

Inhibitors of Rho/MRTF/SRF Transcription Pathway Regulate Mitochondrial Function


Abstract
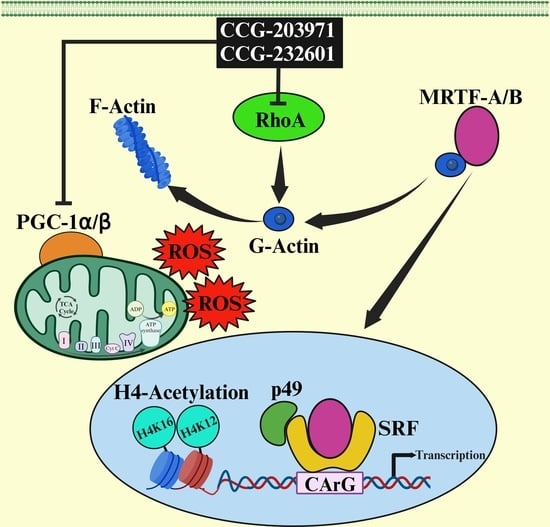
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
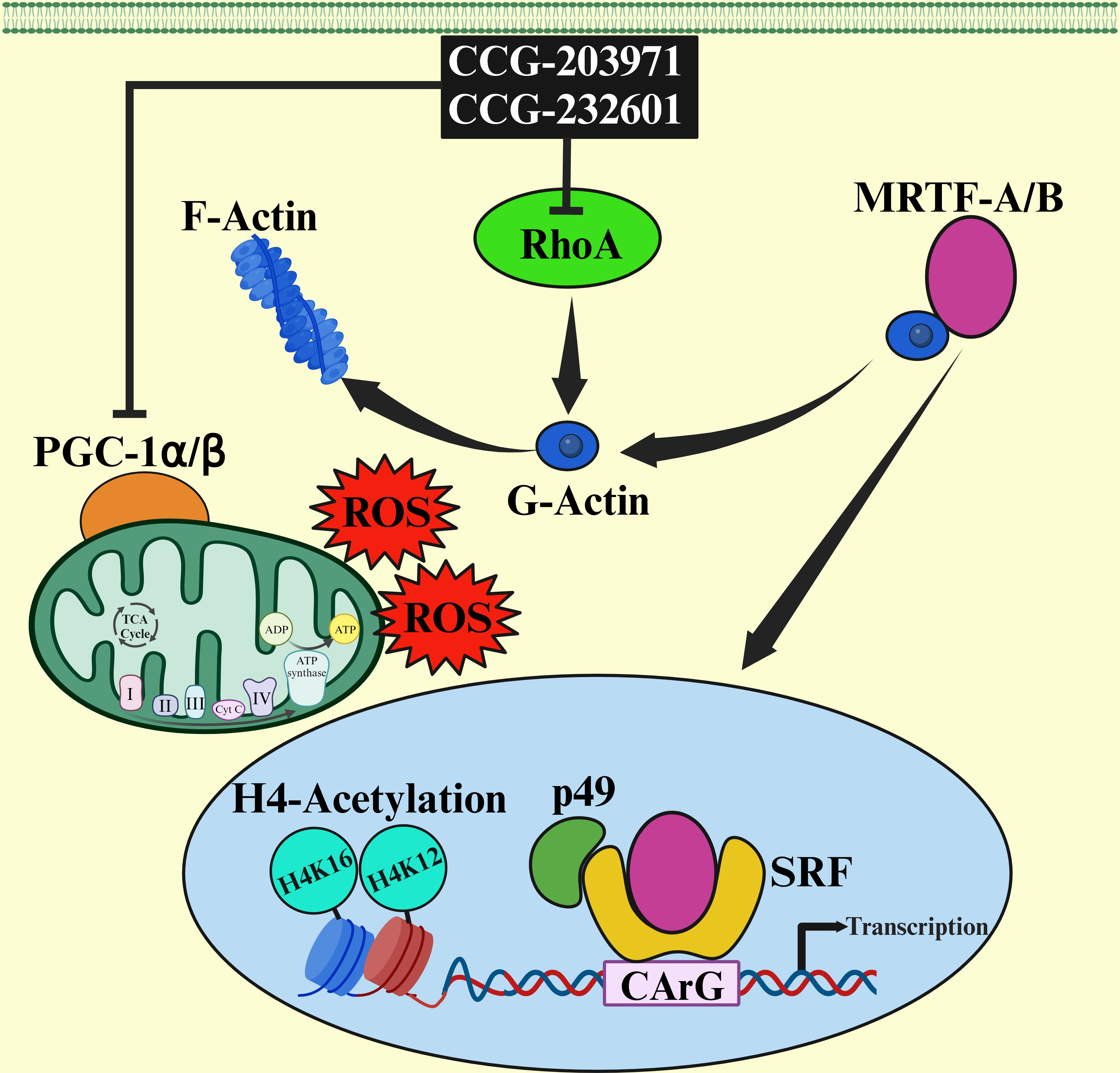
2.1. Small Molecule Inhibitors
2.2. Cells and Culture Conditions
2.3. Cell Viability Assay
2.4. Immunofluorescence Staining
2.5. Quantitative Real-Time Polymerase Chain Reaction
2.6. Western Blot Analysis and Immunoprecipitation
2.7. Extracellular Flux Assays
2.8. High Resolution Respiratory Analysis
2.9. Flow Cytometry
2.10. Statistical Analysis
3. Results
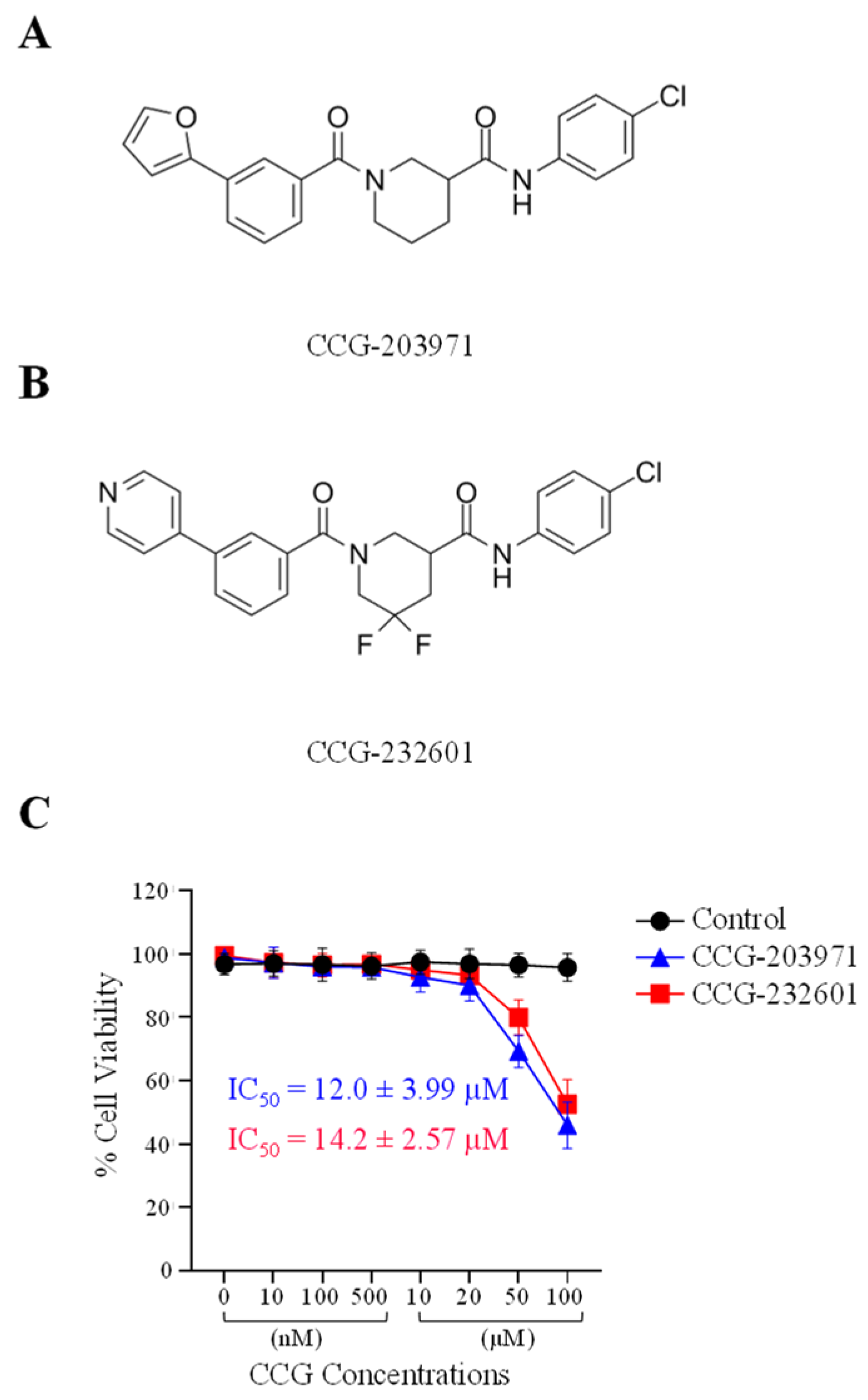
3.1. CCG-203971 and CCG-232601 Exhibit Dose Dependent Cytotoxicity against WI-38 and C2C12 Cell Lines
3.2. CCG-203971 and CCG-232601 Specifically Inhibit Rho/MRTF/SRF Signaling Pathway
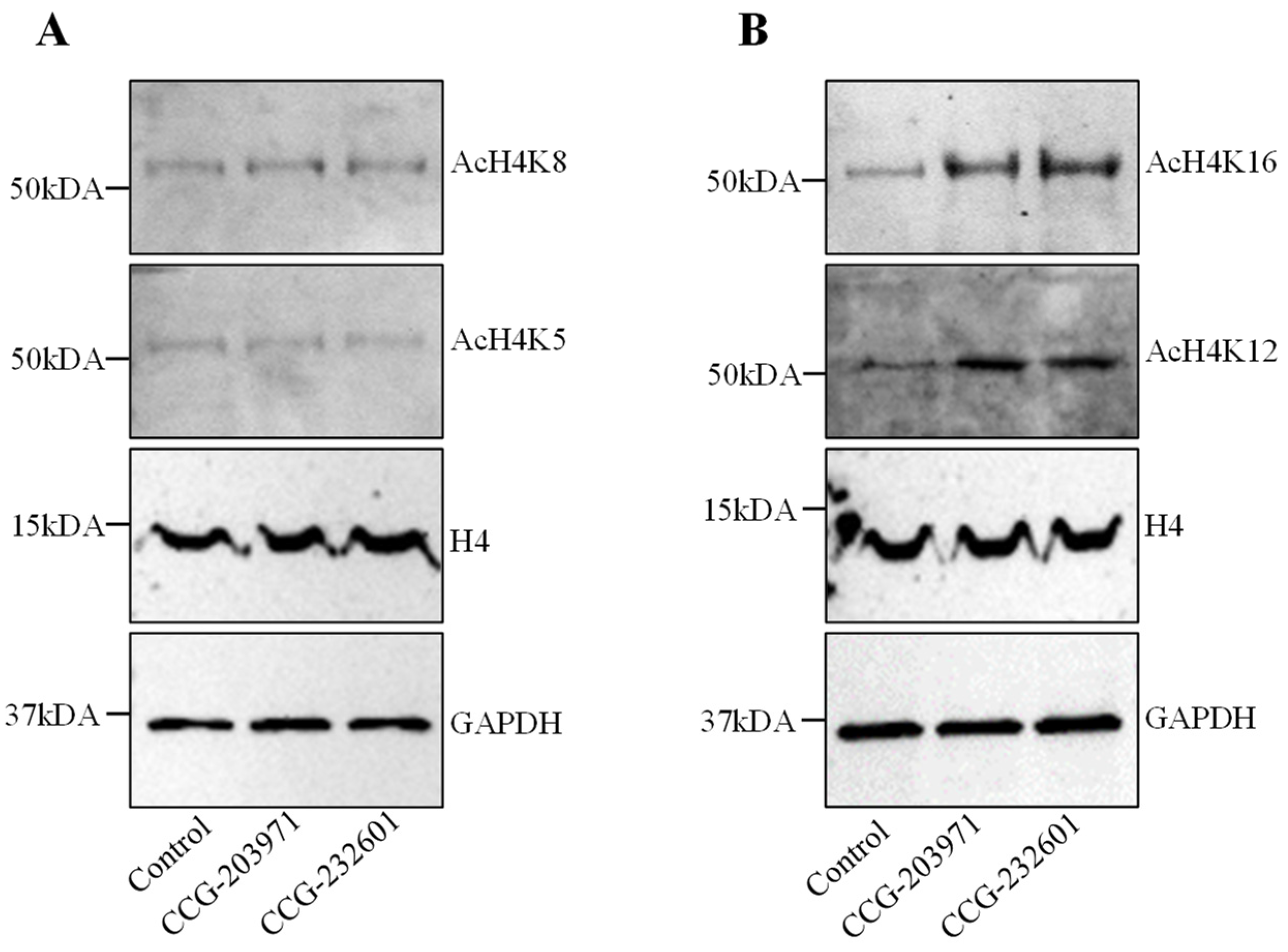
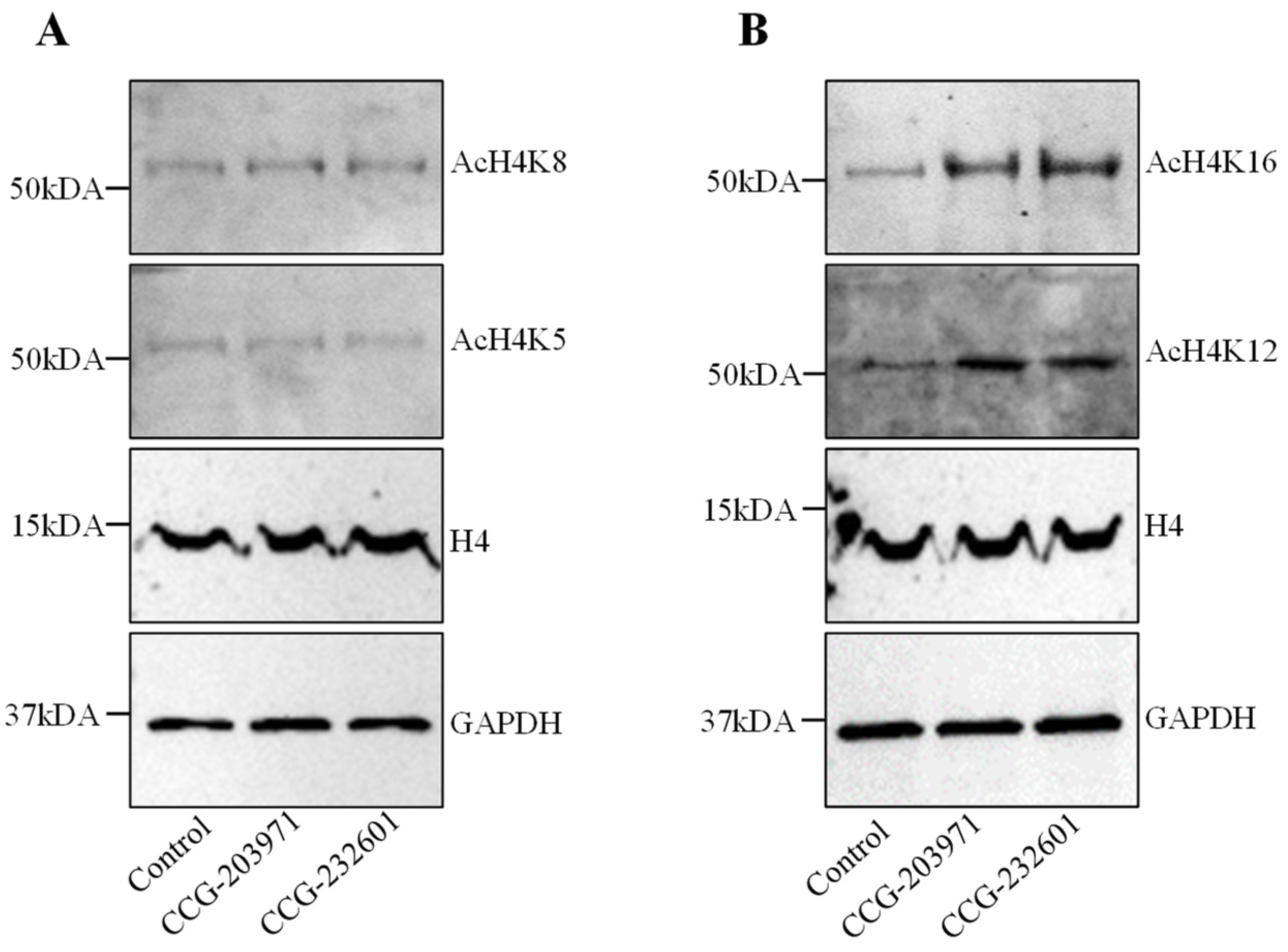
3.3. CCG-203971 and CCG-232601 Induce Histone H4 Hyperacetylation
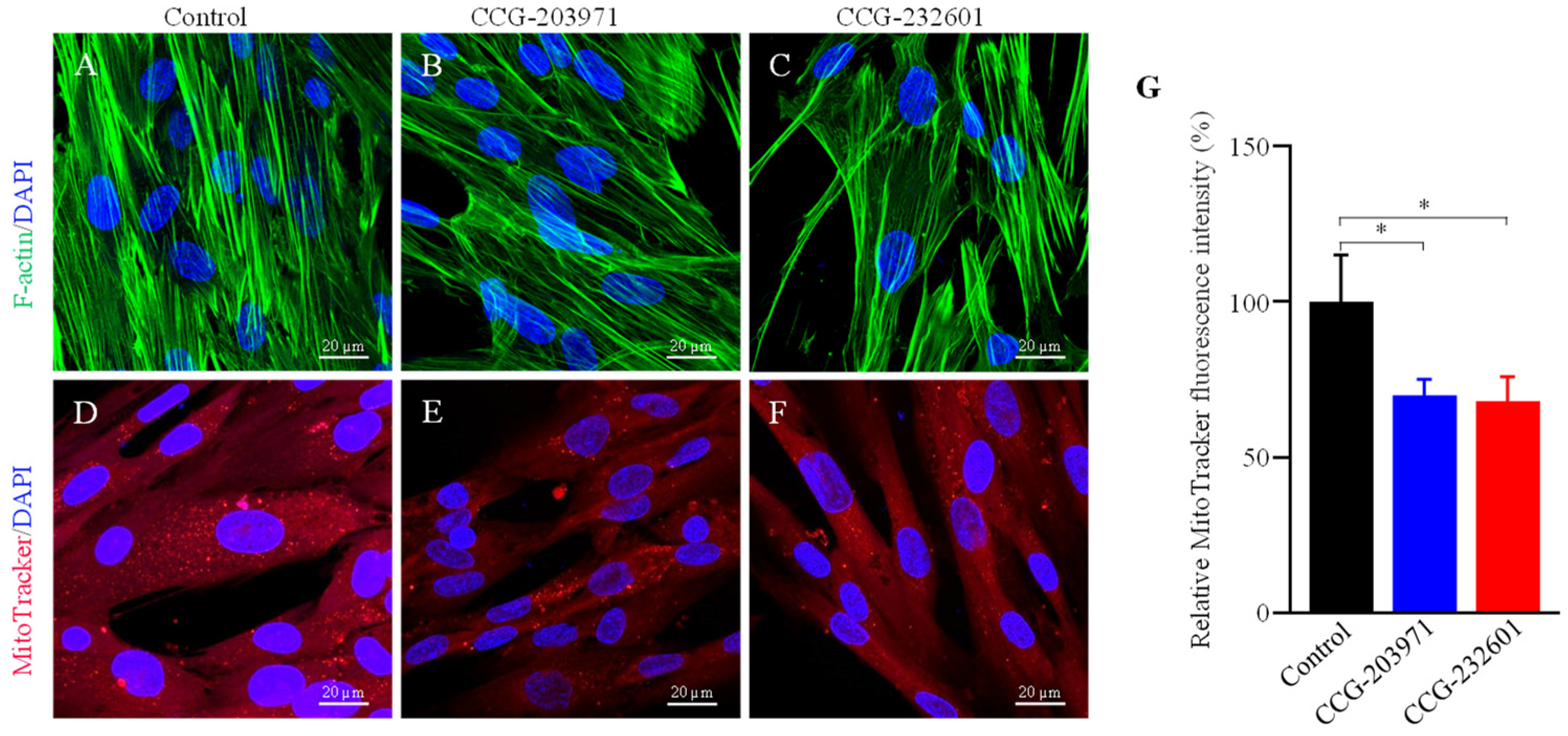
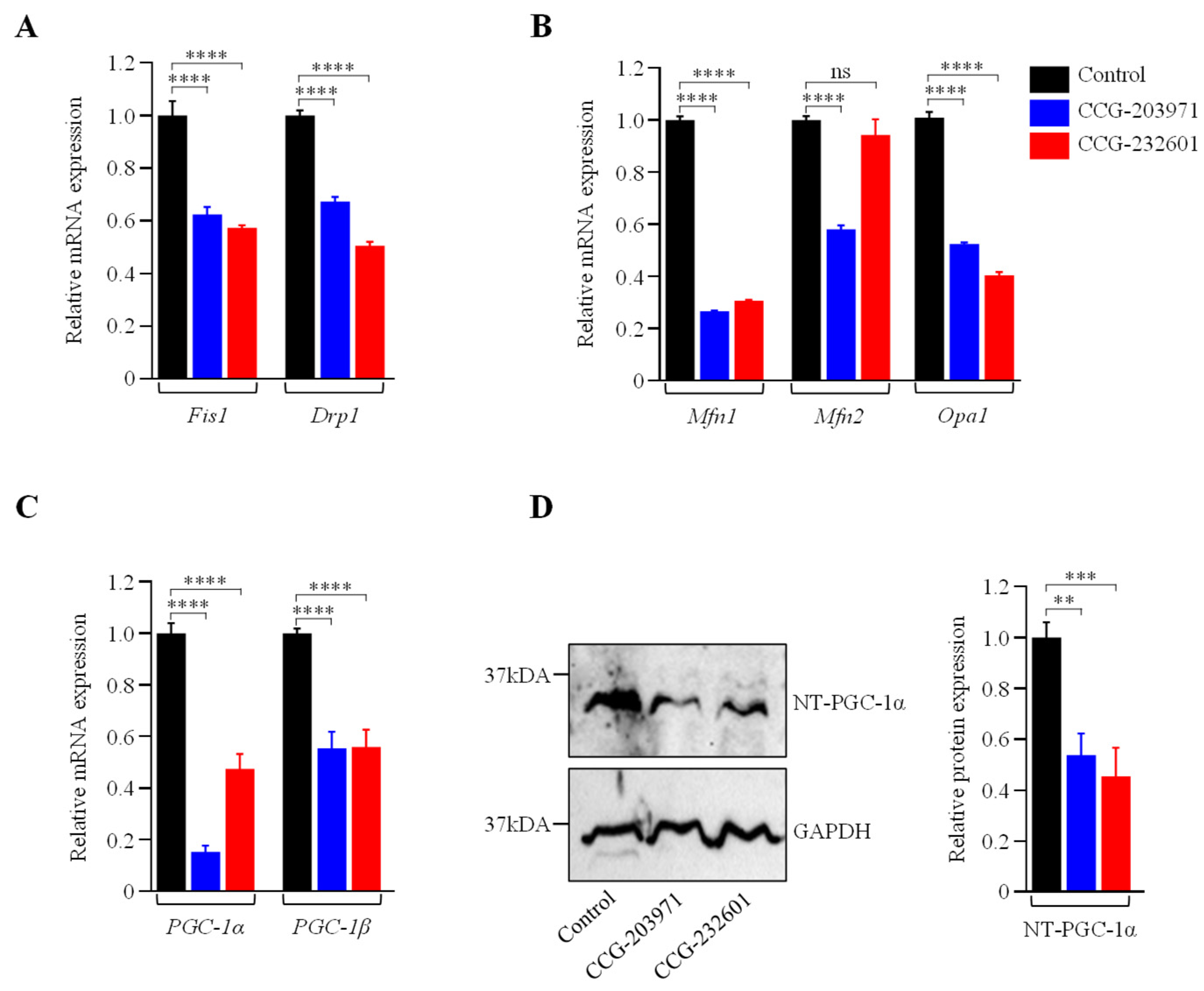
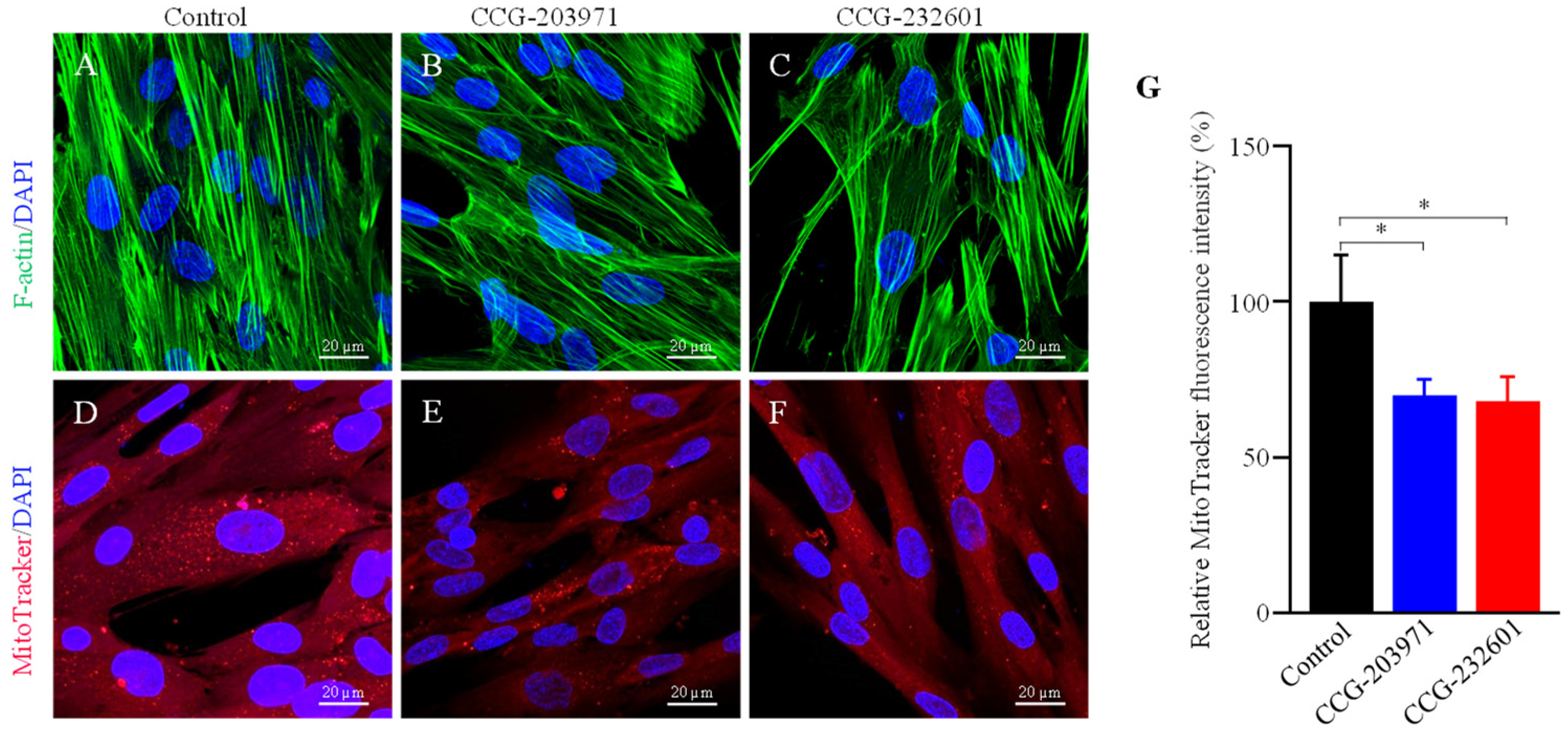
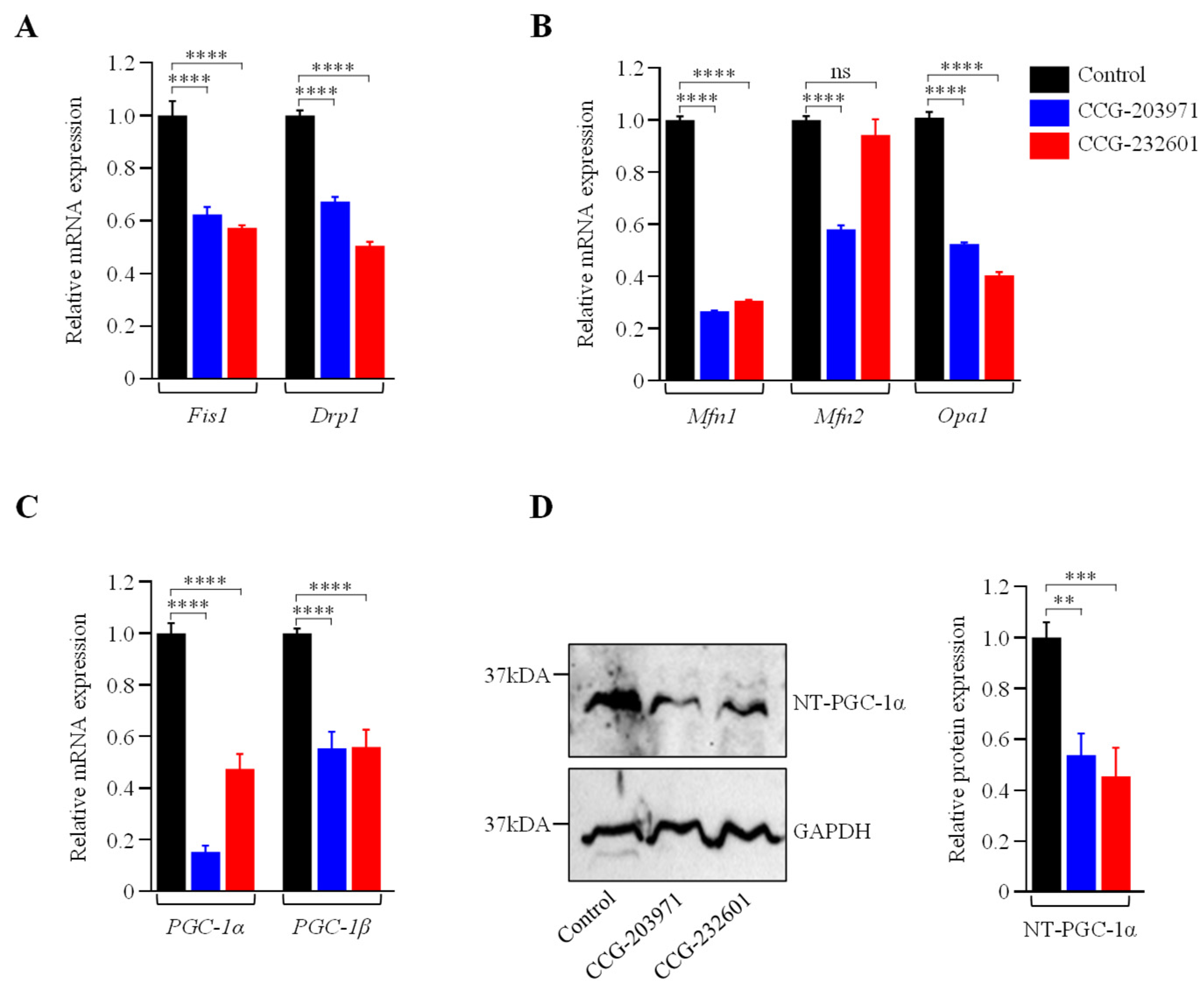
3.4. Actin Dynamics and Mitochondrial Biogenesis Are Regulated by CCG-203971 and CCG-232601
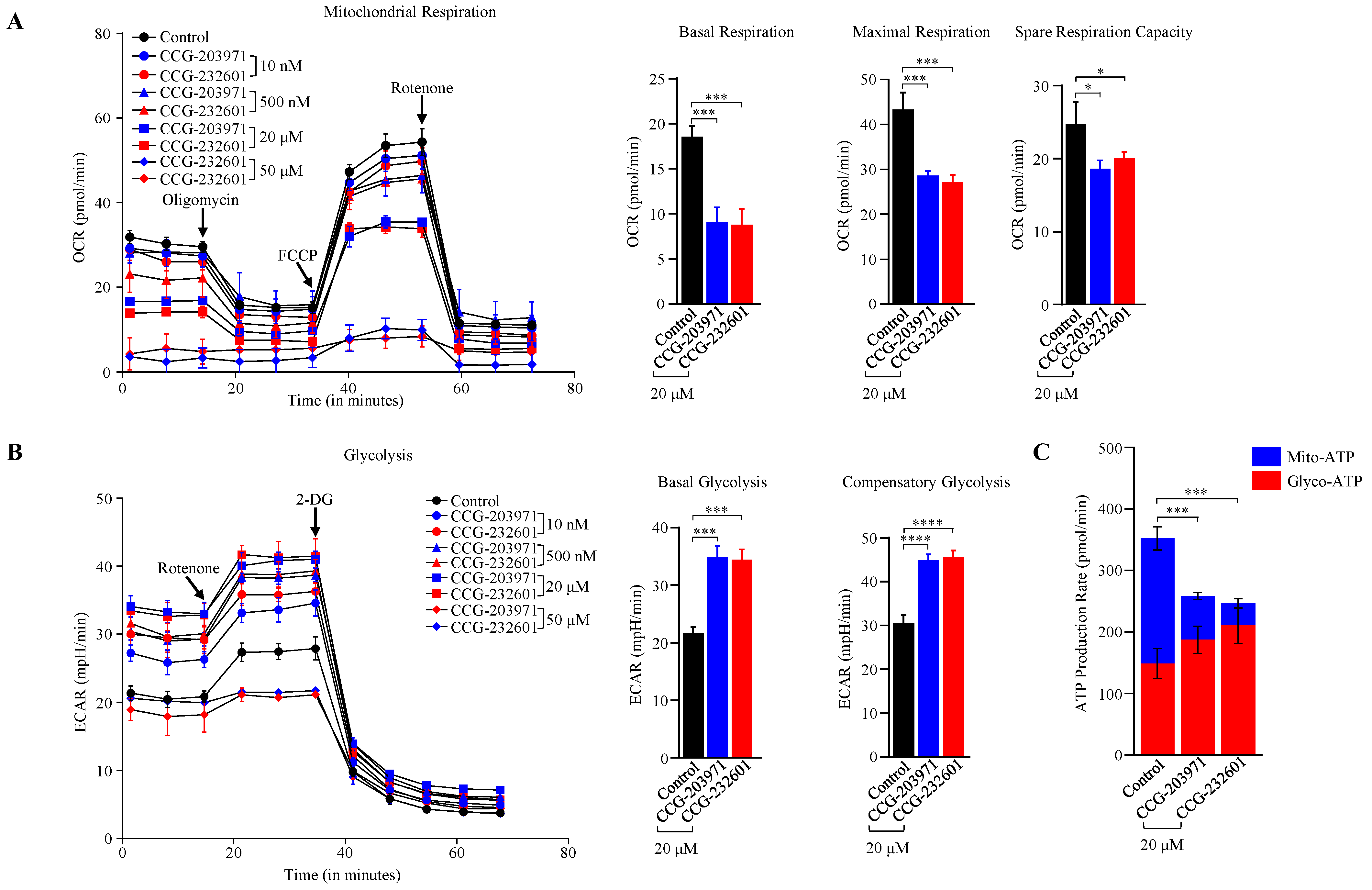
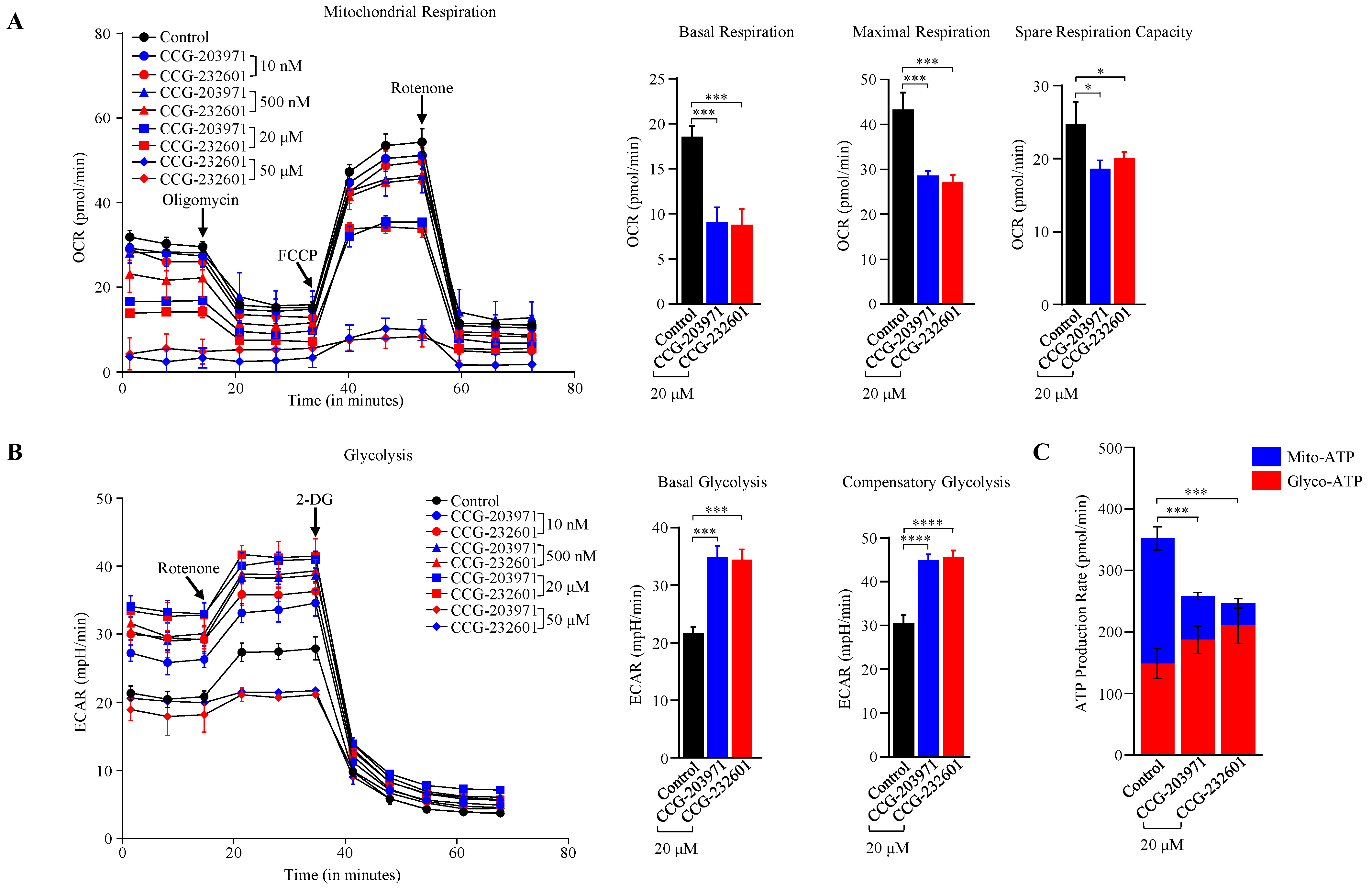
3.5. CCG-203971 and CCG 232601 Induce Reduction in Mitochondrial Respiration and Switch to Non-Oxidative Cellular Bioenergetics
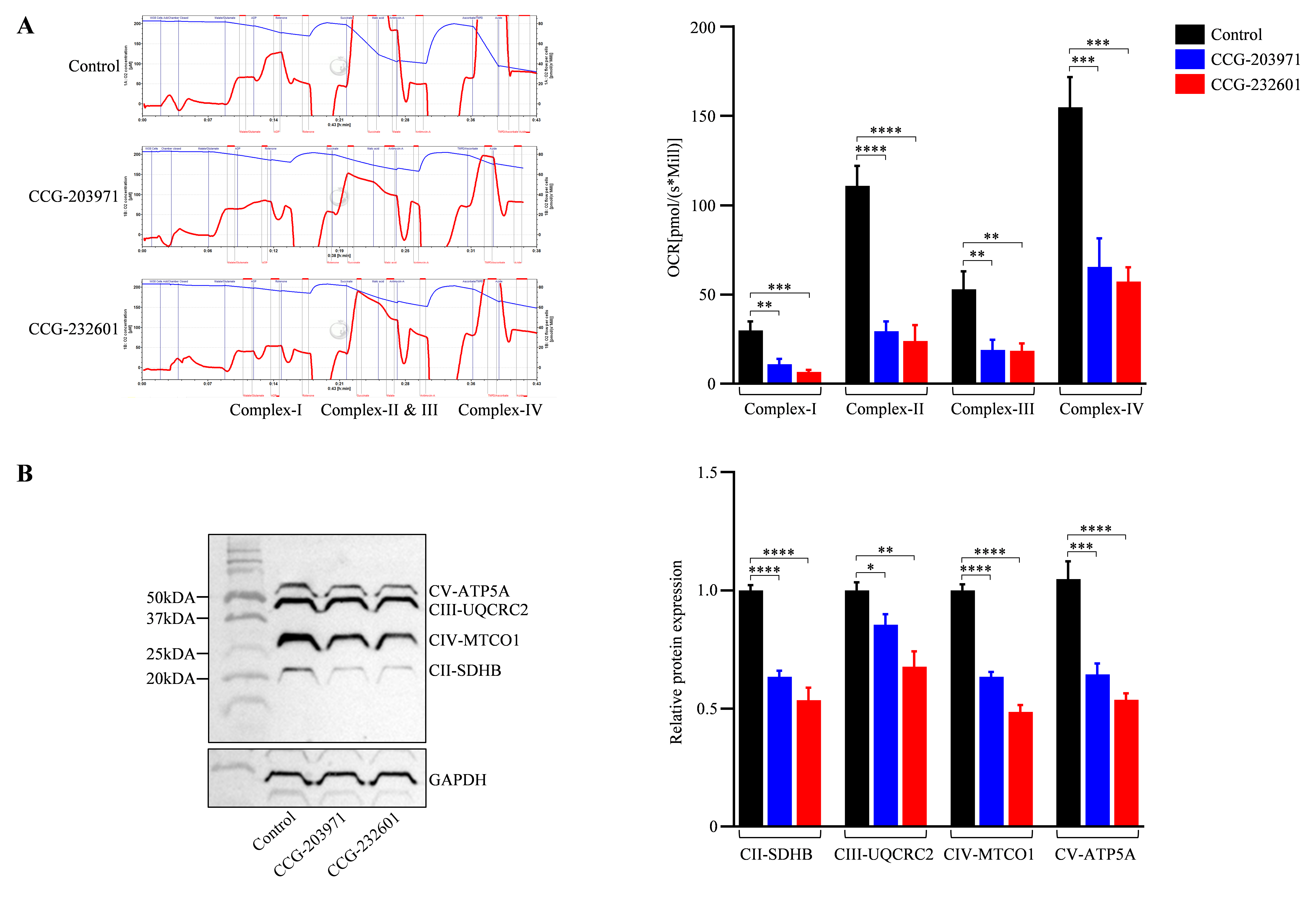
3.6. Functional Analysis of Mitochondrial Respiratory Chain
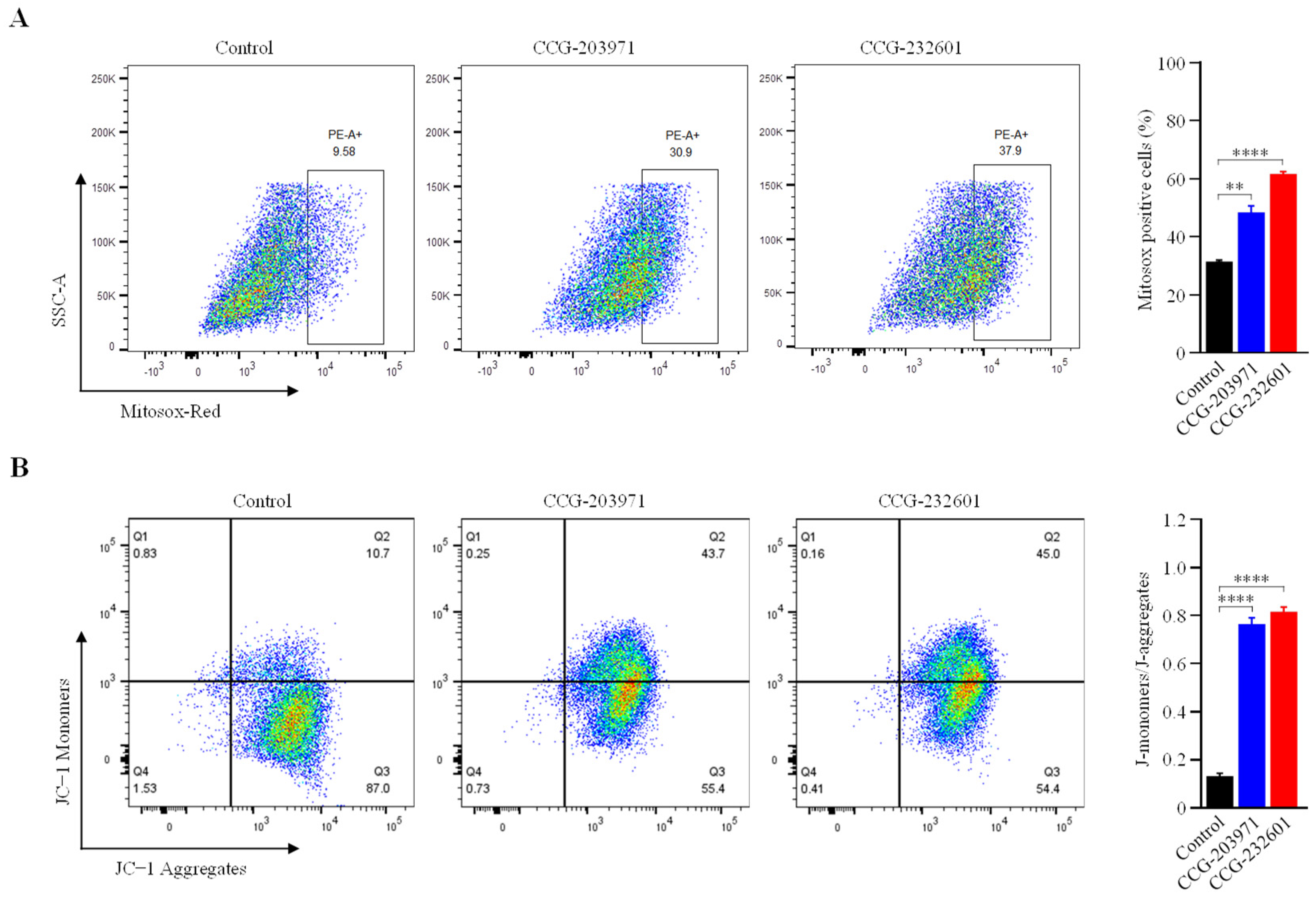
3.7. Increase in Mitochondrial ROS Level and Decrease in Mitochondrial Membrane Potential
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; et al. Targeting Reduced Mitochondrial DNA Quantity as a Therapeutic Approach in Pediatric High-Grade Gliomas. Neuro Oncol. 2020, 22, 139–151. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial Involvement in Migration, Invasion and Metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca2+ Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. The Effect of Reactive Oxygen Species Generated from the Mitochondrial Electron Transport Chain on the Cytochrome c Oxidase Activity and on the Cardiolipin Content in Bovine Heart Submitochondrial Particles. FEBS Lett. 2000, 466, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial Inner-Membrane Fusion and Crista Maintenance Requires the Dynamin-Related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef]
- Smirnova, E.; Shurland, D.-L.; Ryazantsev, S.N.; Van Der Bliek, A.M. A Human Dynamin-Related Protein Controls the Distribution of Mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A Key Regulator of Energy Metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1β-Regulated Mitochondrial Biogenesis and Function in Myotubes Is Mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-Actin Signaling to the MRTF Coactivators Dominates the Immediate Transcriptional Response to Serum in Fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef]
- Gau, D.; Roy, P. SRF’ing and SAP’ing—The Role of MRTF Proteins in Cell Migration. J. Cell Sci. 2018, 131, jcs218222. [Google Scholar] [CrossRef]
- Medjkane, S.; Perez-Sanchez, C.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-Related Transcription Factors and SRF Are Required for Cytoskeletal Dynamics and Experimental Metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef]
- Qu, C.; Yang, W.; Kan, Y.; Zuo, H.; Wu, M.; Zhang, Q.; Wang, H.; Wang, D.; Chen, J. RhoA/ROCK Signaling Regulates Drp1-Mediated Mitochondrial Fission During Collective Cell Migration. Front. Cell Dev. Biol. 2022, 10, 882581. [Google Scholar] [CrossRef]
- Tu, M.; Tan, V.P.; Yu, J.D.; Tripathi, R.; Bigham, Z.; Barlow, M.; Smith, J.M.; Brown, J.H.; Miyamoto, S. RhoA Signaling Increases Mitophagy and Protects Cardiomyocytes against Ischemia by Stabilizing PINK1 Protein and Recruiting Parkin to Mitochondria. Cell Death Differ. 2022, 29, 2472–2486. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Flynn, K.; Lindenberg, K.S.; Schwarz, H.; Bradke, F.; Di Giovanni, S.; Knöll, B. Serum Response Factor (SRF)-Cofilin-Actin Signaling Axis Modulates Mitochondrial Dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, E2523–E2532. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jardin, B.D.; Zhou, P.; Sethi, I.; Akerberg, B.N.; Toepfer, C.N.; Ai, Y.; Li, Y.; Ma, Q.; Guatimosim, S.; et al. Hierarchical and Stage-Specific Regulation of Murine Cardiomyocyte Maturation by Serum Response Factor. Nat. Commun. 2018, 9, 3837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Zhong, Y.; Wei, J.Y. Identification of a Novel Serum Response Factor Cofactor in Cardiac Gene Regulation. J. Biol. Chem. 2004, 279, 55626–55632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Helms, S.; Zhong, Y.; Wei, J.Y. Identification of a Subunit of NADH-Dehydrogenase as a P49/STRAP-Binding Protein. BMC Cell Biol. 2008, 9, 8. [Google Scholar] [CrossRef]
- Cao, D.; Wang, Z.; Zhang, C.-L.; Oh, J.; Xing, W.; Li, S.; Richardson, J.A.; Wang, D.-Z.; Olson, E.N. Modulation of Smooth Muscle Gene Expression by Association of Histone Acetyltransferases and Deacetylases with Myocardin. Mol. Cell. Biol. 2005, 25, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Alberts, A.S.; Geneste, O.; Treisman, R. Activation of SRF-Regulated Chromosomal Templates by Rho-Family GTPases Requires a Signal That Also Induces H4 Hyperacetylation. Cell 1998, 92, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, H.; Chandel, N.S.; Dou, Y.; Yi, R. MOF-Mediated Histone H4 Lysine 16 Acetylation Governs Mitochondrial and Ciliary Functions by Controlling Gene Promoters. Nat. Commun. 2023, 14, 4404. [Google Scholar] [CrossRef]
- Kahl, D.J.; Hutchings, K.M.; Lisabeth, E.M.; Haak, A.J.; Leipprandt, J.R.; Dexheimer, T.; Khanna, D.; Tsou, P.-S.; Campbell, P.L.; Fox, D.A.; et al. 5-Aryl-1,3,4-Oxadiazol-2-Ylthioalkanoic Acids: A Highly Potent New Class of Inhibitors of Rho/Myocardin-Related Transcription Factor (MRTF)/Serum Response Factor (SRF)-Mediated Gene Transcription as Potential Antifibrotic Agents for Scleroderma. J. Med. Chem. 2019, 62, 4350–4369. [Google Scholar] [CrossRef]
- Yu-Wai-Man, C.; Spencer-Dene, B.; Lee, R.M.H.; Hutchings, K.; Lisabeth, E.M.; Treisman, R.; Bailly, M.; Larsen, S.D.; Neubig, R.R.; Khaw, P.T. Local Delivery of Novel MRTF/SRF Inhibitors Prevents Scar Tissue Formation in a Preclinical Model of Fibrosis. Sci. Rep. 2017, 7, 518. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Azhar, G.; Zhang, X.; Patyal, P.; Kc, G.; Sharma, S.; Che, Y.; Wei, J.Y.P. Gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 950. [Google Scholar] [CrossRef]
- Rogers, S.C.; Zhang, X.; Azhar, G.; Luo, S.; Wei, J.Y. Exposure to High or Low Glucose Levels Accelerates the Appearance of Markers of Endothelial Cell Senescence and Induces Dysregulation of Nitric Oxide Synthase. J. Gerontol. Ser. A 2013, 68, 1469–1481. [Google Scholar] [CrossRef]
- Zhang, X.; Williams, E.D.; Azhar, G.; Rogers, S.C.; Wei, J.Y. Does P49/STRAP, a SRF-Binding Protein (SRFBP1), Modulate Cardiac Mitochondrial Function in Aging? Exp. Gerontol. 2016, 82, 150–159. [Google Scholar] [CrossRef]
- Patyal, P.; Nguyen, B.; Zhang, X.; Azhar, G.; Ameer, F.S.; Verma, A.; Crane, J.; Kc, G.; Che, Y.; Wei, J.Y. Rho/SRF Inhibitor Modulates Mitochondrial Functions. Int. J. Mol. Sci. 2022, 23, 11536. [Google Scholar] [CrossRef]
- Mokalled, M.H.; Carroll, K.J.; Cenik, B.K.; Chen, B.; Liu, N.; Olson, E.N.; Bassel-Duby, R. Myocardin-Related Transcription Factors Are Required for Cardiac Development and Function. Dev. Biol. 2015, 406, 109–116. [Google Scholar] [CrossRef]
- McDonald, O.G. Control of SRF Binding to CArG Box Chromatin Regulates Smooth Muscle Gene Expression in Vivo. J. Clin. Investig. 2005, 116, 36–48. [Google Scholar] [CrossRef]
- Etoh, S.; Matsui, H.; Tokuda, M.; Itano, T.; Nakamura, M.; Hatase, O. Purification and Immunohistochemical Study of Actin in Mitochondrial Matrix. Biochem. Int. 1990, 20, 599–606. [Google Scholar] [PubMed]
- Boldogh, I.R.; Yang, H.-C.; Nowakowski, W.D.; Karmon, S.L.; Hays, L.G.; Yates, J.R.; Pon, L.A. Arp2/3 Complex and Actin Dynamics Are Required for Actin-Based Mitochondrial Motility in Yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 3162–3167. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.I.; Blaabjerg, M.; Freude, K.; Meyer, M. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11, 1520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, M.; Zhou, G.; Xie, Z.; Wang, Y.; Han, J.; Li, L.; Wu, Q.; Zhang, S. Role of Rho GTPases in Inflammatory Bowel Disease. Cell Death Discov. 2023, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wu, D.; Gao, B.; Ingram, A.J.; Zhang, B.; Chorneyko, K.; McKenzie, R.; Krepinsky, J.C. RhoA/Rho-Kinase Contribute to the Pathogenesis of Diabetic Renal Disease. Diabetes 2008, 57, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and Roles in Cancer Cell Biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef]
- Barcelo, J.; Samain, R.; Sanz-Moreno, V. Preclinical to Clinical Utility of ROCK Inhibitors in Cancer. Trends Cancer 2023, 9, 250–263. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iñiguez-Lluhí, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A Small-Molecule Inhibitor of RhoA Transcriptional Signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef]
- Bell, J.L.; Haak, A.J.; Wade, S.M.; Kirchhoff, P.D.; Neubig, R.R.; Larsen, S.D. Optimization of Novel Nipecotic Bis(Amide) Inhibitors of the Rho/MKL1/SRF Transcriptional Pathway as Potential Anti-Metastasis Agents. Bioorg. Med. Chem. Lett. 2013, 23, 3826–3832. [Google Scholar] [CrossRef]
- Hutchings, K.M.; Lisabeth, E.M.; Rajeswaran, W.; Wilson, M.W.; Sorenson, R.J.; Campbell, P.L.; Ruth, J.H.; Amin, A.; Tsou, P.-S.; Leipprandt, J.R.; et al. Pharmacokinetic Optimitzation of CCG-203971: Novel Inhibitors of the Rho/MRTF/SRF Transcriptional Pathway as Potential Antifibrotic Therapeutics for Systemic Scleroderma. Bioorg. Med. Chem. Lett. 2017, 27, 1744–1749. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Krysztofiak, A.; Szymonowicz, K.; Hlouschek, J.; Xiang, K.; Waterkamp, C.; Larafa, S.; Goetting, I.; Vega-Rubin-de-Celis, S.; Theiss, C.; Matschke, V.; et al. Metabolism of Cancer Cells Commonly Responds to Irradiation by a Transient Early Mitochondrial Shutdown. iScience 2021, 24, 103366. [Google Scholar] [CrossRef]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.S.; Zhou, Y.; et al. Inhibition of Myocardin-Related Transcription Factor/Serum Response Factor Signaling Decreases Lung Fibrosis and Promotes Mesenchymal Cell Apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef]
- Damiani, E.; Solorio, J.A.; Doyle, A.P.; Wallace, H.M. How reliable are in vitro IC50 values? Values vary with cytotoxicity assays in human glioblastoma cells. Toxicol. Lett. 2019, 302, 28–34. [Google Scholar] [CrossRef]
- Haak, A.J.; Tsou, P.-S.; Amin, M.A.; Ruth, J.H.; Campbell, P.; Fox, D.A.; Khanna, D.; Larsen, S.D.; Neubig, R.R. Targeting the Myofibroblast Genetic Switch: Inhibitors of Myocardin-Related Transcription Factor/Serum Response Factor–Regulated Gene Transcription Prevent Fibrosis in a Murine Model of Skin Injury. J. Pharmacol. Exp. Ther. 2014, 349, 480–486. [Google Scholar] [CrossRef]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho Family of Ras-Like GTPases in Eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Huang, X.; Cheng, J.; Li, Z.; De Folter, S.; Huang, Z.; Jiang, X.; Pang, H.; Tao, S. Conservation and Evolution in and among SRF- and MEF2-Type MADS Domains and Their Binding Sites. Mol. Biol. Evol. 2011, 28, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Sural, S.; Liang, C.-Y.; Wang, F.-Y.; Ching, T.-T.; Hsu, A.-L. HSB-1/HSF-1 Pathway Modulates Histone H4 in Mitochondria to Control mtDNA Transcription and Longevity. Sci. Adv. 2020, 6, eaaz4452. [Google Scholar] [CrossRef] [PubMed]
- Shoulars, K.; Brown, T.; Alejandro, M.A.; Crowley, J.; Markaverich, B.M. Identification of nuclear type II [3H]estradiol binding sites as histone H4. Biochem. Biophys. Res. Commun. 2002, 296, 1083–1090. [Google Scholar] [CrossRef]
- Georgieva, E.I.; Sendra, R. Mobility of acetylated histones in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 1999, 269, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Pollak, M. Overcoming Drug Development Bottlenecks with Repurposing: Repurposing Biguanides to Target Energy Metabolism for Cancer Treatment. Nat. Med. 2014, 20, 591–593. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patyal, P.; Zhang, X.; Verma, A.; Azhar, G.; Wei, J.Y. Inhibitors of Rho/MRTF/SRF Transcription Pathway Regulate Mitochondrial Function. Cells 2024, 13, 392. https://doi.org/10.3390/cells13050392
Patyal P, Zhang X, Verma A, Azhar G, Wei JY. Inhibitors of Rho/MRTF/SRF Transcription Pathway Regulate Mitochondrial Function. Cells. 2024; 13(5):392. https://doi.org/10.3390/cells13050392
Chicago/Turabian StylePatyal, Pankaj, Xiaomin Zhang, Ambika Verma, Gohar Azhar, and Jeanne Y. Wei. 2024. "Inhibitors of Rho/MRTF/SRF Transcription Pathway Regulate Mitochondrial Function" Cells 13, no. 5: 392. https://doi.org/10.3390/cells13050392
APA StylePatyal, P., Zhang, X., Verma, A., Azhar, G., & Wei, J. Y. (2024). Inhibitors of Rho/MRTF/SRF Transcription Pathway Regulate Mitochondrial Function. Cells, 13(5), 392. https://doi.org/10.3390/cells13050392