

The CDK Inhibitor Dinaciclib Improves Cisplatin Response in Nonseminomatous Testicular Cancer: A Preclinical Study

 , , ,
, , ,  ,
,  ,
,  , and
, and
Abstract
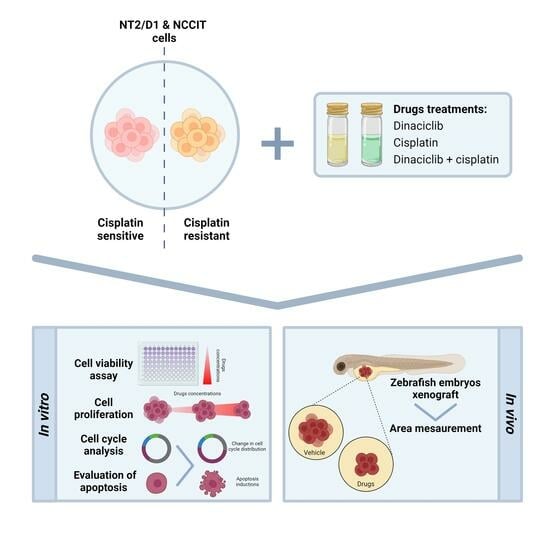
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Viability and Cell Proliferation Assay
2.3. Combined Drug Treatment: CP plus Dinaciclib
2.4. Cell-Cycle Analysis
2.5. Tumor Xenograft
2.6. Western Blot
2.7. Statistical Analysis
3. Results
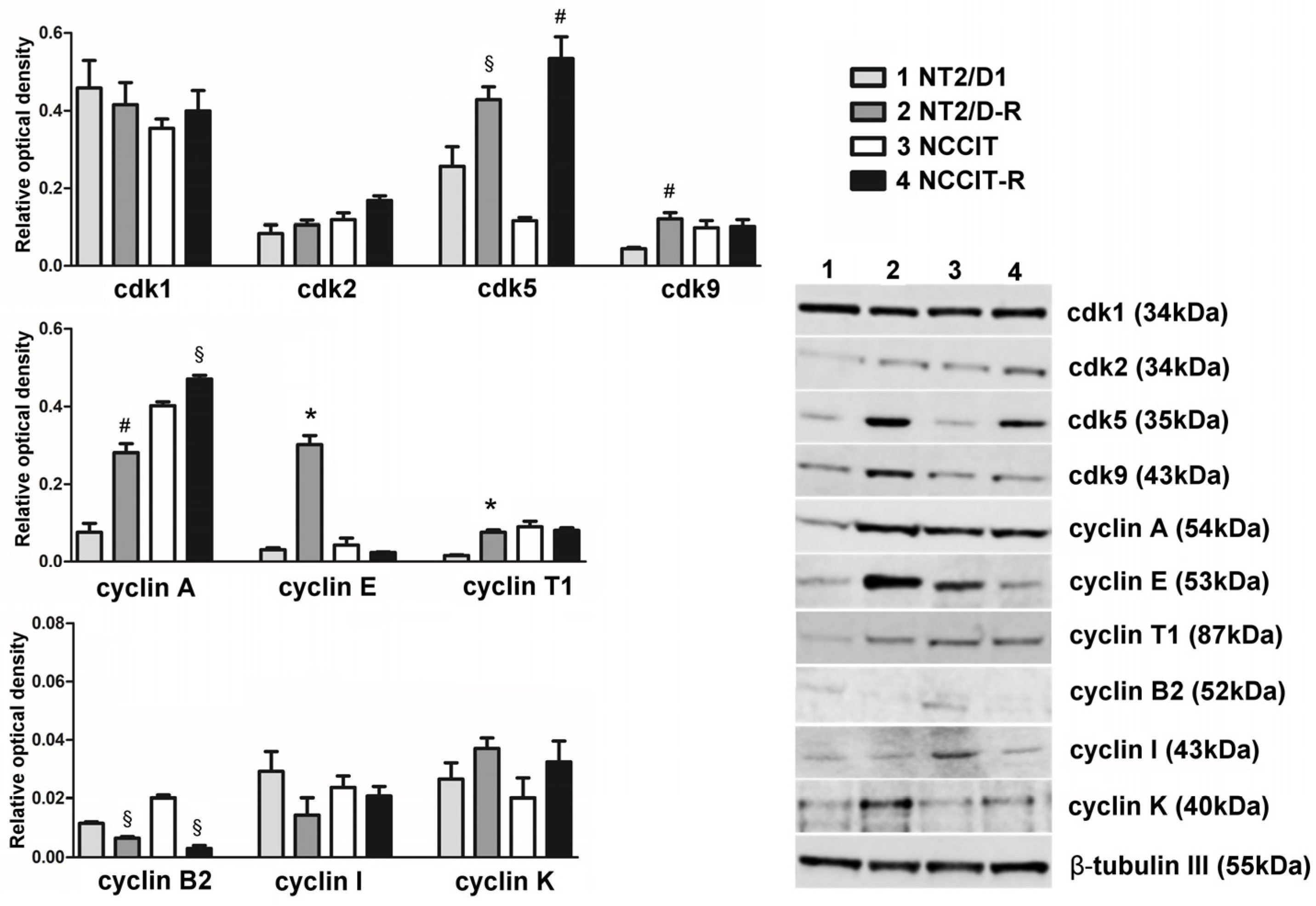
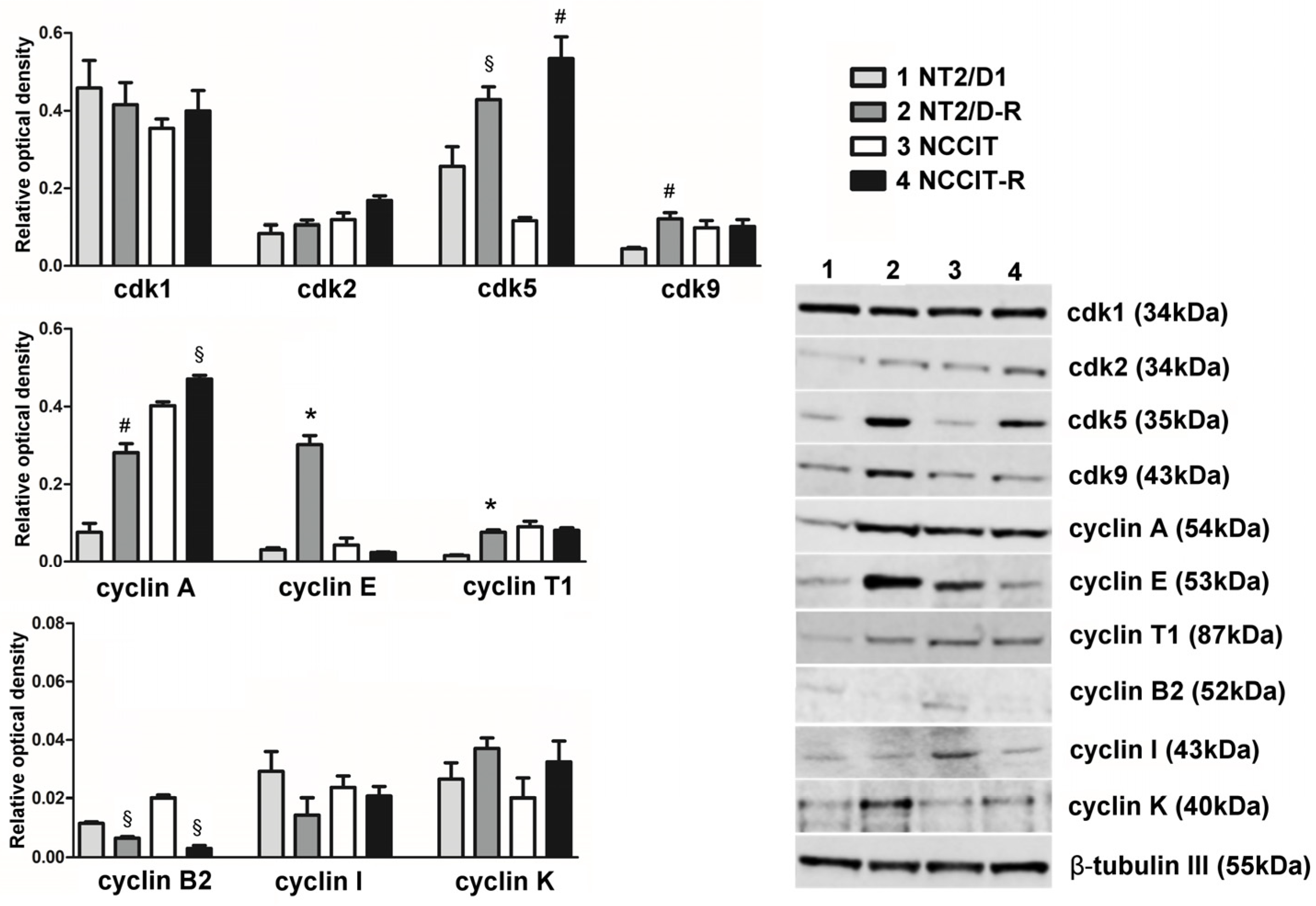
3.1. Protein Expression of CDKs Targeted by Dinaciclib and Their Related Cyclins
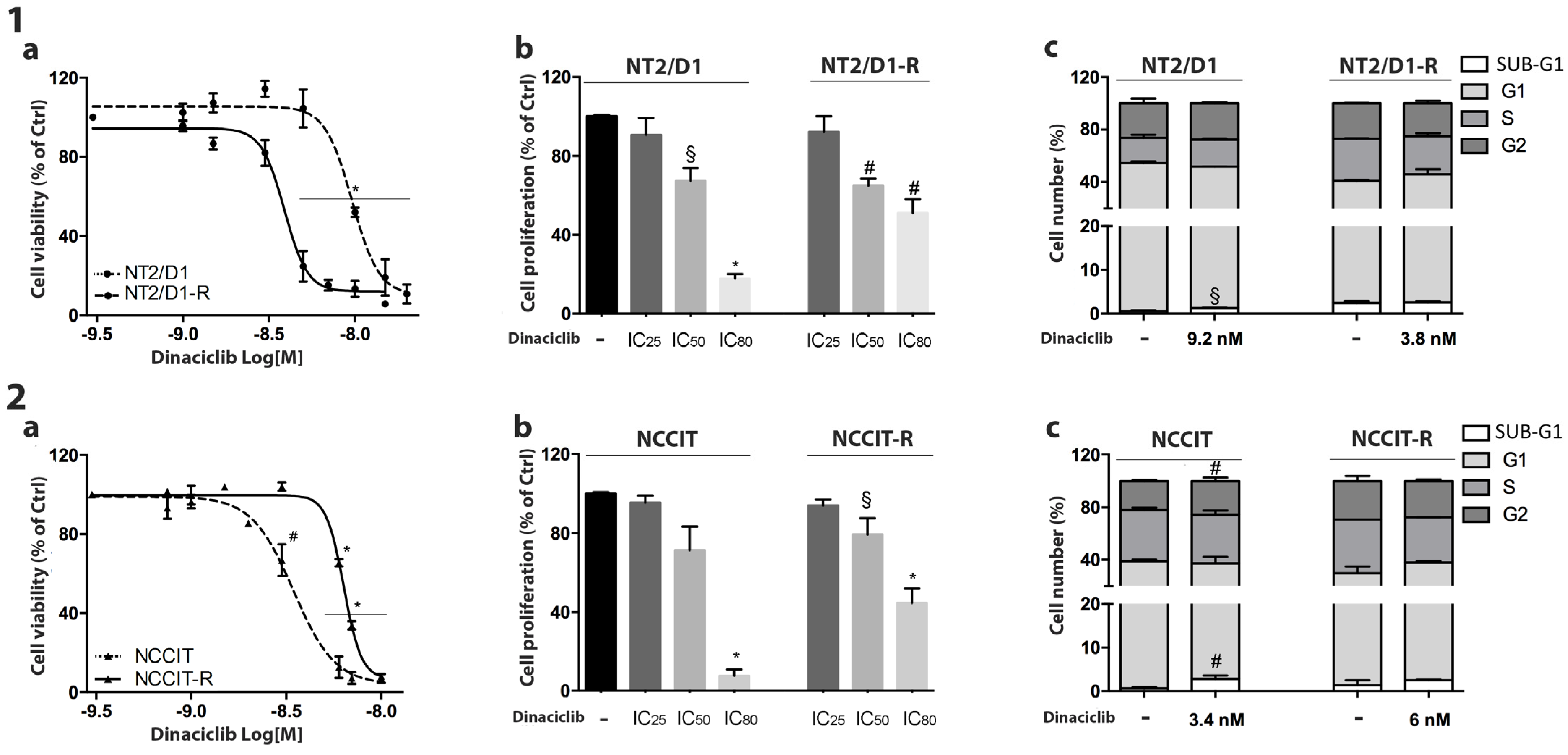
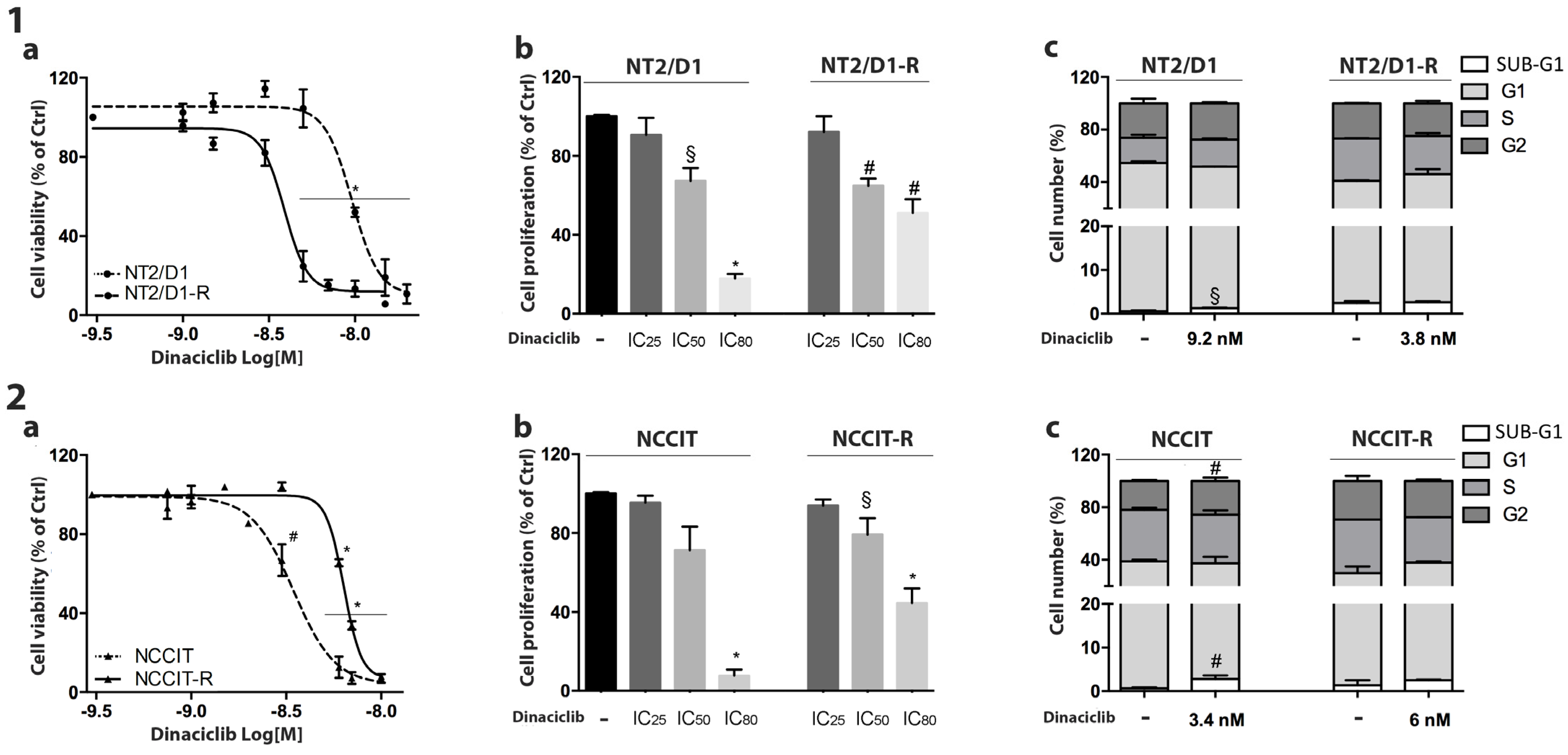
3.2. Effect of Dinaciclib Treatment on NT2/D1/-R and NCCIT/-R Cells
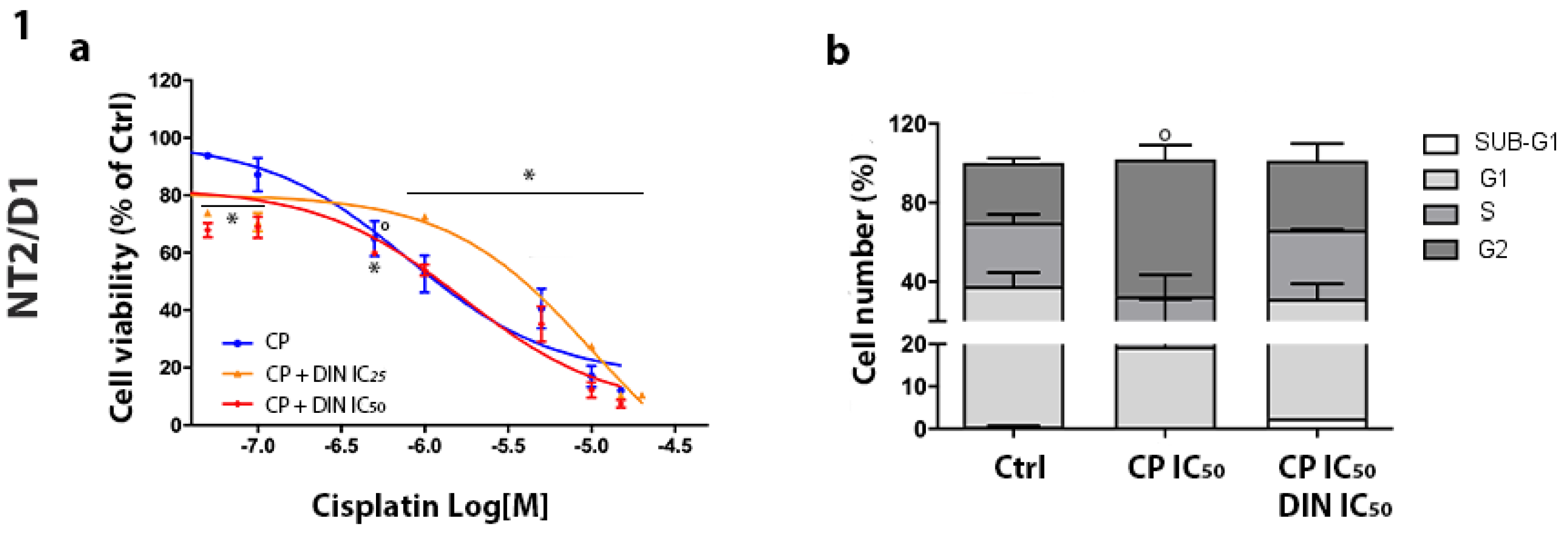
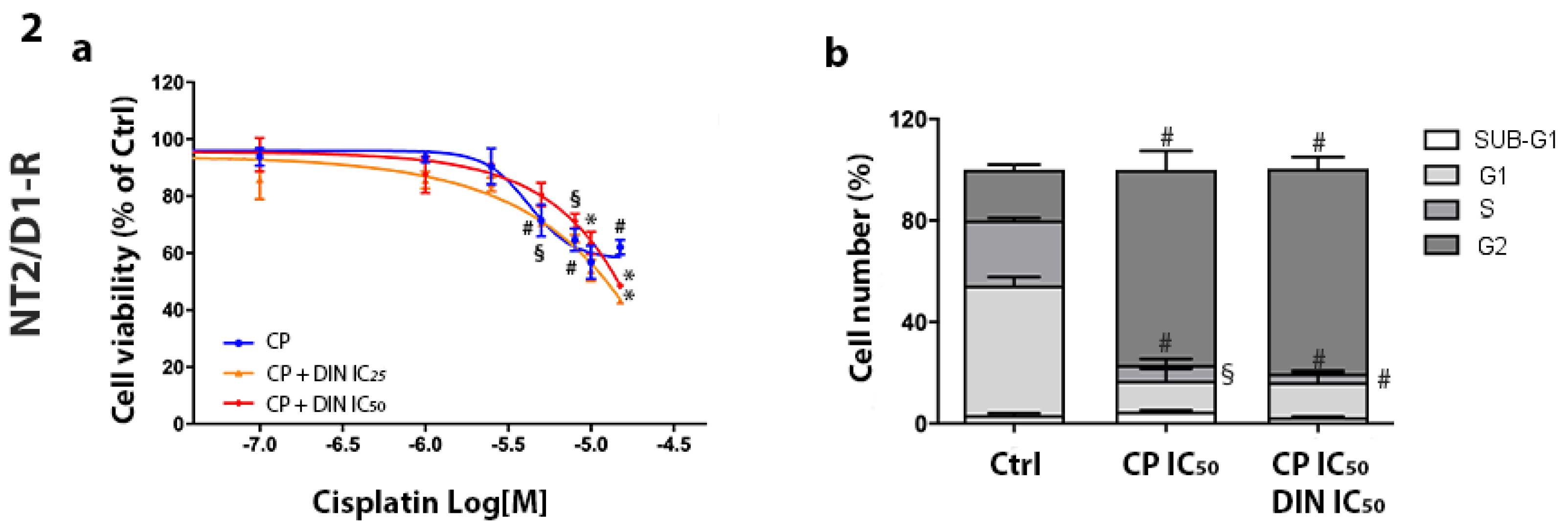
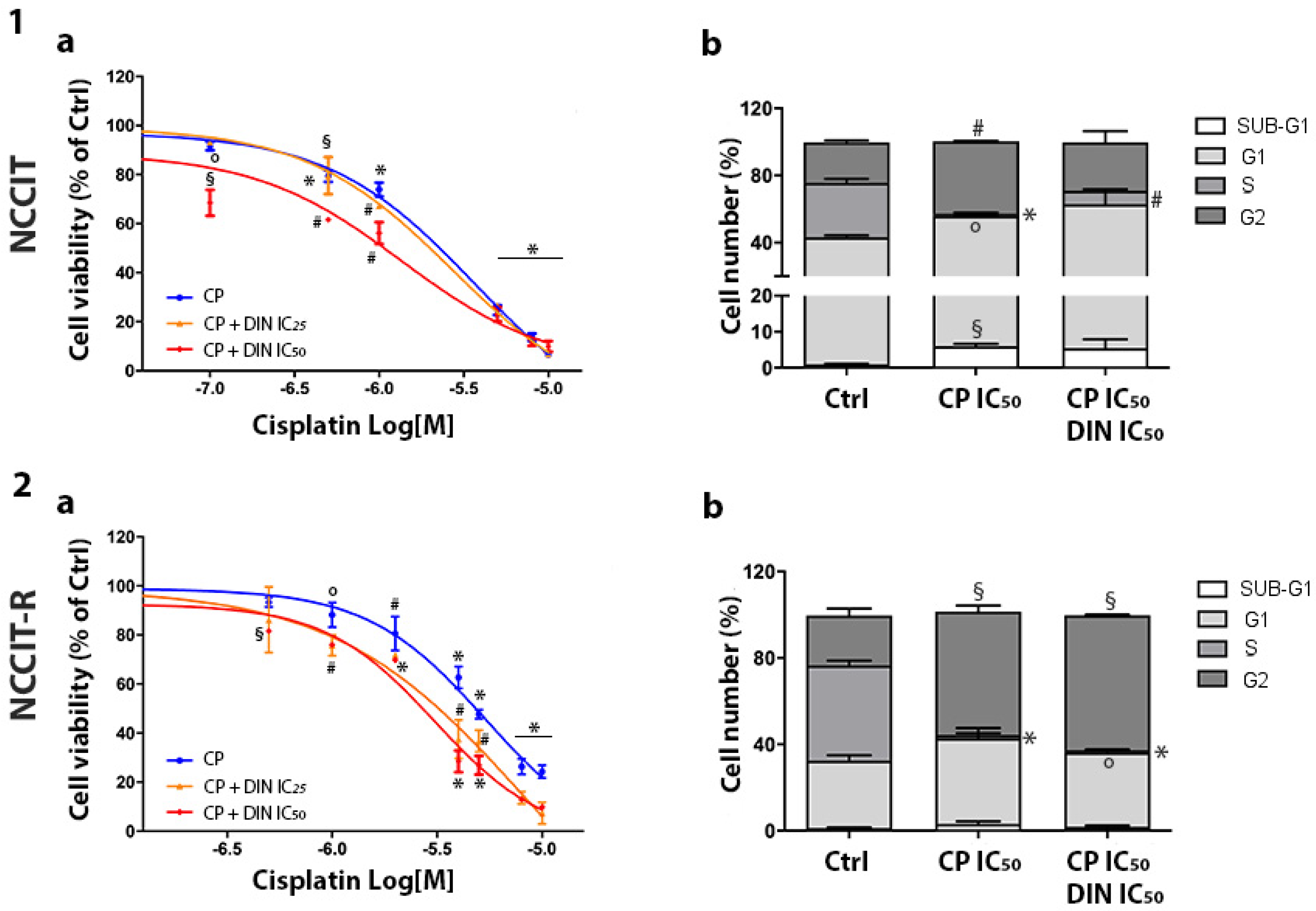
3.3. Effect of Combined Treatment of Dinaciclib/CP in NT2/D1 and NCCIT-Sensitive and CP-Resistant Cells
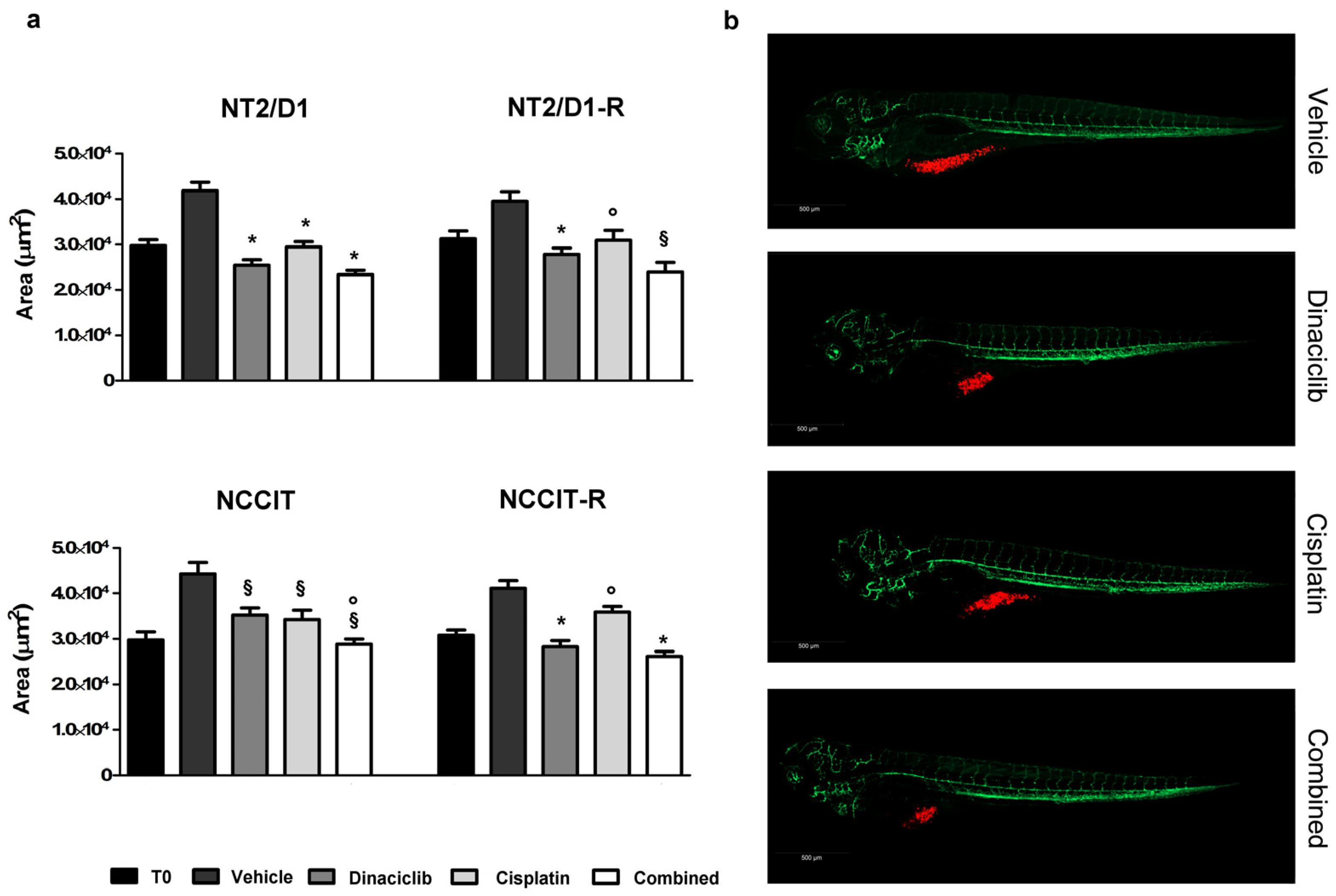
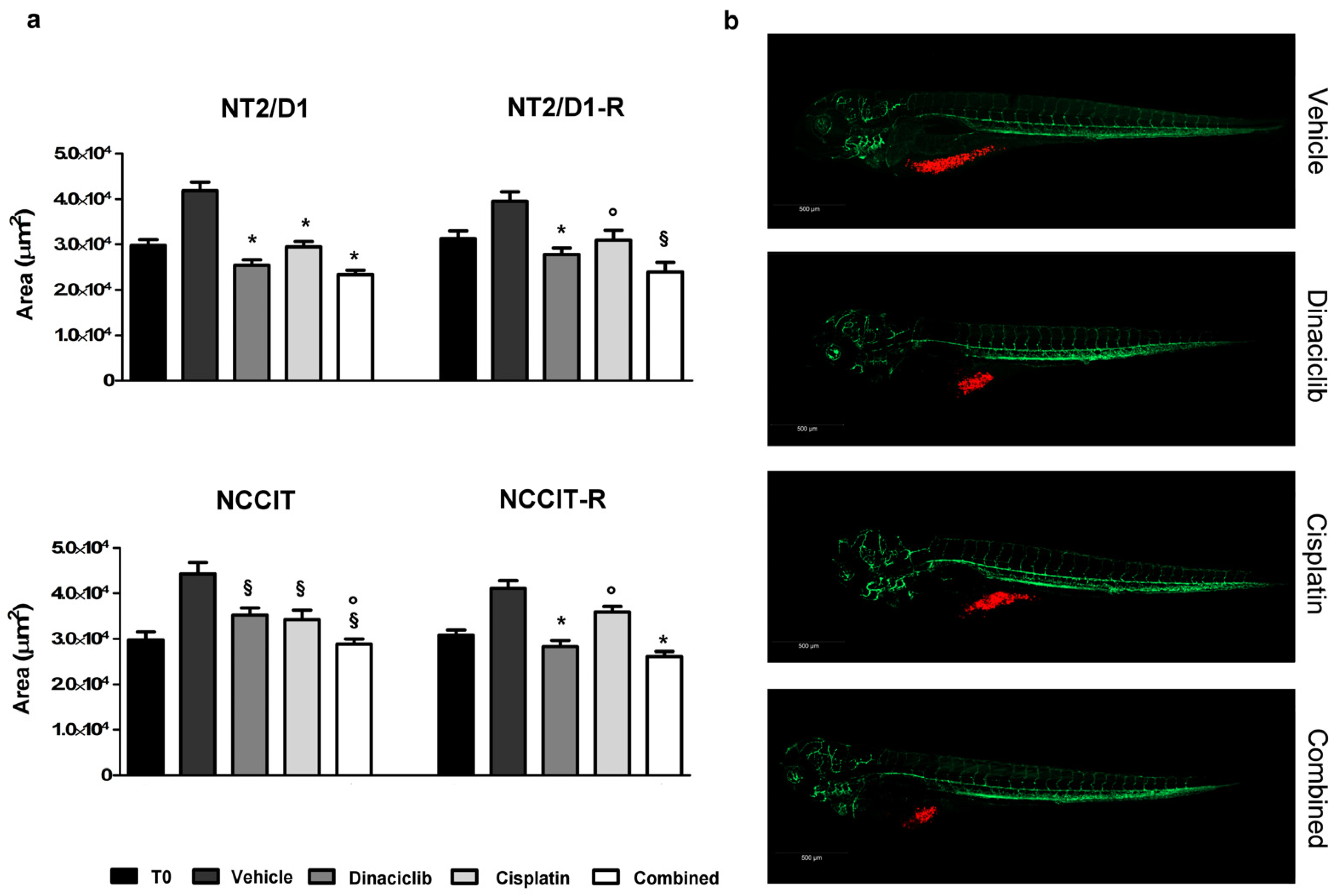
3.4. Effect of Dinaciclib Alone or Combined with Cisplatin in the Zebrafish/Tumor Xenograft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, J.; Adra, N.; Einhorn, L.H. Testicular Cancer: Biology to Bedside. Cancer Res. 2021, 81, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: Genetic and environmental aspects. Hum. Reprod. Update 2006, 12, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Fosså, S.D.; Nuver, J.; Heidenreich, A.; Schmoll, H.J.; Bokemeyer, C.; Horwich, A.; Beyer, J.; Kataja, V. Testicular seminoma and non-seminoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), vi125–vi132. [Google Scholar] [CrossRef]
- Lobo, J.; Costa, A.L.; Vilela-Salgueiro, B.; Rodrigues, Â.; Guimarães, R.; Cantante, M.; Lopes, P.; Antunes, L.; Jerónimo, C.; Henrique, R. Testicular germ cell tumors: Revisiting a series in light of the new WHO classification and AJCC staging systems, focusing on challenges for pathologists. Hum. Pathol. 2018, 82, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.; Lucia, J.; Foster, R.S.; Ulbright, T.M. The pathology of late recurrence of testicular germ cell tumors. Am. J. Surg. Pathol. 2000, 24, 257–273. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, M.J.; Feldman, D.R.; Carver, B.S.; Sheinfeld, J. Late Relapse of Testicular Germ Cell Tumors. Urol. Clin. N. Am. 2015, 42, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J. Guidelines on Testicular Cancer: 2015 Update. Eur. Urol. 2015, 68, 1054–1068. [Google Scholar] [CrossRef]
- Bakardjieva-Mihaylova, V.; Skvarova Kramarzova, K.; Slamova, M.; Svaton, M.; Rejlova, K.; Zaliova, M.; Dobiasova, A.; Fiser, K.; Stuchly, J.; Grega, M.; et al. Molecular Basis of Cisplatin Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 1316. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Song, Y.S.; Li, Y.; Jia, Y.H.; Zhao, H.D. Cdk5 links with DNA damage response and cancer. Mol. Cancer 2017, 16, 60. [Google Scholar] [CrossRef]
- Li, R.; Liu, G.Z.; Luo, S.Y.; Chen, R.; Zhang, J.X. Cyclin I promotes cisplatin resistance via Cdk5 activation in cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4533–4541. [Google Scholar]
- Ehrlich, S.M.; Liebl, J.; Ardelt, M.A.; Lehr, T.; De Toni, E.N.; Mayr, D.; Brandl, L.; Kirchner, T.; Zahler, S.; Gerbes, A.L.; et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma—A novel therapeutic approach. J. Hepatol. 2015, 63, 102–113. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Z.; Mao, W.; Ahmed, A.A.; Yang, H.; Zhou, J.; Jennings, N.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Miranda, R.; et al. CDK5 Regulates Paclitaxel Sensitivity in Ovarian Cancer Cells by Modulating AKT Activation, p21Cip1- and p27Kip1-Mediated G1 Cell Cycle Arrest and Apoptosis. PLoS ONE 2015, 10, e0131833. [Google Scholar] [CrossRef] [PubMed]
- Funke, K.; Düster, R.; Wilson, P.D.; Arévalo, L.; Geyer, M.; Schorle, H. Transcriptional CDK Inhibitors as Potential Treatment Option for Testicular Germ Cell Tumors. Cancers 2022, 14, 1690. [Google Scholar] [CrossRef] [PubMed]
- Rossini, E.; Bosatta, V.; Abate, A.; Fragni, M.; Salvi, V.; Basnet, R.M.; Zizioli, D.; Bosisio, D.; Piovani, G.; Valcamonico, F.; et al. Cisplatin Cytotoxicity in Human Testicular Germ Cell Tumor Cell Lines Is Enhanced by the CDK4/6 Inhibitor Palbociclib. Clin. Genitourin. Cancer 2021, 19, 316–324. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar] [PubMed]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259. [Google Scholar] [CrossRef]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef]
- Chen, X.X.; Xie, F.F.; Zhu, X.J.; Lin, F.; Pan, S.S.; Gong, L.H.; Qiu, J.G.; Zhang, W.J.; Jiang, Q.W.; Mei, X.L.; et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget 2015, 6, 14926–14939. [Google Scholar] [CrossRef]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef]
- Oechsle, K.; Honecker, F.; Cheng, T.; Mayer, F.; Czaykowski, P.; Winquist, E.; Wood, L.; Fenner, M.; Glaesener, S.; Hartmann, J.T.; et al. Preclinical and clinical activity of sunitinib in patients with cisplatin-refractory or multiply relapsed germ cell tumors: A Canadian Urologic Oncology Group/German Testicular Cancer Study Group cooperative study. Ann. Oncol. 2011, 22, 2654–2660. [Google Scholar] [CrossRef]
- Fenske, A.E.; Glaesener, S.; Bokemeyer, C.; Thomale, J.; Dahm-Daphi, J.; Honecker, F.; Dartsch, D.C. Cisplatin resistance induced in germ cell tumour cells is due to reduced susceptibility towards cell death but not to altered DNA damage induction or repair. Cancer Lett. 2012, 324, 171–178. [Google Scholar] [CrossRef]
- Rossini, E.; Tamburello, M.; Abate, A.; Beretta, S.; Fragni, M.; Cominelli, M.; Cosentini, D.; Hantel, C.; Bono, F.; Grisanti, S.; et al. Cytotoxic Effect of Progesterone, Tamoxifen and Their Combination in Experimental Cell Models of Human Adrenocortical Cancer. Front. Endocrinol. 2021, 12, 669426. [Google Scholar] [CrossRef]
- Abate, A.; Rossini, E.; Tamburello, M.; Laganà, M.; Cosentini, D.; Grisanti, S.; Fiorentini, C.; Tiberio, G.A.M.; Scatolini, M.; Grosso, E.; et al. Ribociclib Cytotoxicity Alone or Combined with Progesterone and/or Mitotane in in Vitro Adrenocortical Carcinoma Cells. Endocrinology 2022, 163, bqab248. [Google Scholar] [CrossRef] [PubMed]
- Basnet, R.M.; Zizioli, D.; Muscò, A.; Finazzi, D.; Sigala, S.; Rossini, E.; Tobia, C.; Guerra, J.; Presta, M.; Memo, M. Caffeine Inhibits Direct and Indirect Angiogenesis in Zebrafish Embryos. Int. J. Mol. Sci. 2021, 22, 4856. [Google Scholar] [CrossRef] [PubMed]
- Tamburello, M.; Abate, A.; Rossini, E.; Basnet, R.M.; Zizioli, D.; Cosentini, D.; Hantel, C.; Laganà, M.; Tiberio, G.A.M.; Grisanti, S.; et al. Preclinical Evidence of Progesterone as a New Pharmacological Strategy in Human Adrenocortical Carcinoma Cell Lines. Int. J. Mol. Sci. 2023, 24, 6829. [Google Scholar] [CrossRef] [PubMed]
- Gianoncelli, A.; Guarienti, M.; Fragni, M.; Bertuzzi, M.; Rossini, E.; Abate, A.; Basnet, R.M.; Zizioli, D.; Bono, F.; Terzolo, M.; et al. Adrenocortical Carcinoma Xenograft in Zebrafish Embryos as a Model To Study the In Vivo Cytotoxicity of Abiraterone Acetate. Endocrinology 2019, 160, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef]
- Einhorn, L.H. Curing metastatic testicular cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 4592–4595. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef]
- Rosales, J.L.; Lee, K.Y. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays 2006, 28, 1023–1034. [Google Scholar] [CrossRef]
- Session, D.R.; Fautsch, M.P.; Avula, R.; Jones, W.R.; Nehra, A.; Wieben, E.D. Cyclin-dependent kinase 5 is expressed in both Sertoli cells and metaphase spermatocytes. Fertil. Steril. 2001, 75, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Musa, F.R.; Tokuda, M.; Kuwata, Y.; Ogawa, T.; Tomizawa, K.; Konishi, R.; Takenaka, I.; Hatase, O. Expression of cyclin-dependent kinase 5 and associated cyclins in Leydig and Sertoli cells of the testis. J. Androl. 1998, 19, 657–666. [Google Scholar] [CrossRef]
- Rosales, J.L.; Lee, B.C.; Modarressi, M.; Sarker, K.P.; Lee, K.Y.; Jeong, Y.G.; Oko, R.; Lee, K.Y. Outer dense fibers serve as a functional target for Cdk5.p35 in the developing sperm tail. J. Biol. Chem. 2004, 279, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Mori, T.; Anazawa, Y.; Matsui, K.; Fukuda, S.; Nakamura, Y.; Arakawa, H. Cyclin K as a direct transcriptional target of the p53 tumor suppressor. Neoplasia 2002, 4, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Anshabo, A.T.; Milne, R.; Wang, S.; Albrecht, H. CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front. Oncol. 2021, 11, 678559. [Google Scholar] [CrossRef]
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010, 11, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Sergio, C.M.; Kang, J.; Muthukaruppan, A.; Boersma, M.N.; Stone, A.; Barraclough, J.; Lee, C.S.; Black, M.A.; Miller, L.D.; et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol. Cancer Ther. 2012, 11, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. A dual role of Cdk2 in DNA damage response. Cell Div. 2009, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Vilella, F.; Atkinson, S.P.; Lloret, M.; Passos, J.F.; von Zglinicki, T.; O’Connor, J.E.; Burks, D.; Jones, R.; Armstrong, L.; et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells 2011, 29, 651–659. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 knockout mice are viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef]
- Ortega, S.; Prieto, I.; Odajima, J.; Martín, A.; Dubus, P.; Sotillo, R.; Barbero, J.L.; Malumbres, M.; Barbacid, M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 2003, 35, 25–31. [Google Scholar] [CrossRef]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef]
- Cybulski, M.; Jarosz, B.; Nowakowski, A.; Jeleniewicz, W.; Kutarska, E.; Bednarek, W.; Stepulak, A. Cyclin A correlates with YB1, progression and resistance to chemotherapy in human epithelial ovarian cancer. Anticancer Res. 2015, 35, 1715–1721. [Google Scholar]
- Liao, H.; Ji, F.; Geng, X.; Xing, M.; Li, W.; Chen, Z.; Shen, H.; Ying, S. CDK1 promotes nascent DNA synthesis and induces resistance of cancer cells to DNA-damaging therapeutic agents. Oncotarget 2017, 8, 90662–90673. [Google Scholar] [CrossRef]
- Hsu, W.H.; Zhao, X.; Zhu, J.; Kim, I.K.; Rao, G.; McCutcheon, J.; Hsu, S.T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef]
- Howard, D.; James, D.; Murphy, K.; Garcia-Parra, J.; Pan-Castillo, B.; Rex, S.; Moul, A.; Jones, E.; Bilbao-Asensio, M.; Michue-Seijas, S.; et al. Dinaciclib, a Bimodal Agent Effective against Endometrial Cancer. Cancers 2021, 13, 1135. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Nooter, K.; Boersma, A.W.; Kortland, C.J.; Stoter, G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int. J. Cancer 1997, 73, 592–599. [Google Scholar] [CrossRef]
- Desai, B.M.; Villanueva, J.; Nguyen, T.T.; Lioni, M.; Xiao, M.; Kong, J.; Krepler, C.; Vultur, A.; Flaherty, K.T.; Nathanson, K.L.; et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS ONE 2013, 8, e59588. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, M.; Morlando, S.; Solomos, D.; Mehmood, A.; Cox, A.W.I.; Chiesa, M.; D’Alessandra, Y.; Garofalo, M.; Topham, C.H.; Di Leva, G. Pre-therapeutic efficacy of the CDK inhibitor dinaciclib in medulloblastoma cells. Sci. Rep. 2021, 11, 5374. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Sadowska, M.; Walker, A.; Feldman, E.J.; Iyer, S.P.; Baer, M.R.; Sausville, E.A.; Lapidus, R.G.; Zhang, D.; Zhu, Y.; et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother. Pharmacol. 2013, 72, 897–908. [Google Scholar] [CrossRef]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef]
- Guha, M. Cyclin-dependent kinase inhibitors move into Phase III. Nat. Rev. Drug Discov. 2012, 11, 892–894. [Google Scholar] [CrossRef]
- Julve, M.; Clark, J.J.; Lythgoe, M.P. Advances in cyclin-dependent kinase inhibitors for the treatment of melanoma. Expert Opin. Pharmacother. 2021, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | CP | Dinaciclib |
|---|---|---|
| NT2/D1 | 0.7 µM (95% CI: 0.35–1.43) | 9.2 nM (95% CI: 8.3–10.1) |
| NT2/D1-R | 6.1 µM (95% CI: 2.3–16.0) | 3.8 nM (95% CI: 3.3–4.5) |
| NCCIT | 2.7 µM (95% CI: 1.1–6.7) | 3.4 nM (95% CI: 2.9–4.0) |
| NCCIT-R | 4.1 µM (95% CI: 2.1–8.7) | 6.0 nM (95% CI: 5.3–6.8) |
| Cell Line | CP | CP + Dinaciclib IC25 | CP + Dinaciclib IC50 |
|---|---|---|---|
| NT2/D1 | 87.81% ± 0.27% | 89.47% ± 1.22% | 92.53% ± 1.99% |
| NT2/D1-R | 37.90% ± 3.61% | 56.86% ± 0.19% (°) | 51.42% ± 1.63% (°) |
| NCCIT | 93.51% ± 2.09% | 93.26% ± 0.34% | 90.06% ± 3.07% |
| NCCIT-R | 75.70% ± 8.28% | 92.63% ± 6.29% (°) | 90.11% ± 1.48% (°) |
| Cell Line | Vehicle (T3) | Dinaciclib (T3) | Cisplatin (T3) | Combined (T3) |
|---|---|---|---|---|
| NT2/D1 | 39,426 (35,902–43,297) | 28,086 (25,649–30,754) p < 0.01 vs vehicle | 23,597 (21,741–25,612) p < 0.01 vs vehicle | 22,192 (20,238–24,334) p < 0.01 vs vehicle p < 0.01 vs cisplatin |
| NT2/D1-R | 36,301 (32,866–40,096) | 30,668 (27,332–34,411) p < 0.01 vs vehicle | 25,436 (23,305–27,762) p= 0.030 vs vehicle | 19,732 (17,814–21,856) p < 0.01 vs vehicle p < 0.01 vs cisplatin |
| NCCIT | 41,991 (37,004–47,651) | 32,358 (28,149–37,196) p= 0.023 vs vehicle | 34,113 (30,061–38,711) p < 0.01 vs vehicle | 28,004 (24,678–31,778) p < 0.01 vs vehicle p = 0.132 vs cisplatin |
| NCCIT-R | 41,494 (37,533–45,873) | 35,879 (32,789–39,261) p < 0.01 vs vehicle | 25,960 (23,965–28,122) p = 0.035 vs vehicle | 24,588 (22,455–26,924) p < 0.01 vs vehicle p < 0.01 vs cisplatin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossini, E.; Tamburello, M.; Abate, A.; Zini, S.; Ribaudo, G.; Gianoncelli, A.; Calza, S.; Valcamonico, F.; Suardi, N.R.; Mirabella, G.; et al. The CDK Inhibitor Dinaciclib Improves Cisplatin Response in Nonseminomatous Testicular Cancer: A Preclinical Study. Cells 2024, 13, 368. https://doi.org/10.3390/cells13050368
Rossini E, Tamburello M, Abate A, Zini S, Ribaudo G, Gianoncelli A, Calza S, Valcamonico F, Suardi NR, Mirabella G, et al. The CDK Inhibitor Dinaciclib Improves Cisplatin Response in Nonseminomatous Testicular Cancer: A Preclinical Study. Cells. 2024; 13(5):368. https://doi.org/10.3390/cells13050368
Chicago/Turabian StyleRossini, Elisa, Mariangela Tamburello, Andrea Abate, Silvia Zini, Giovanni Ribaudo, Alessandra Gianoncelli, Stefano Calza, Francesca Valcamonico, Nazareno R. Suardi, Giuseppe Mirabella, and et al. 2024. "The CDK Inhibitor Dinaciclib Improves Cisplatin Response in Nonseminomatous Testicular Cancer: A Preclinical Study" Cells 13, no. 5: 368. https://doi.org/10.3390/cells13050368
APA StyleRossini, E., Tamburello, M., Abate, A., Zini, S., Ribaudo, G., Gianoncelli, A., Calza, S., Valcamonico, F., Suardi, N. R., Mirabella, G., Berruti, A., & Sigala, S. (2024). The CDK Inhibitor Dinaciclib Improves Cisplatin Response in Nonseminomatous Testicular Cancer: A Preclinical Study. Cells, 13(5), 368. https://doi.org/10.3390/cells13050368