Abstract
The mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that functions via its discrete binding partners to form two multiprotein complexes, mTOR complex 1 and 2 (mTORC1 and mTORC2). Rapamycin-sensitive mTORC1, which regulates protein synthesis and cell growth, is tightly controlled by PI3K/Akt and is nutrient-/growth factor-sensitive. In the brain, mTORC1 is also sensitive to neurotransmitter signaling. mTORC2, which is modulated by growth factor signaling, is associated with ribosomes and is insensitive to rapamycin. mTOR regulates stem cell and cancer stem cell characteristics. Aberrant Akt/mTOR activation is involved in multistep tumorigenesis in a variety of cancers, thereby suggesting that the inhibition of mTOR may have therapeutic potential. Rapamycin and its analogues, known as rapalogues, suppress mTOR activity through an allosteric mechanism that only suppresses mTORC1, albeit incompletely. ATP-catalytic binding site inhibitors are designed to inhibit both complexes. This review describes the regulation of mTOR and the targeting of its complexes in the treatment of cancers, such as glioblastoma, and their stem cells.
1. Introduction
Mechanistic target of rapamycin (mTOR; also known as mammalian target of rapamycin), an atypical serine/threonine (S/T) protein kinase, is a member of the phosphoinositide 3-kinase-related kinases (PI3K), which are conserved in all eukaryotes, localized in chromosome 1p36.22 [1]. The name mTOR originates from its inhibitor rapamycin, also known as sirolimus, which forms a complex with FK506-binding protein 12 (FKBP12) to inhibit its activity [2,3]. mTOR is a 289 kDa protein that regulates multiple cellular processes, including protein translation and metabolism. The deregulation of mTOR and associated proteins in its signaling pathway results in aberrant cellular growth, proliferation, migration, and survival, contributing to both the pathogenesis and therapy resistance of many cancers [2,3,4,5,6]. Structurally, the C-terminus contains a kinase domain, placing mTOR in the PI3K family, and includes the FKBP-rapamycin-binding (FRB) domain. Meanwhile the middle segment is categorized by a FAT (FRAP/ATM/TRRAP) domain, and the N-terminus mediates most interactions with the associated proteins via its numerous HEAT (Huntingtin; elongation factor 3, EF3; protein phosphatase 2A, PP2A; and kinase TOR1) repeats (See Figure 1A) [7,8].
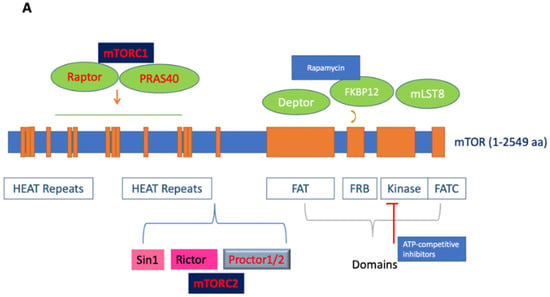
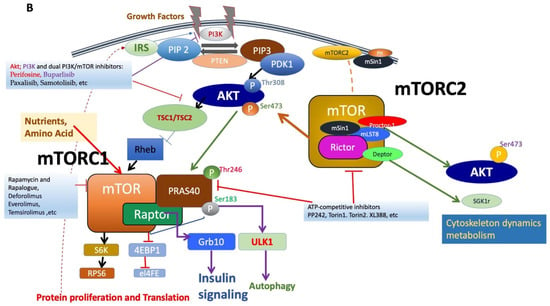
Figure 1.
(A). Figure depicting the mTOR structure, consisting of 2549 amino acids, which has several domains essential for the activity of mTORC1 and mTORC2. mTORC1 has several binding partners, including Raptor and PRAS40, while mTORC2 has Rictor, Sin1, and Protor1/2. In addition, the figure shows FKBP12, a rapamycin-binding partner. See text for additional details. (B). Illustration of the mTORC1 and mTORC2 signaling pathway. Activated PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to form PIP3. PIP3 binds to PDK1/AKT via the PH-domains to mediate Akt phosphorylation, which is further facilitated by the activation of mTORC2. Activated Akt promotes both the phosphorylation of PRAS40 on Thr246 and the inhibition of TSC1/TSC2 complex activity, resulting in increased GTP-bound Rheb levels and mTORC1 activation. Activated mTORC1 then acts on multiple protein substrates, including 4E-BP1, S6K, and PRAS40. The phosphorylation of 4E-BP1 and S6K by activated mTORC1 regulates multiple functions, including mRNA translation, cellular growth, and proliferation. Furthermore, S6K provides negative feedback to inhibit the insulin-signaling pathway via IRS, which is disinhibited along with subsequent mitogenic pathways following the prolonged inhibition of mTORC1 (dotted line). Representative inhibitors: rapamycin and rapalogues; ATP binding inhibitors; Akt, PI3K, and dual PI3K/mTOR inhibitors are presented (for details, see Table 1 and text).
mTOR functions by forming two major multiprotein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (See Figure 1B). mTORC1 is formed by the regulatory associated protein of mTOR (Raptor), proline-rich Akt substrate of 40 kDa (PRAS40), mammalian lethal with Sec-13 protein 8 (mLST8; also known as GßL), and DEP domain TOR-binding protein (Deptor) [3,9,10]. While Raptor does not possess any innate enzymatic activity, it is integral to the kinase activity of mTORC1 via its promotion of complex formation [11,12]. PRAS40 contains an mTOR signaling motif, and its overexpression competes with other mTORC1 targets for phosphorylation. PRAS40 responds to growth factor depletion to suppress mTORC1 activation [3,9]. Patients who underwent treatment with rapamycin had elevated activated PRAS40 expression and displayed levels of activated Akt as well as therapeutic resistance, suggesting the significance of activated PRAS40 as a surrogate marker for activated Akt [13]. Another component of mTORC1, mLST8, is limited in most mechanisms of mTORC1 activation but contributes to its activation by amino acids. While the upstream regulation of Deptor remains largely unknown, it is a component of mTORC1 and mTORC2, capable of inhibiting the activity of both complexes [14]. mTORC1 senses and controls cellular growth in response to various cellular signals, such as insulin or growth factors. Its targets have been previously characterized, particularly ribosomal S6 kinases and eukaryotic initiation factor 4E-binding proteins (eIF4E-BP1; also known as 4E-BP1). mTORC1 promotes the initiation of protein translation by associating with the eukaryotic initiation factor 3 (eIF3) complexes to phosphorylate these substrates. mTORC1 promotes protein translation via the activation of p70 S6 kinase (S6K) and the inhibition of 4E-BP1 and enhances RNA translation through the S6 ribosomal protein [2,15]. S6K exists as two distinct isoforms, S6K1 and S6K2; S6K1 consists of a 70 kDa cytoplasmic isoform and an 85 kDa nuclear isoform. Activated S6K appears to phosphorylate the 40S ribosomal subunit to increase the translational efficiency of a specific class of polypyrimidine mRNA transcripts, thereby regulating protein synthesis. S6K may also phosphorylate eukaryotic initiation factor 4B (eIF4B) at Ser422, allowing the association between eIF4B and eIF3, thus regulating translation via the promotion of eukaryotic initiation factor 4F (eIF4F) complex formation. Moreover, the phosphorylation of 4E-BP1 by mTOR regulates protein synthesis and promotes cap-dependent translation by releasing eIF4E, allowing its association with eukaryotic initiation factor 4G (eIF4G) among other factors. The translational control of nuclear S6K has been shown to regulate the transition from G1 to S phase in DNA synthesis [9].
In mTORC1’s downstream pathway, S6K performs various functions, including the phosphorylation of several targets, such as tumor suppressor programmed cell death protein 4 (PDCD4) or insulin receptor substrate-1 (IRS-1), thereby leading to its degradation and inhibiting PI3K and Akt in a known negative feedback loop [16,17]. Alternatively, Akt inhibition does not occur exclusively in the presence of insulin-like growth factor 1 (IGF-1); thus, mTOR may inhibit Akt via various mechanisms in the presence of different growth factors [18]. Recently, growth factor receptor-bound protein 10 (GRB10) has also been identified as an mTORC1 substrate [18,19,20]. mTORC1 simultaneously phosphorylates and stabilizes GRB10, further contributing to the feedback inhibition of the PI3K/Akt signaling pathway [21].
Notably, the mechanism by which rapamycin inhibits mTORC1, particularly mTORC1, activity is via the formation of a dimer with the small 12 kDa FKBP12, which then directly binds and potently inhibits mTOR [22,23]. Meanwhile, upstream of mTORC1 and mTORC2, Ras homolog enriched in brain (Rheb), a GTPase, also directly binds mTOR but results in the activation of mTORC1 [24]. This represents a critical mechanism by which extracellular and intracellular stimuli influence mTORC1 activity [25]. The tuberous sclerosis complex (TSC1/2), composed of TSC1 (hamartin) and TSC2 (tuberin), regulates Rheb1 activity via its GTPase-activating protein (GAP) activity [26,27]. The phosphorylation of TSC2 through the activation of the upstream PI3K/Akt and extracellular signal-regulated kinase (ERK1/2)-mitogen-activated protein kinase (MAPK) signaling pathways disables the GAP activity of TSC1/2, allowing for the disinhibition of Rheb1 and the activation of mTORC1 [2,28,29,30].
mTORC2 is composed of rapamycin-insensitive companion of mTOR (Rictor), stress-activated protein kinase interacting protein 1 (Sin1), and protein observed with Rictor (Protor) [10,31,32,33,34]. Structurally, mTORC2 is a homodimer of mTOR, Rictor, Sin1, and mLST8 heterotetramers [3,35]. Rictor and Raptor are mutually exclusive in their binding to mTOR. Notably, Rictor makes mTORC2 insensitive to rapamycin by masking the FRB domain of mTOR; therefore, FKBP12-rapamycin is unable to bind the Rictor-containing mTOR complex, and thus, it does not affect S6K [35]. The main function of Rictor is to provide scaffolding and regulate further substrate recruitment by mTORC2, while Sin1 determines the subcellular localization of mTORC2 and contributes to the substrate binding site [36,37]. Although the stability of the complex is dependent on other mTORC2 components, Rictor facilitates the binding of Protor 1 and 2 to mTORC2. Protor 1 and 2 are not involved in the catalytic function of mTORC2, but they play a significant role in the regulation of serum/glucocorticoid-regulated kinase 1 (SGK1) activity [38]. Sin1, another binding partner, is critical for the regulation of mTORC2 substrate specificity secondary to its promotion of Rictor–mTOR binding [38,39]. For example, the N-terminus of Sin1 is essential to aid in the binding of Rictor to mLST8 [35]. In addition, the Pleckstrin homology (PH)-domain of Sin1 has been shown to mediate the association of mTORC2 with cellular membranes [40]. The mutation of the Sin1 key domain has recently been implicated in cancer development due to its disruption of mTORC2 with resultant sustained Akt activation [41]. Furthermore, the kinase domain of mTOR controls the integrity of mTORC2 via the phosphorylation of Sin1, maintaining its protein stability and preventing its lysosomal degradation [42].
There are several overlaps in the mTORC1 and mTORC2 subunits, including mTOR, Deptor, and mLST8. Deptor aids in the regulation of these complexes by binding to mTOR via its PDZ domain to inhibit mTORC1 and mTORC2 [43]. On the other hand, mLST8 may play an important role in regulating the dynamic equilibrium between mTORC1 and mTORC2 in cells due to its integral role in maintaining mTORC2’s Rictor–mTOR interaction [36].
Among other members of the S/T protein kinase (SGK) family, Akt, also known as protein kinase B, is a major substrate of mTORC2 [10,31,44,45]. mTORC2 phosphorylates the C-terminal hydrophobic motif of these kinases in response to growth factor signaling. Akt was originally discovered as a proto-oncogene but has since demonstrated critical involvement in the regulation of various cellular functions, including transcription, protein synthesis, metabolism, cellular growth, proliferation, and survival. Akt lies in the interface between PI3K and mTOR, which is a part of the canonical PI3K/mTOR pathway. Although mutations in Akt are rare, ample mutations in upstream effectors of Akt, such as PTEN and PI3K, have been identified. mTORC2 directly phosphorylates Akt at the site responsible for maximal Akt activation, its hydrophobic motif Ser473 [46]. Akt is subsequently phosphorylated at Thr308 by phosphatidylinositol-dependent protein kinase 1 (PDK1), resulting in its full activation. The Akt phosphorylation of protein kinase C α (PKCα) regulates the actin cytoskeleton and various cellular functions; this pathway is crucial for the maintenance of normal and cancer cells through its involvement in multiple physiological functions, including cell cycle progression, transcription, translation, cellular differentiation, metabolism, motility, and apoptosis [12,31,34,45]. Additionally, upstream signaling by various stimuli, including growth factors and insulin, increases PI3K signaling, thus leading to the activation of mTORC2 by promoting its association with ribosomes; as a result, mTORC2 phosphorylates Akt at Thr450 in addition to its well-known site, AktSer473 [47]. This pathway, stimulated by PI3K signaling in cancer cells, can be disrupted by inducing apoptosis in phosphatase and tensin homolog (PTEN)-deficient cells [47]. The interaction between mTORC1 and mTORC2 is further illustrated as both complexes influence one another; Akt regulates PRAS40 phosphorylation, disinhibiting mTORC1 activity, and S6K regulates Sin1 to modulate mTORC2 activity. Recently, it has been suggested that mTORC2 activation can also occur via a PI3K-independent mechanism, involving the Yes-associated protein (YAP)/Hippo, Notch, and small G-protein Rac1 pathway [48].
mTOR is primarily localized in the cytoplasm; however, in response to certain growth factors, it can transport to the nucleus [33]. While its role in the nucleus is not fully understood, mTOR has been suggested to function via nucleocytoplasmic signaling [49]. Activated mTOR has been localized in subnuclear structures that resemble polymorphonuclear (PML) bodies, which are associated with Akt activation and control cell proliferation, apoptosis, and cellular senescence [50]. The presence of S6K has been shown in the nucleus as well as in cytoplasm [51]. Recent studies have shown that the nucleocytoplasmic shuttling of mTOR commonly occurs, and that phosphorylated S6K and Raptor play an important role in it [52]. In quiescent glioblastoma (GBM) cells, the platelet-derived growth factor (PDGF)-induced localization of mTOR in the nucleus was reduced by pretreatment with rapamycin [33].
Rapamycin and its analogues, termed rapalogues, act as allosteric inhibitors of mTOR; however, due to their incomplete mTORC1 inhibition or the loss of negative feedback loops resulting in unexpected mTORC2 activation, they have generally been ineffective in GBM clinical trials [53]. These types of inhibitors were called “first-generation mTOR inhibitors,” and their drawbacks led to the discovery of novel ATP-binding inhibitors of both complexes, which simultaneously suppress mTORC1 activity while effectively inhibiting mTORC2 activity, as demonstrated by the complete dephosphorylation of the mTORC1 downstream substrate pS6KSer235/236 and the mTORC2 substrate pAKTSer473 (See Figure 1B). These ATP-binding inhibitors were termed “second-generation mTOR inhibitors,” also known as “TORKinib” or “TORKi.” These concurrent mTORC1 and mTORC2 inhibitors have effectively targeted the proliferation and self-renewal of GBM cancer stem cells. Thus, the effectiveness of mTOR inhibitors in cancer therapy can be evaluated by their ability to suppress both complexes along with their degree of interference in both cellular proliferation and migration.
A “third generation” of mTOR inhibitors, namely, RapaLink-1, was invented to overcome resistance to rapalogues and TORKi. RapaLink-1 is a bivalent mTOR inhibitor formed by linking rapamycin with MLN0128, an ATP-binding inhibitor, thus consisting of an FRB domain linked to a TORKi. Consequently, with its targeting of both the FRB and the kinase domains of mTOR, RapaLink-1 has demonstrated the ability to target breast cancer cells with somatic mutations in mTOR, FRB, or the kinase domain, which typically confer drug resistance [54]. RapaLink-1 potently inhibits the mTORC1 pathway via its inhibition of 4E-BP1 phosphorylation, resulting in growth inhibition at levels comparable to rapamycin alone or in combination with MLN0128 both in vitro and in vivo.
2. mTOR Pathways in Cancer
mTOR has been shown to frequently undergo aberrant activation in cancer [3,55]. In fact, mTORC1 has been found to play a prominent role in the growth of established tumor cells [56]. The activation of mTOR is often the result of mutations in upstream regulators, such as a gain-of-function of PI3K or epidermal growth factor receptor (EGFR) and/or loss-of-function of tumor suppressor gene PTEN [57,58,59,60,61,62,63,64]. The activation of mTOR signaling drives the variations seen in cancer cell metabolism, including pathways for amino acid, glucose, nucleotide, fatty acid, and lipid metabolism [3,8]. In addition, other signaling molecules with oncogenic potential, such as RAS, can also stimulate mTOR signaling [65]. With its regulation of protein translation, ribosome biogenesis, cell proliferation, metabolism, and survival, mTOR has emerged as a promising chemotherapeutic target, used solely or combined with other chemotherapeutic agents [2,3,66,67].
As previously described, rapamycin and rapalogues act as the partial inhibitors of downstream effectors in the mTORC1 pathway, mainly 4E-BP1, while concurrently causing a compensatory increase in mTORC2/Akt activity [33]. Interestingly, rapamycin can inhibit or activate mTORC2 in certain cancer cells; while the exact mechanism of the latter remains to be elucidated, rapamycin may inhibit mTORC2 by stalling its assembly [33,68]. The PI3K/Akt pathway has been implicated in pathophysiology and resistance to chemotherapy in many solid tumors [10,69]. Although the regulation of mTORC2 is only recently becoming more understood, mTORC1 is known to be activated by both extracellular as well as intracellular stimuli, including growth factors and amino acids [62]. As discussed previously, Rheb1 and its disinhibition by upstream Akt activation represent one such mechanism by which mTORC1 is activated via these signals. Moreover, a major obstacle in the targeting of mTORC1 is its sensitivity to nutrients, and variations in cancer cell metabolism may complicate mTORC1 suppression by contributing to a state of persistent activation. Nevertheless, in tumors with mTORC1 activation, such as subependymal giant cell astrocytomas (SEGA) of tuberous sclerosis (TS), rapalogues have shown significant efficacy. For example, everolimus has demonstrated a 75% response rate in SEGA [70]. Numerous early clinical trials in recent years have investigated the safety and efficacy of rapamycin, rapalogues, and ATP-binding mTOR inhibitors as monotherapy or in combination with other agents in the management of various malignancies are presented in Supplementary Table S1.
3. Targeting mTOR and PI3K in GBM
GBM, the most common primary brain tumor in adults, has an incidence of approximately 10,000 cases per year in the United States [71]. The Cancer Genome Atlas (TCGA) network classifies GBM into four molecular subtypes based on specific gene alterations: proneural, neural, classical, and mesenchymal transcriptomic [72,73,74]. The signal transduction cascade of EGFR is often altered in GBM, with the extensive genomic analyses of human GBM samples demonstrating the genetic mutations of EGFR in approximately 57% of GBM patients [72,75]. Aberrant signaling of mTOR is linked to tumorigenesis of numerous malignancies, including GBM. The mutation in tumor suppressor PTEN as well as loss of heterozygosity on chromosome 10q (LOH10q) occurs frequently in both primary as well as secondary GBM [76]. Also, mutations in PTEN were seen in up to 36–60% of GBM [72]. Aberrant EGFR signaling and the loss of PTEN both lead to the activation of the PI3K/Akt/mTOR pathway, thus suggesting a potential therapeutic advantage of inhibition of this pathway [72,77,78]. Further, genetic studies have identified the activation of receptor tyrosine kinase/PI3K in 86% of GBM samples [72]. This increased the activation of the Akt/mTOR pathway that stimulates cellular growth, proliferation, migration, and survival, which are major hallmarks of GBM cells [2,79]. It is possible that mTOR activation is the major cause of GBM’s relentless growth and dissemination [79,80]. Consequently, multiple clinical trials have been initiated to investigate the therapeutic response, toxicities, changes in metabolism and biomarkers, and predictors of response to PI3K/mTOR inhibitors in GBM. These trials have predominantly been conducted in recurrent GBM with a smaller subset of newly diagnosed GBM. Further, mTOR inhibitors were evaluated in a variety of combinations, such as monotherapy, in conjunction with standard of care therapy, and/or in combination with other pathway inhibitors. Table 1 represents the ongoing clinical trials using PI3K, Akt, and mTOR inhibitors in GBM. Here, we discuss some of these trials.

Table 1.
Clinical trials of PI3K/Akt/mTOR signaling inhibitors in glioblastoma.
Multiple phase II clinical trials using rapalogues showed limited success in producing meaningful clinical results, attributable to the concurrent inhibition of negative feedback loops in addition to crosstalk with other mitogenic pathways (NCT00515086, NCT00016328, NCT00022724) [81]. As PI3K is negatively regulated by PTEN, one trial by Cloughesy et al. in 2008 treated 15 patients with PTEN-deficient recurrent GBM with one week of oral sirolimus daily prior to re-resection and continued postoperatively until progression [13]. Of the 14 tumor samples with adequate tissue, rapamycin was detected in all 14 during re-resection. While 7 of 14 patients demonstrated decreased tumor cell proliferation according to the Ki-67 proliferative index, the remaining patients were noted to have Akt activation with increased PRAS40 phosphorylation, thought to be secondary to the loss of the negative feedback loop. The compensatory Akt activation in this subset of patients was correlated with a shorter progression-free survival (PFS), suggesting the failure of sirolimus in the treatment of PTEN-deficient GBM.
Temsirolimus (CCI-779), another mTOR inhibitor, was trialed in 43 recurrent GBM patients at a weekly intravenous dose of 250 mg. This dose was tolerated without any serious toxicities; however, median time to progression was only 9 weeks, indicating limited efficacy as a monotherapy using temsirolimus [81]. A larger phase II trial of 65 recurrent GBM patients showed more promising results in a subset of patients with higher baseline tumor levels of phosphorylated p70S6K. These patients had a significantly longer PFS of 5.4 months compared to 1.9 months. Overall, 36% of patients demonstrated radiographic improvement, and there was a 51% incidence of grade 3–5 toxicities [82].
A phase I study was conducted on 10 recurrent GBM patients with four days of mTOR inhibitor ridaforolimus administered daily at an intravenous dose of 12.5 to 15 mg prior to re-resection and continued postoperatively [83]. Ridaforolimus demonstrated its ability to cross the blood–brain barrier and inhibit mTOR activity, as evidenced by a decrease in its downstream effectors in tumor specimens; pS6 levels were reduced, and phosphorylated 4E-BP1(p4E-BP1) was reduced by >80% compared to patient serum baseline [81]. A subsequent study demonstrated a dramatic down-regulation of pS6K with combined PI3K/mTOR inhibition [33]. These studies, while not clinically successful, laid the foundation for the possible utility of combination PI3K/mTOR therapy [13].
Several phase I and phase II trials have also evaluated the efficacy of mTOR inhibitors as an adjunct to the standard of care treatment for GBM. Standard temozolomide (TMZ) and radiation therapy (RT) were supplemented with mTOR inhibitor everolimus in a phase 1 trial of 18 newly diagnosed GBM patients. Over a median follow-up of 8.4 months, nine patients (50%) developed grade 3/4 toxicities. Stable disease occurred in 14 patients, and 4 patients had partial response, as shown by imaging analysis [84]. In an additional study, 100 newly diagnosed GBM patients were administered everolimus for 1 week prior to conventional TMZ/RT and continued until progression. Overall survival for one year was seen in 64%, while median PFS was 6.4 months, demonstrating no appreciable survival benefit of standard therapy in conjunction with everolimus compared to controls [85]. Another phase 1 dose-escalation trial evaluating temsirolimus in 12 GBM patients in combination with TMZ/RT observed grade 4/5 infections in 25% of patients. Confining temsirolimus to the initial radiation phase instead of continuing it with TMZ adjuvant therapy and adding prophylactic antibiotics reduced the infection rate, though 2 of 13 patients in the second cohort exhibited worsening of pre-existing viral and fungal infections [86].
PI3K inhibitors may be isoform-selective or may target all four isoforms. Buparlisib (BKM120) is a pan-PI3K inhibitor, an ATP-competitive inhibitor targeting all isoforms, and it was evaluated in a phase I clinical trial in conjunction with standard therapy for newly diagnosed GBM patients, but the trial was discontinued due to significant toxicities [87]. In patients with recurrent GBM, phase II clinical trials demonstrated adequate brain tissue penetration of BKM120; however, it failed to render a significant clinical response as monotherapy or in conjunction with re-resection [58,87]. Another PI3K inhibitor is the combined PI3K/mTOR inhibitor, voxtalisib, which underwent a phase I trial in the treatment of high-grade gliomas. When administered in conjunction with TMZ with or without RT, stable disease was seen in 68% of patients, and partial response was achieved in 4% of patients. Lymphopenia (13%) and thrombocytopenia (9%) were the most frequent serious adverse events [88].
Despite advances in mTOR-targeting therapies, it is thought that the activation of mitogenic pathways and RAS/ERK1/2 via feedback loops contributes to the resistance of GBM [53]. The goal of active site inhibitors is to simultaneously target mTOR’s function in cell growth and proliferation along with its feedback loops. Second-generation mTOR inhibitors, termed “TORKinibs”, inhibit both mTORC1 and mTORC2 via allosteric interactions with the ATP-binding pocket [89,90]. Several small molecules have been identified that act as ATP-competitive inhibitors of mTOR, including PP242, KU0063794, AZD3147, and eCF309, among others. Pyrazolopyrimidines, PP242 and PP30, are potent mTOR inhibitors that display a high degree of selectivity towards mTOR relative to PI3Ks and other protein kinases. Meanwhile, KU0063794 also showed promise in suppressing cell cycle and proliferation compared to the selective PI3K inhibitor, LY294002, or combined PI3K/mTORC1 inhibitor, PI-103 [91].
Novel ATP-competitive mTOR inhibitor, Torin 1, of the quinoline class, inhibits both mTOR complexes [92]. Drawbacks of Torin 1 include its water insolubility and rapid metabolism by the liver that result in poor bioavailability and a relatively shorter half-life [93]. Consequently, the related compound Torin 2, which was created by Liu et al., exhibits improved water-solubility with increased oral bioavailability and a longer half-life [93]. Torin 2 has also emerged as a potent mTOR inhibitor capable of suppressing cellular proliferation and migration in GBM [94,95]. This recent study demonstrates that Torin 2 is the only mTOR inhibitor with the ability to suppress the self-renewal of cancer stem cells (CSCs) in GBM. Another recently discovered ATP-competitive mTOR inhibitor, XL388, of the benzoxazepine class, works similarly to Torin 1 and Torin 2. As with Torin 2, this drug demonstrates sufficient oral bioavailability and efficacy at low concentrations, as well as selectivity of mTOR over PI3K [96].
As opposed to mTORC1 inhibitors, the inhibition of AktSer473 phosphorylation by combined mTORC1 and mTORC2 inhibitors produces superior outcomes in GBM treatment. Compared to rapamycin, these novel inhibitors also demonstrate the superior suppression of p70S6K phosphorylation, and PP242 displays the enhanced suppression of GBM cell proliferation and migration [97,98]. Similarly, in vivo studies investigating the ATP-competitive mTOR inhibitor, AZD8055, demonstrate the reduced phosphorylation of both S6 and Akt with the subsequent reduction of tumor growth [99,100]. Clinical trials are now evaluating these dual inhibitors, including AZD8055 and sapanisertib (MLN0128, TAK228).
4. The mTOR Pathway and Stem Cells
mTOR functions via two distinct protein complexes, mTORC1 and mTORC2, which are a part of the key intracellular signaling pathway of PI3K/Akt/mTOR. These complexes respond to different signals, including growth factors and cellular energy to regulate cell growth and shape by influencing cytoskeletal remodeling, determining when and where cells grow as dictated by mTORC1 and mTORC2, respectively [8,101,102]. mTORC1 regulation of protein synthesis, via S6K and 4E-BP1, and initiation of protein translation have played a critical role in the maintenance of stem cells [103]. The mTOR pathway functions in normal neural stem cells (NSCs) by altering gene transcription for normal cell growth and migration processes [31]. Furthermore, studies have shown that mTORC1 is essential for the proliferation and survival of neural stem cells and appears to regulate their postnatal differentiation in the subventricular zone (SVZ) [104]. For example, insulin encourages neural differentiation via the PI3K/Akt/mTOR pathway by inducing mTOR phosphorylation [105,106,107]. Also, the activation of mTOR via suppression of its negative regulators, such as PTEN or TSC1, was shown to promote axonal regeneration [108]. These observations imply that alterations in the mTOR pathway can lead to severe deficits in nervous system development, resulting in various abnormalities, such as tumors, autism, and seizures [8]. Suppression of mTORC1 may inhibit NSC differentiation and decrease the population of neural progenitor cells. Alternatively, the hyperactivation of mTORC1 may also lead to the depletion of NSC progenitor cells due to accelerated differentiation [109,110]. Also, one observation revealed the gradual loss of NSCs in the neonatal SVZ which led to terminal differentiation and not proliferation after mTORC1 hyperactivation [111]. Furthermore, reduction in neural differentiation of NSCs following mTOR inhibition with rapamycin was seen without changes in proliferation [107]. Studies have shown that the mTOR pathway is also associated with dendrite formation and signal transduction between neurons, an additional function of mTOR besides neural regulation, as demonstrated in the development of newly born olfactory bulb neurons and their dendrites [104]. In addition, both mTOR complexes are shown to be involved in the dendritogenesis of SVZ-derived neurons [104]. These studies support the pivotal role of the mTOR pathway in neurogenesis via its influence on NSC function.
Recent studies have identified small subpopulations of cells present within the tumor and surrounding areas, termed CSCs, that are capable of self-renewal and play a critical role in tumor initiation, progression, maintenance, and recurrence. Additionally, these CSCs remain resistant to conventional chemotherapy and radiation therapy; therefore, they are the prime cause of therapy failure as well as cancer recurrence. Thus, understanding the origin and characteristics of CSCs will elucidate the mechanism that regulates stemness and drug resistance, leading to effective treatment of cancers, by targeting CSCs [112]. The existence of CSCs has been well established in brain tumors, such as GBM and medulloblastoma [112,113,114,115,116,117]. It is thought that the resilience of the CSC populations allows for the recurrence and invasion of GBM despite aggressive chemotherapy [114]. Particularly, studies have demonstrated CSCs in GBM can enter a quiescent state, rendering a refractory status with lower susceptibility to therapies [118]. Further, CSCs have been identified beyond the tumor margin of GBM; thus, the presence of these CSCs in the peritumoral area has emerged as an important prognostic marker, as measured by the expression of stem cell marker Nestin in relation to c-Jun N-terminal kinase (JNK)/MAPK [119].
GBM stem cells and CSCs are under the influence of several signaling pathways, namely, the PI3K/Akt/mTOR and EGFR pathways [120]. Nevertheless, the regulation of the molecular aspects of GBM stem cells remains elusive. Ample evidence demonstrated that the mTOR pathway regulates CSCs and has a significant role in the persistent growth and invasion of GBM stem cells. The deregulated mTOR pathway has been implicated in multiple cancer types, such as breast and renal cancer, and the inhibition of this pathway has shown therapeutic potential [3,10,121,122]. A study evaluating the interference of this pathway in CSC by rapamycin or rapalogues demonstrated that the proliferation of stem cells was significantly curtailed. Further, the Akt/mTOR signaling pathway has been implicated in maintaining the stem cell properties of CSCs in glioma [123]. Therefore, combined targeting of this pathway has been evaluated for the potential targeting of GBM stem cells and the improved treatment of GBM [124,125,126].
The role of mTOR in GBM stem cell regulation is just beginning to be achieved. However, there is evidence that the mTOR pathway can regulate hematopoietic stem cells, via its role in controlling autophagy [127]. Thus, a benefit of mTOR inhibition is the induction of autophagy in GBM stem cells with resultant antiproliferative and antidifferentiation effects [128]. Rapamycin and its analogues have been shown to incompletely inhibit mTORC1 from executing its pro-growth functions [7,23]. Therefore, approaches to inhibit multiple deregulated pathways by targeting both mTORC1 and mTORC2 complexes may lead to the further suppression of stem cell survival and proliferation [80,129]. While the previously introduced small molecule inhibitors PP242 and Torin 1 did not show clinical benefits in trial, Torin 2 showed some clinical success [79,92,93,96,97]. Our recent findings have demonstrated that Torin 2 was more effective in suppressing stem cell growth and self-renewal as demonstrated by reduced neurosphere sizes [94,95,130]. In fact, while Torin 1 and XL388 delayed the self-renewal of GBM stem cells, Torin 2 completely halted stem cell self-renewal [130]. With these findings, mTOR inhibition emerges as a promising strategy to target CSCs in the treatment of GBM. A recent study utilizing Pim-1, another S/T kinase, demonstrated its potential influence in the regulation of GBM stem cells as its inhibition led to the eradication of stem-like neurosphere cells, and this effect can interact with Akt/mTOR to control the size and cell viability of neural stem cell neurospheres [131]. Future investigation is required to further define the role of the PI3K/Akt/mTOR pathway in stem cell quiescence and evaluate its significance in GBM treatment.
5. Conclusions
While substantial progress has been made in understanding the mechanisms of the mTOR complexes since their discovery nearly three decades ago, numerous molecular and cellular properties of mTOR are yet to be discerned, particularly the non-canonical regulation of these complexes. Furthermore, the regulation and activation of mTORC2 are still being investigated. As the deregulation of mTOR is seen in multiple tumor types, the mTOR pathway represents a promising therapeutic target. Allosteric inhibitors of mTOR, rapamycin, and its derivatives incompletely inhibit mTORC1 and result in the activation of mitogenic pathways. Although “second-generation” ATP-competitive compounds have shown promise in inhibiting both mTORC1 and mTORC2, a third-generation inhibitor of mTOR, RapaLink-1, effectively targets multiple domains in both complexes. The success of these compounds in targeting GBM CSC and treating GBM remains to be further investigated.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells13050409/s1, Table S1: Clinical trials of PI3K/Akt/mTOR signaling inhibitors in cancers.
Author Contributions
Conceptualization, M.J.-U.; writing—original draft preparation, M.J.-U., S.L.Z., E.S. and M.D.; writing—review and editing, M.J.-U., S.L.Z., E.S., M.D., S.J.H. and C.D.G.; visualization, M.J.-U. and S.L.Z.; supervision, M.J.-U., S.J.H. and C.D.G.; project administration, M.J.-U.; funding acquisition, M.J.-U. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Advanced Research Foundation and The Rockefeller Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell. Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Darb-Esfahani, S.; Faggad, A.; Noske, A.; Weichert, W.; Buckendahl, A.C.; Müller, B.; Budczies, J.; Röske, A.; Dietel, M.; Denkert, C. Phospho-mTOR and phospho-4EBP1 in endometrial adenocarcinoma: Association with stage and grade in vivo and link with response to rapamycin treatment in vitro. J. Cancer Res. Clin. Oncol. 2009, 135, 933–941. [Google Scholar] [CrossRef]
- Rosenwald, I.B. The role of translation in neoplastic transformation from a pathologist’s point of view. Oncogene 2004, 23, 3230–3247. [Google Scholar] [CrossRef]
- Chan, S. Targeting the mammalian target of rapamycin (mTOR): A new approach to treating cancer. Br. J. Cancer 2004, 91, 1420–1424. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef]
- Russell, R.C.; Fang, C.; Guan, K.L. An emerging role for TOR signaling in mammalian tissue and stem cell physiology. Development 2011, 138, 3343–3356. [Google Scholar] [CrossRef]
- Volarević, S.; Thomas, G. Role of S6 phosphorylation and S6 kinase in cell growth. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 65, 101–127. [Google Scholar]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122 Pt 20, 3589–3594. [Google Scholar] [CrossRef]
- Reiling, J.H.; Sabatini, D.M. Stress and mTORture signaling. Oncogene 2006, 25, 6373–6383. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Gulati, N.; Karsy, M.; Albert, L.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar] [PubMed]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Hwang, Y.; Kim, L.C.; Song, W.; Edwards, D.N.; Cook, R.S.; Chen, J. Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth In Vivo. Cancer Res. 2019, 79, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.; Ouyang, X.; Liu, H.; Li, S.; Jin, J.; Tang, W.; Xia, Y.; Su, B. Sin1-mediated mTOR signaling in cell growth, metabolism and immune response. Natl. Sci. Rev. 2019, 6, 1149–1162. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef]
- Liu, Q.; Qiu, J.; Liang, M.; Golinski, J.; van Leyen, K.; Jung, J.E.; You, Z.; Lo, E.H.; Degterev, A.; Whalen, M.J. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 2014, 5, e1084. [Google Scholar] [CrossRef]
- Chen, C.H.; Sarbassov, D.D. The mTOR (mammalian target of rapamycin) kinase maintains integrity of mTOR complex 2. J. Biol. Chem. 2011, 286, 40386–40394. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Álvarez, J.; Andersen, J.K. mTORC2: The other mTOR in autophagy regulation. Aging Cell 2021, 20, e13431. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.A.; Kim, J.H.; Wu, A.L.; Park, I.H.; Chen, J. A nuclear transport signal in mammalian target of rapamycin is critical for its cytoplasmic signaling to S6 kinase 1. J. Biol. Chem. 2006, 281, 7357–7363. [Google Scholar] [CrossRef]
- Salomoni, P.; Pandolfi, P.P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar] [CrossRef]
- Rosner, M.; Hengstschläger, M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 2011, 30, 4509–4522. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Owens, R.J.; Stubbs, C.D.; Parker, A.W.; Hitchman, R.; Yadav, R.B.; Dumoux, M.; Hawes, C.; Botchway, S.W. Direct imaging of the recruitment and phosphorylation of S6K1 in the mTORC1 pathway in living cells. Sci. Rep. 2019, 9, 3408. [Google Scholar] [CrossRef]
- Albert, L.; Karsy, M.; Murali, R.; Jhanwar-Uniyal, M. Inhibition of mTOR Activates the MAPK Pathway in Glioblastoma Multiforme. Cancer Genom. Proteom. 2009, 6, 255–261. [Google Scholar]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Mosley, J.D.; Poirier, J.T.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/ErbB2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; DeAngelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.; Drappatz, J.; et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-Oncology 2012, 14, 1511–1518. [Google Scholar] [CrossRef]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients with Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832. [Google Scholar] [CrossRef]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F.; et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Inhibitors: Rationale and Importance to Inhibiting These Pathways in Human Health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. The pharmacology of mTOR inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Follo, M.Y.; Manzoli, L.; Poli, A.; McCubrey, J.A.; Cocco, L. PLC and PI3K/Akt/mTOR signalling in disease and cancer. Adv. Biol. Regul. 2015, 57, 10–16. [Google Scholar] [CrossRef]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Muñoz-Hidalgo, L.; San-Miguel, T.; Megías, J.; Monleón, D.; Navarro, L.; Roldán, P.; Cerdá-Nicolás, M.; López-Ginés, C. Somatic copy number alterations are associated with EGFR amplification and shortened survival in patients with primary glioblastoma. Neoplasia 2020, 22, 10–21. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef]
- Pitz, M.W.; Eisenhauer, E.A.; MacNeil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncology 2015, 17, 1270–1274. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Albert, L.; McKenna, E.; Karsy, M.; Rajdev, P.; Braun, A.; Murali, R. Deciphering the signaling pathways of cancer stem cells of glioblastoma multiforme: Role of Akt/mTOR and MAPK pathways. Adv. Enzym. Regul. 2011, 51, 164–170. [Google Scholar] [CrossRef]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investig. New Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef]
- Reardon, D.A.; Wen, P.Y.; Alfred Yung, W.K.; Berk, L.; Narasimhan, N.; Turner, C.D.; Clackson, T.; Rivera, V.M.; Vogelbaum, M.A. Ridaforolimus for patients with progressive or recurrent malignant glioma: A perisurgical, sequential, ascending-dose trial. Cancer Chemother. Pharmacol. 2012, 69, 849–860. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; Jaeckle, K.A.; Buckner, J.C. North Central Cancer Treatment Group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Dietz, A.B.; Kaufmann, T.J.; Gustafson, M.P.; Brown, P.D.; Uhm, J.H.; Rao, R.D.; Doyle, L.; et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin. Cancer Res. 2010, 16, 5573–5580. [Google Scholar] [CrossRef]
- Wen, P.; Rodon, J.; Mason, W.; Beck, J.; DeGroot, J.; Donnet, V.; Mills, D.; El-Hashimy, M.; Rosenthal, M. Phase I, open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open 2020, 5, e000673. [Google Scholar] [CrossRef]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro-Oncology 2015, 17, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a Highly Potent, Selective Mammalian Target of Rapamycin (mTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Ahmed, T.; Sabatini, D.M.; Gray, N.S. Discovery of 9-(6-Aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a Potent, Selective, and Orally Available Mammalian Target of Rapamycin (mTOR) Inhibitor for Treatment of Cancer. J. Med. Chem. 2011, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.G.; Jeong, S.W.; Gillick, J.L.; Sursal, T.; Murali, R.; Gandhi, C.D.; Jhanwar-Uniyal, M. Targeting the mTOR pathway using novel ATP-competitive inhibitors, Torin1, Torin2 and XL388, in the treatment of glioblastoma. Int. J. Oncol. 2021, 59, 83. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Dominguez, J.F.; Mohan, A.L.; Tobias, M.E.; Gandhi, C.D. Disentangling the signaling pathways of mTOR complexes, mTORC1 and mTORC2, as a therapeutic target in glioblastoma. Adv. Biol. Regul. 2022, 83, 100854. [Google Scholar] [CrossRef]
- Takeuchi, C.S.; Kim, B.G.; Blazey, C.M.; Ma, S.; Johnson, H.W.B.; Anand, N.K.; Arcalas, A.; Baik, T.G.; Buhr, C.A.; Cannoy, J.; et al. Discovery of a Novel Class of Highly Potent, Selective, ATP-Competitive, and Orally Bioavailable Inhibitors of the Mammalian Target of Rapamycin (mTOR). J. Med. Chem. 2013, 56, 2218–2234. [Google Scholar] [CrossRef]
- Neil, J.; Shannon, C.; Mohan, A.; Laurent, D.; Murali, R.; Jhanwar-Uniyal, M. ATP-site binding inhibitor effectively targets mTORC1 and mTORC2 complexes in glioblastoma. Int. J. Oncol. 2016, 48, 1045–1052. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M. Mighty RapaLink-1 vanquishes undruggable mutant mTOR in glioblastoma. Transl. Cancer Res. 2017, 6, S1143–S1148. [Google Scholar] [CrossRef]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef]
- Marshall, G.; Howard, Z.; Dry, J.; Fenton, S.; Heathcote, D.; Gray, N.; Keen, H.; Logie, A.; Holt, S.; Smith, P.; et al. Benefits of mTOR kinase targeting in oncology: Pre-clinical evidence with AZD8055. Biochem. Soc. Trans. 2011, 39, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Frank, A.R.; Jewell, J.L. mTOR signaling in stem and progenitor cells. Development 2018, 145, dev152595. [Google Scholar] [CrossRef] [PubMed]
- Skalecka, A.; Liszewska, E.; Bilinski, R.; Gkogkas, C.; Khoutorsky, A.; Malik, A.R.; Sonenberg, N.; Jaworski, J. mTOR kinase is needed for the development and stabilization of dendritic arbors in newly born olfactory bulb neurons. Dev. Neurobiol. 2016, 76, 1308–1327. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef]
- Han, J.; Wang, B.; Xiao, Z.; Gao, Y.; Zhao, Y.; Zhang, J.; Chen, B.; Wang, X.; Dai, J. Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol. Cell. Neurosci. 2008, 39, 118–124. [Google Scholar] [CrossRef]
- Lee, J.-H.S.; Vo, T.-T.; Fruman, D.A. Targeting mTOR for the treatment of B cell malignancies. Br. J. Clin. Pharmacol. 2016, 82, 1213–1228. [Google Scholar] [CrossRef]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef]
- Mahoney, C.; Feliciano, D.M.; Bordey, A.; Hartman, N.W. Switching on mTORC1 induces neurogenesis but not proliferation in neural stem cells of young mice. Neurosci. Lett. 2016, 614, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Ferguson, L.P.; Diaz, E.; Reya, T. The Role of the Microenvironment and Immune System in Regulating Stem Cell Fate in Cancer. Trends Cancer 2021, 7, 624–634. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Huang, G.H.; Xu, Q.F.; Cui, Y.H.; Li, N.; Bian, X.W.; Lv, S.Q. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncology 2022, 24, 669–682. [Google Scholar] [CrossRef]
- Mangiola, A.; Lama, G.; Giannitelli, C.; De Bonis, P.; Anile, C.; Lauriola, L.; La Torre, G.; Sabatino, G.; Maira, G.; Jhanwar-Uniyal, M.; et al. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: Possible prognostic implications. Clin. Cancer Res. 2007, 13, 6970–6977. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar]
- Voss, M.H.; Bastos, D.A.; Karlo, C.A.; Ajeti, A.; Hakimi, A.A.; Feldman, D.R.; Hsieh, J.J.; Molina, A.M.; Patil, S.; Motzer, R.J. Treatment outcome with mTOR inhibitors for metastatic renal cell carcinoma with nonclear and sarcomatoid histologies. Ann. Oncol. 2014, 25, 663–668. [Google Scholar] [CrossRef]
- Friedman, M.D.; Jeevan, D.S.; Tobias, M.; Murali, R.; Jhanwar-Uniyal, M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol. Rep. 2013, 30, 1645–1650. [Google Scholar] [CrossRef]
- Colapietro, A.; Yang, P.; Rossetti, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Conway, T.L.; Chakraborty, S.; Marampon, F.; Mattei, V.; et al. The Botanical Drug PBI-05204, a Supercritical CO2 Extract of Nerium Oleander, Inhibits Growth of Human Glioblastoma, Reduces Akt/mTOR Activities, and Modulates GSC Cell-Renewal Properties. Front. Pharmacol. 2020, 11, 552428. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef]
- Gallia, G.L.; Tyler, B.M.; Hann, C.L.; Siu, I.M.; Giranda, V.L.; Vescovi, A.L.; Brem, H.; Riggins, G.J. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol. Cancer Ther. 2009, 8, 386–393. [Google Scholar] [CrossRef]
- Borsa, M.; Obba, S.; Richter, F.C.; Zhang, H.; Riffelmacher, T.; Carrelha, J.; Alsaleh, G.; Jacobsen, S.E.W.; Simon, A.K. Autophagy preserves hematopoietic stem cells by restraining MTORC1-mediated cellular anabolism. Autophagy 2024, 20, 45–57. [Google Scholar] [CrossRef]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Ezell, S.A.; Mayo, M.; Bihani, T.; Tepsuporn, S.; Wang, S.; Passino, M.; Grosskurth, S.E.; Collins, M.; Parmentier, J.; Reimer, C.; et al. Synergistic induction of apoptosis by combination of BTK and dual mTORC1/2 inhibitors in diffuse large B cell lymphoma. Oncotarget 2014, 5, 4990–5001. [Google Scholar] [CrossRef][Green Version]
- Jhanwar-Uniyal, M.; Gellerson, O.; Bree, J.; Das, M.; Kleinman, G.; Gandhi, C.D. Defining the role of mTOR pathway in the regulation of stem cells of glioblastoma. Adv. Biol. Regul. 2023, 88, 100946. [Google Scholar] [CrossRef]
- Seifert, C.; Balz, E.; Herzog, S.; Korolev, A.; Gaßmann, S.; Paland, H.; Fink, M.A.; Grube, M.; Marx, S.; Jedlitschky, G.; et al. PIM1 Inhibition Affects Glioblastoma Stem Cell Behavior and Kills Glioblastoma Stem-like Cells. Int. J. Mol. Sci. 2021, 22, 11126. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).