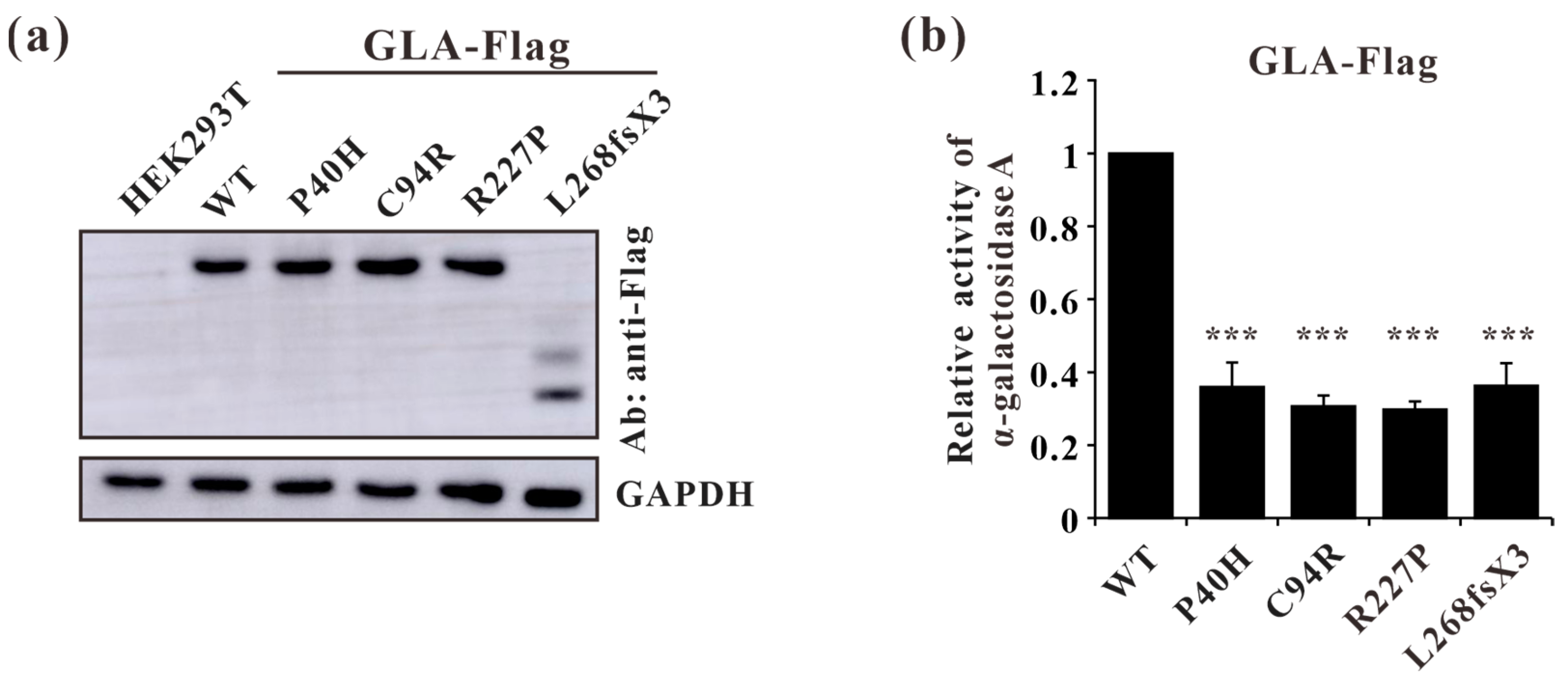
3.7. Impaired Enzyme Activities of GLA Mutants
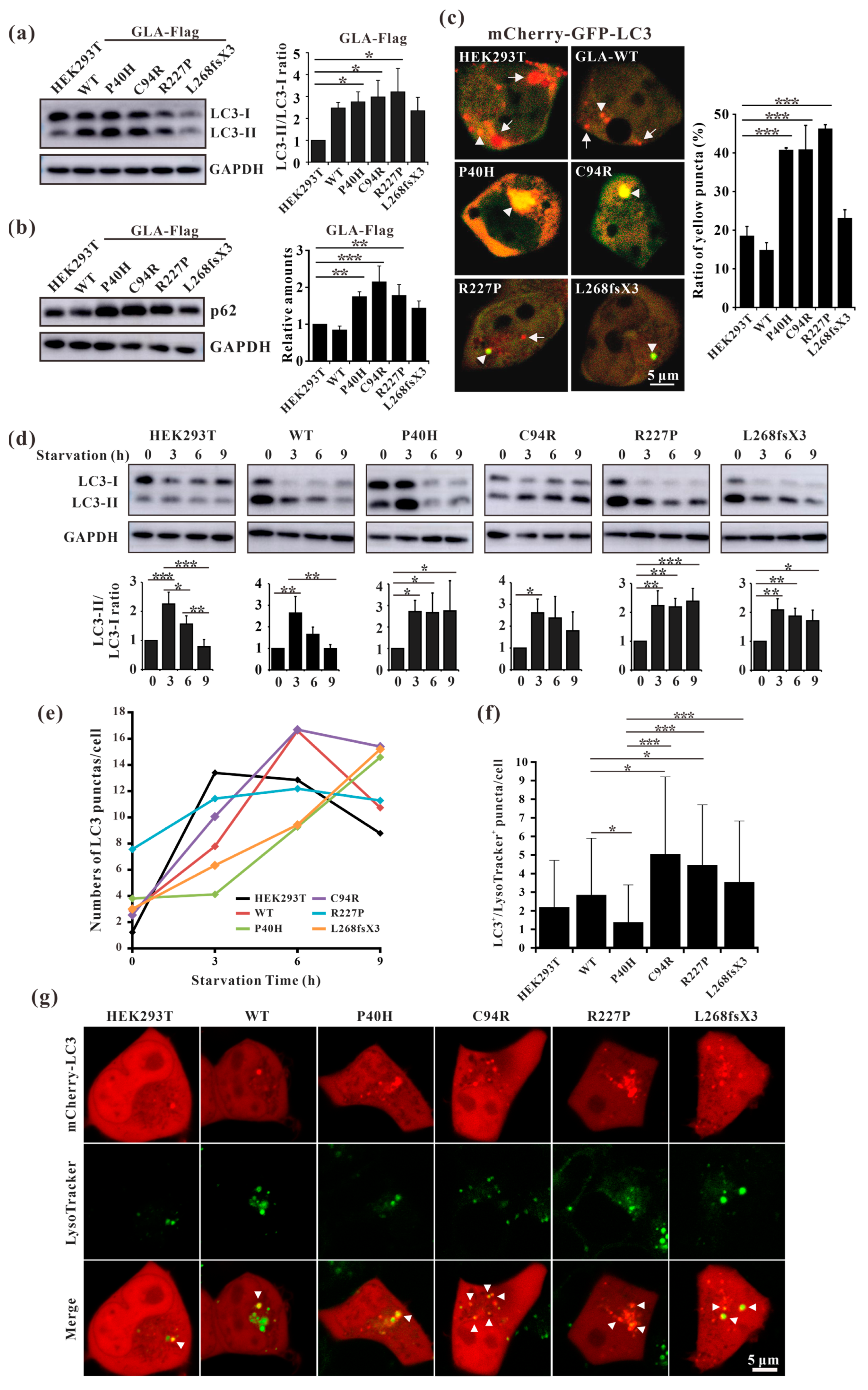
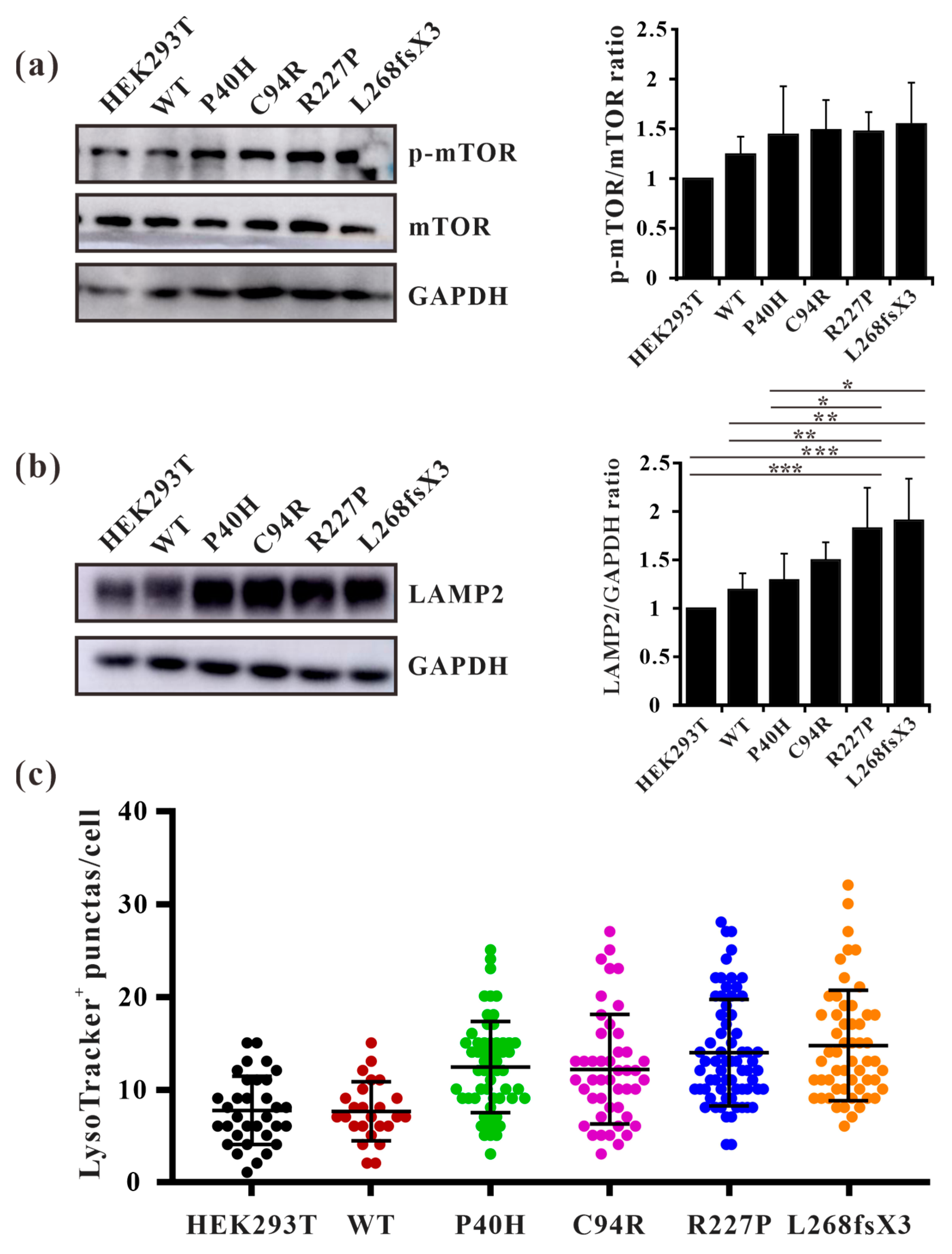
3.8. GLA Mutants Cause Intracellular Accumulation of Autophagosomes
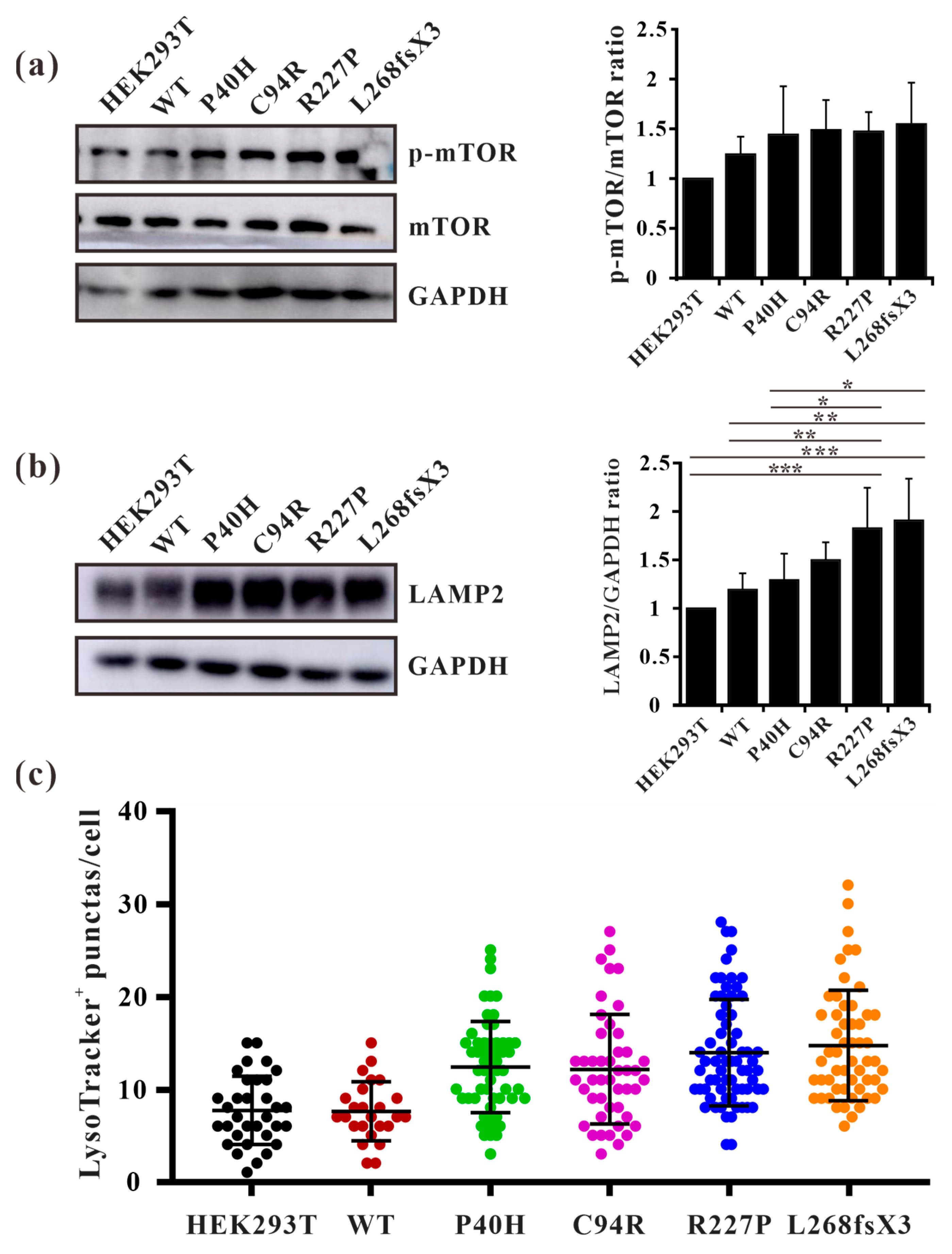
3.9. GLA Mutation Leads to an Increase of Lysosome Numbers in Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Desnick, R.J.; Wasserstein, M.P.; Banikazemi, M. Fabry disease (alpha-galactosidase A deficiency): Renal involvement and enzyme replacement therapy. Contrib. Nephrol. 2001, 136, 174–192. [Google Scholar] [CrossRef]
- Hopkin, R.J.; Bissler, J.; Banikazemi, M.; Clarke, L.; Eng, C.M.; Germain, D.P.; Lemay, R.; Tylki-Szymanska, A.; Wilcox, W.R. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr. Res. 2008, 64, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Vedder, A.C.; Linthorst, G.E.; van Breemen, M.J.; Groener, J.E.; Bemelman, F.J.; Strijland, A.; Mannens, M.M.; Aerts, J.M.; Hollak, C.E. The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels. J. Inherit. Metab. Dis. 2007, 30, 68–78. [Google Scholar] [CrossRef]
- Yokoyama, T.; Manabe, S.; Horita, S.; Kataoka, H.; Mochizuki, T.; Nitta, K. The origin of urinary mulberry cells in Fabry disease. Kidney Int. 2021, 99, 1246. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Tondel, C.; Svarstad, E.; Gubler, M.C.; Oliveira, J.P.; Mauer, M. Accumulation of Globotriaosylceramide in Podocytes in Fabry Nephropathy Is Associated with Progressive Podocyte Loss. J. Am. Soc. Nephrol. 2020, 31, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, A.; Camporeale, A.; Pieroni, M.; Pieruzzi, F.; Figliozzi, S.; Lusardi, P.; Spada, M.; Mignani, R.; Burlina, A.; Carubbi, F.; et al. Atrial Dysfunction Assessed by Cardiac Magnetic Resonance as an Early Marker of Fabry Cardiomyopathy. JACC Cardiovasc. Imaging 2020, 13, 2262–2264. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagege, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Chimenz, R.; Chirico, V.; Cuppari, C.; Ceravolo, G.; Concolino, D.; Monardo, P.; Lacquaniti, A. Fabry disease and kidney involvement: Starting from childhood to understand the future. Pediatr. Nephrol. 2022, 37, 95–103. [Google Scholar] [CrossRef]
- Kornreich, R.; Desnick, R.J.; Bishop, D.F. Nucleotide sequence of the human alpha-galactosidase A gene. Nucleic Acids Res. 1989, 17, 3301–3302. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.F.; Calhoun, D.H.; Bernstein, H.S.; Hantzopoulos, P.; Quinn, M.; Desnick, R.J. Human alpha-galactosidase A: Nucleotide sequence of a cDNA clone encoding the mature enzyme. Proc. Natl. Acad. Sci. USA 1986, 83, 4859–4863. [Google Scholar] [CrossRef] [PubMed]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Whybra, C.; Kampmann, C.; Willers, I.; Davies, J.; Winchester, B.; Kriegsmann, J.; Bruhl, K.; Gal, A.; Bunge, S.; Beck, M. Anderson-Fabry disease: Clinical manifestations of disease in female heterozygotes. J. Inherit. Metab. Dis. 2001, 24, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma lyso-Gb3: A biomarker for monitoring fabry patients during enzyme replacement therapy. Clin. Exp. Nephrol. 2018, 22, 843–849. [Google Scholar] [CrossRef]
- Campbell, K.N.; Tumlin, J.A. Protecting Podocytes: A Key Target for Therapy of Focal Segmental Glomerulosclerosis. Am. J. Nephrol. 2018, 47 (Suppl. S1), 14–29. [Google Scholar] [CrossRef]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Zhao, N.; Xiong, Q.; Zhou, Y.A.; Wu, C.; Xiao, H. A Novel alpha-Galactosidase A Splicing Mutation Predisposes to Fabry Disease. Front. Genet. 2019, 10, 60. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Xiong, Q.; Wang, Z.; Cui, X.; Zhou, Y.A.; Wang, Y.; Xiao, H.; Wu, C. Functional evaluation of a novel GLA causative mutation in Fabry disease. Mol. Genet. Genom. Med. 2019, 7, e864. [Google Scholar] [CrossRef]
- Ferri, L.; Malesci, D.; Fioravanti, A.; Bagordo, G.; Filippini, A.; Ficcadenti, A.; Manna, R.; Antuzzi, D.; Verrecchia, E.; Donati, I.; et al. Functional and pharmacological evaluation of novel GLA variants in Fabry disease identifies six (two de novo) causative mutations and two amenable variants to the chaperone DGJ. Clin. Chim. Acta 2018, 481, 25–33. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Samad, A.; Shah, A.A.; Wadood, A.; Alkathiri, A.; Alshehri, M.A.; Alam, M.Z.; Hussain, T.; He, P.; Umair, M. A novel biallelic variant in the Popeye domain-containing protein 1 (POPDC1) underlies limb girdle muscle dystrophy type 25. Clin. Genet. 2023, 103, 219–225. [Google Scholar] [CrossRef]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D., Jr.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Dickson, C.J.; Walker, R.C.; Gould, I.R. Lipid21: Complex Lipid Membrane Simulations with AMBER. J. Chem. Theory Comput. 2022, 18, 1726–1736. [Google Scholar] [CrossRef]
- Xie, H.; Li, Y.; Yu, F.; Xie, X.; Qiu, K.; Fu, J. An Investigation of Molecular Docking and Molecular Dynamic Simulation on Imidazopyridines as B-Raf Kinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 27350–27361. [Google Scholar] [CrossRef]
- Ajmal, A.; Ali, Y.; Khan, A.; Wadood, A.; Rehman, A.U. Identification of novel peptide inhibitors for the KRas-G12C variant to prevent oncogenic signaling. J. Biomol. Struct. Dyn. 2023, 41, 8866–8875. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Ajmal, A.; Mahmood, A.; Khurshid, B.; Li, P.; Jan, S.M.; Rehman, A.U.; He, P.; Abdalla, A.N.; Umair, M.; et al. Identification of novel inhibitors for SARS-CoV-2 as therapeutic options using machine learning-based virtual screening, molecular docking and MD simulation. Front. Mol. Biosci. 2023, 10, 1060076. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.W.; Whitehair, O.; Wu, M.J.; O’Meara, M.; Lien, Y.H. Analysis of splice-site mutations of the alpha-galactosidase A gene in Fabry disease. Clin. Genet. 2003, 63, 476–482. [Google Scholar] [CrossRef]
- Samad, A.; Khurshid, B.; Mahmood, A.; Rehman, A.U.; Khalid, A.; Abdalla, A.N.; Algarni, A.S.; Wadood, A. Identification of novel peptide inhibitors for oncogenic KRAS G12D as therapeutic options using mutagenesis-based remodeling and MD simulations. J. Biomol. Struct. Dyn. 2023, 41, 13425–13437. [Google Scholar] [CrossRef]
- Arshia, A.H.; Shadravan, S.; Solhjoo, A.; Sakhteman, A.; Sami, A. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Comput. Biol. Med. 2021, 139, 104967. [Google Scholar] [CrossRef]
- Sharma, A.; Sartori, M.; Zaragoza, J.J.; Villa, G.; Lu, R.; Faggiana, E.; Brocca, A.; Di Lullo, L.; Feriozzi, S.; Ronco, C. Fabry’s disease: An example of cardiorenal syndrome type 5. Heart Fail. Rev. 2015, 20, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Hossain, M.A.; Miyajima, T.; Nagao, K.; Miyashita, T.; Eto, Y. Dysregulated DNA methylation of GLA gene was associated with dysfunction of autophagy. Mol. Genet. Metab. 2019, 126, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Chevrier, M.; Brakch, N.; Celine, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerriere, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef]
- Song, H.Y.; Chien, C.S.; Yarmishyn, A.A.; Chou, S.J.; Yang, Y.P.; Wang, M.L.; Wang, C.Y.; Leu, H.B.; Yu, W.C.; Chang, Y.L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef]
- Melick, C.H.; Jewell, J.L. Regulation of mTORC1 by Upstream Stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.M.; do Monte, S.J.; do Nascimento, F.F.; de Castro, J.A.; Sousa, J.L.; Filho, H.C.; da Silva, R.N.; Labilloy, A.; Monte Neto, J.T.; da Silva, A.S. Lysosome-associated protein 1 (LAMP-1) and lysosome-associated protein 2 (LAMP-2) in a larger family carrier of Fabry disease. Gene 2014, 536, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Precision medicine in Fabry disease. Nephrol. Dial. Transplant. 2021, 36, 14–23. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Xi, Y.; Zhang, Y.; Samad, A.; Lan, W.; Wu, Y.; Zhao, J.; Chen, G.; Wu, C.; Xiong, Q. GLA Mutations Suppress Autophagy and Stimulate Lysosome Generation in Fabry Disease. Cells 2024, 13, 437. https://doi.org/10.3390/cells13050437
Li P, Xi Y, Zhang Y, Samad A, Lan W, Wu Y, Zhao J, Chen G, Wu C, Xiong Q. GLA Mutations Suppress Autophagy and Stimulate Lysosome Generation in Fabry Disease. Cells. 2024; 13(5):437. https://doi.org/10.3390/cells13050437
Chicago/Turabian StyleLi, Ping, Yuqian Xi, Yanping Zhang, Abdus Samad, Wenli Lan, Ya Wu, Jiayu Zhao, Guangxin Chen, Changxin Wu, and Qiuhong Xiong. 2024. "GLA Mutations Suppress Autophagy and Stimulate Lysosome Generation in Fabry Disease" Cells 13, no. 5: 437. https://doi.org/10.3390/cells13050437
APA StyleLi, P., Xi, Y., Zhang, Y., Samad, A., Lan, W., Wu, Y., Zhao, J., Chen, G., Wu, C., & Xiong, Q. (2024). GLA Mutations Suppress Autophagy and Stimulate Lysosome Generation in Fabry Disease. Cells, 13(5), 437. https://doi.org/10.3390/cells13050437