
Optogenetic Control of the Mitochondrial Protein Import in Mammalian Cells


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
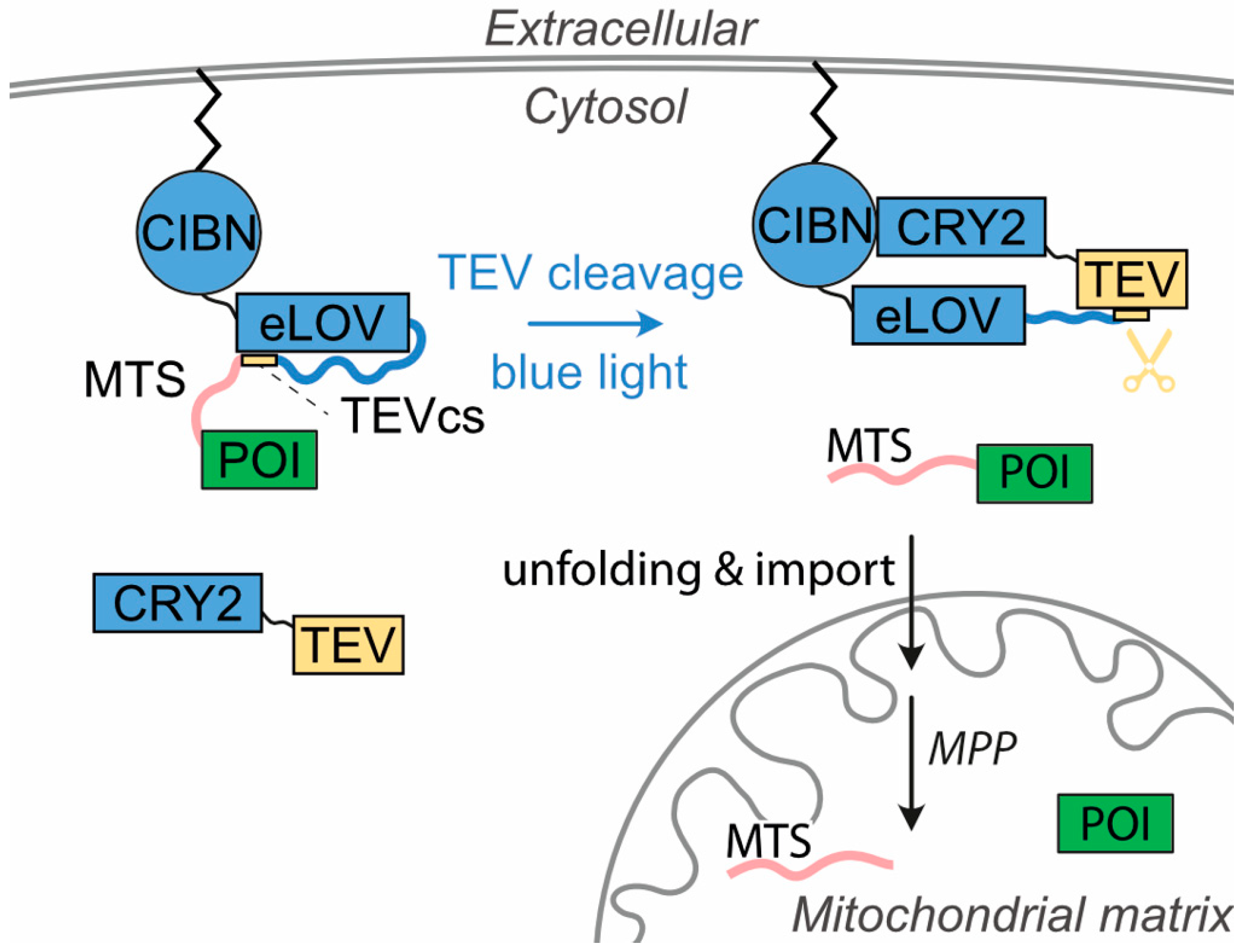
1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Cell Culture and Transfection
2.3. Illumination
2.4. Cell Lysis
2.5. Mitochondrial Fractionation and Proteinase K Digestion
2.6. Depletion of the Membrane Potential
2.7. SDS PAGE and Western Blotting
2.8. Fluorescence Microscopy
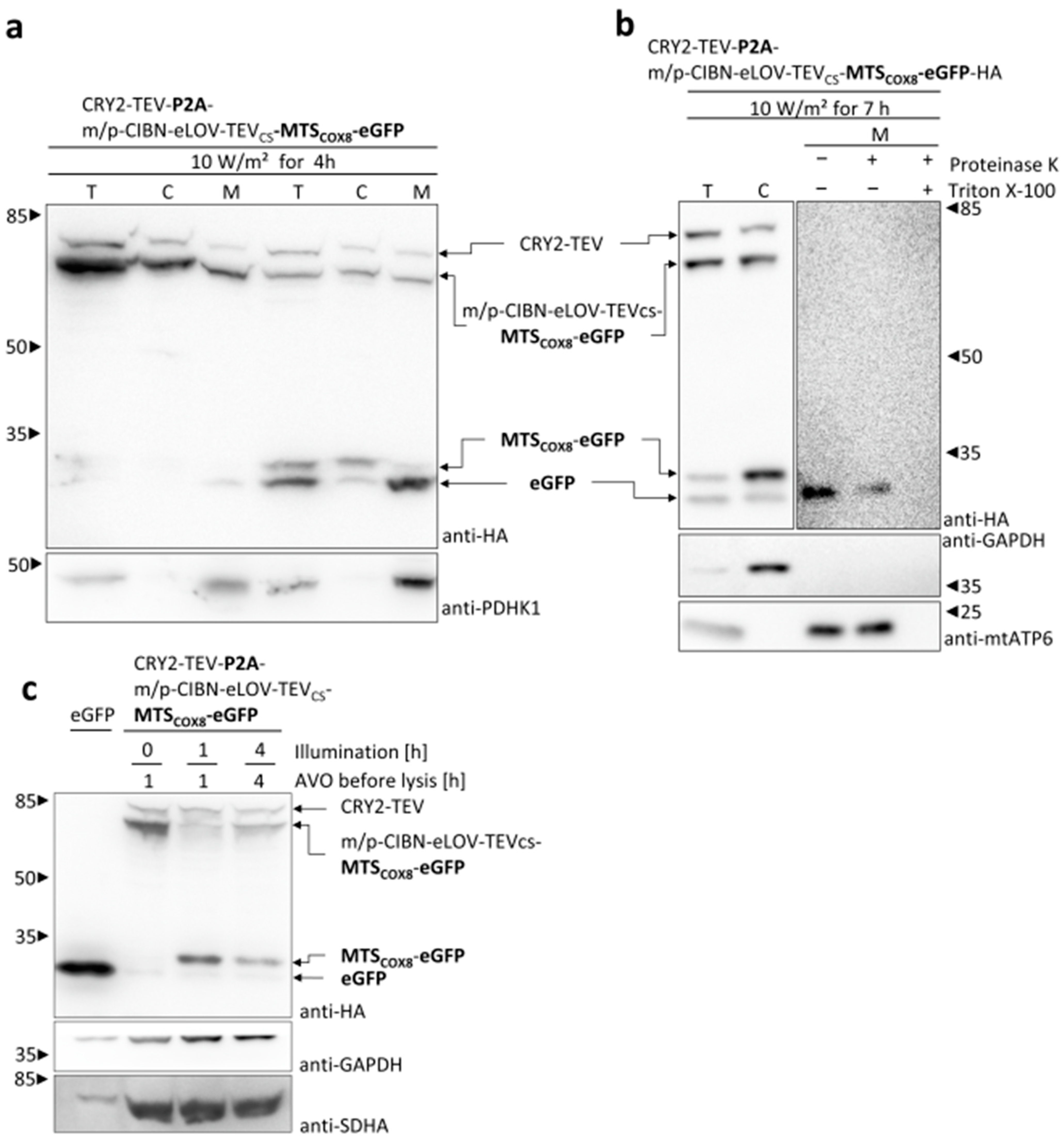
3. Results
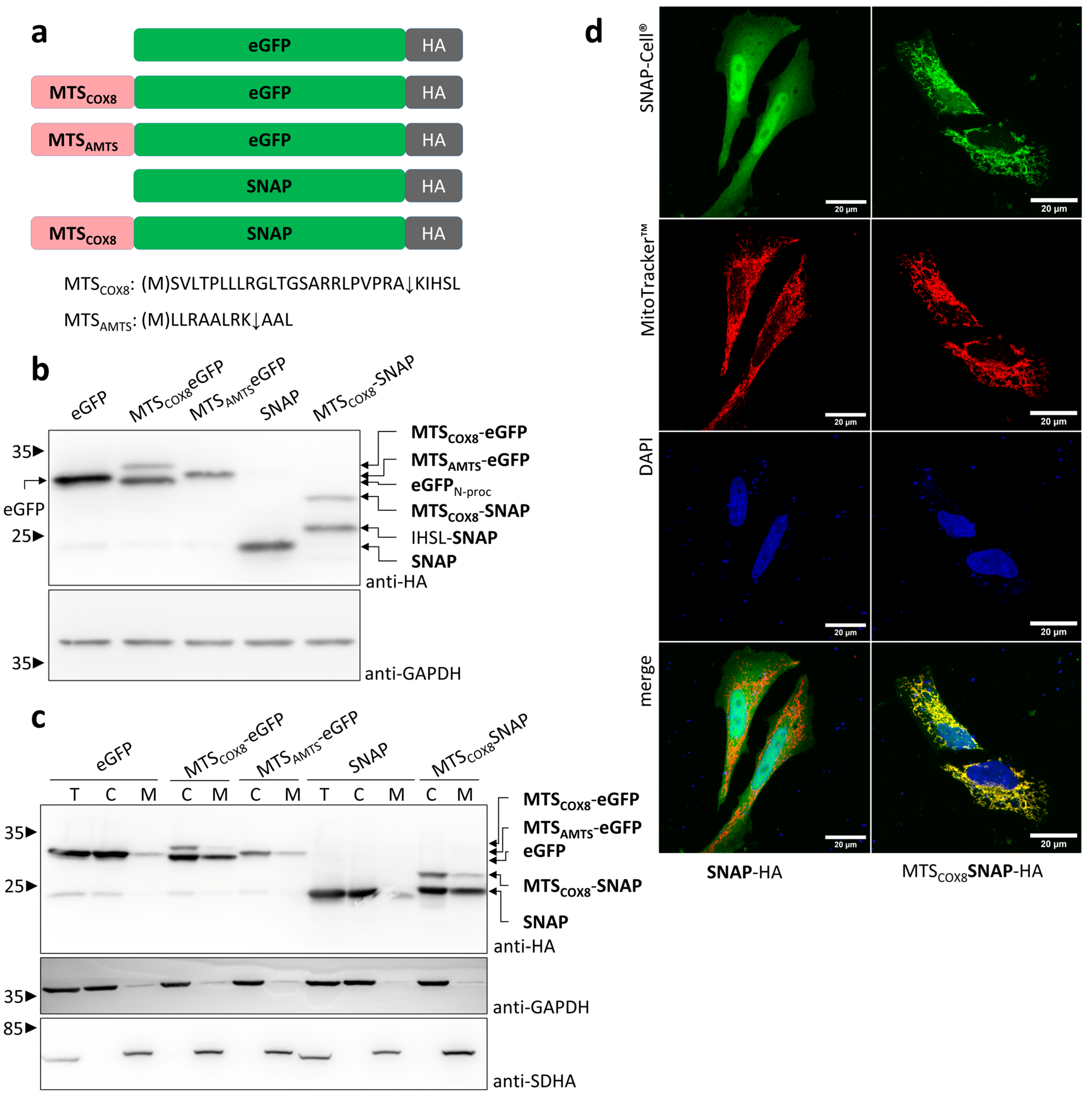
3.1. Characterization of Mitochondrial Signal Sequences
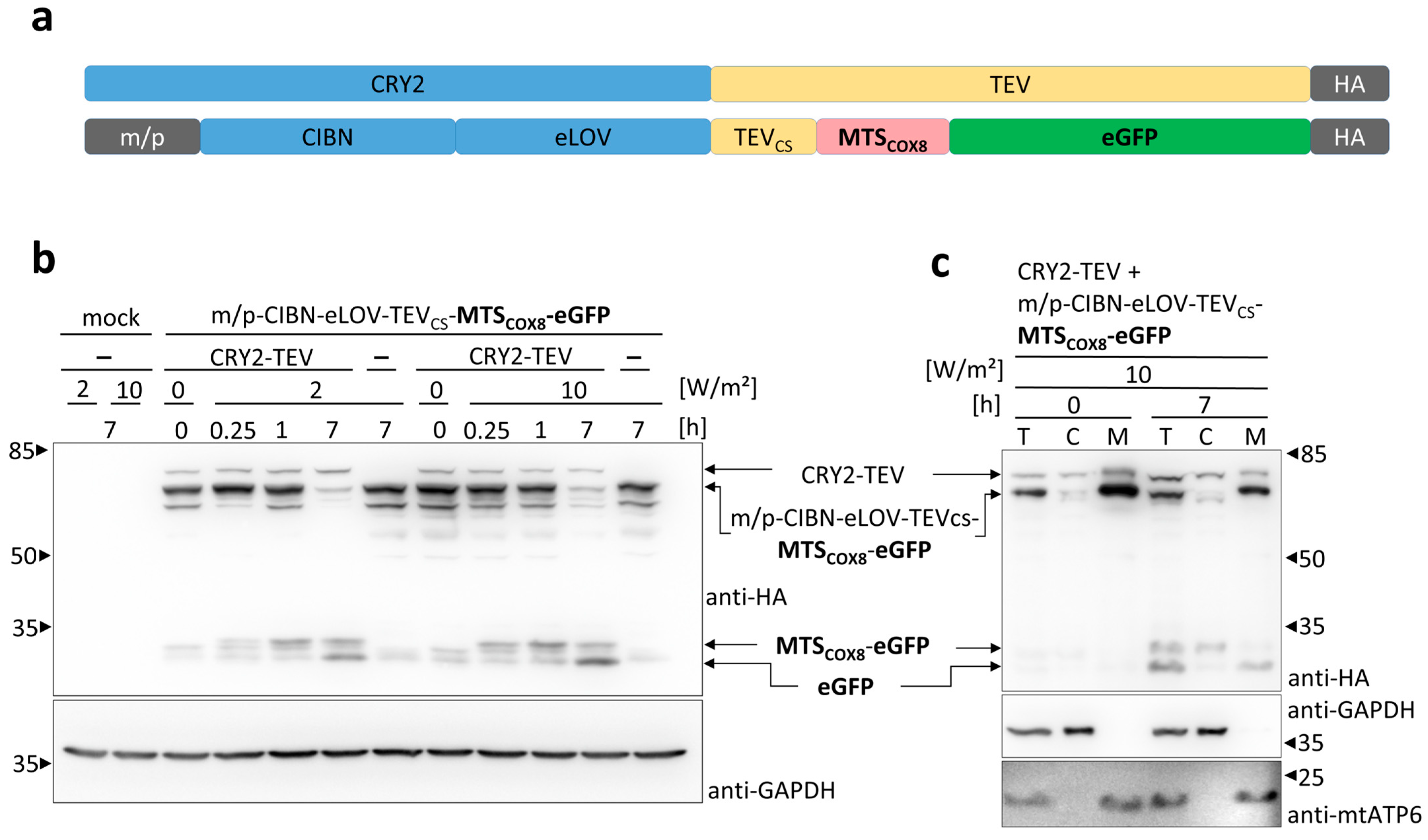
3.2. Light-Induced Mitochondrial Import Based on the Two Plasmids System
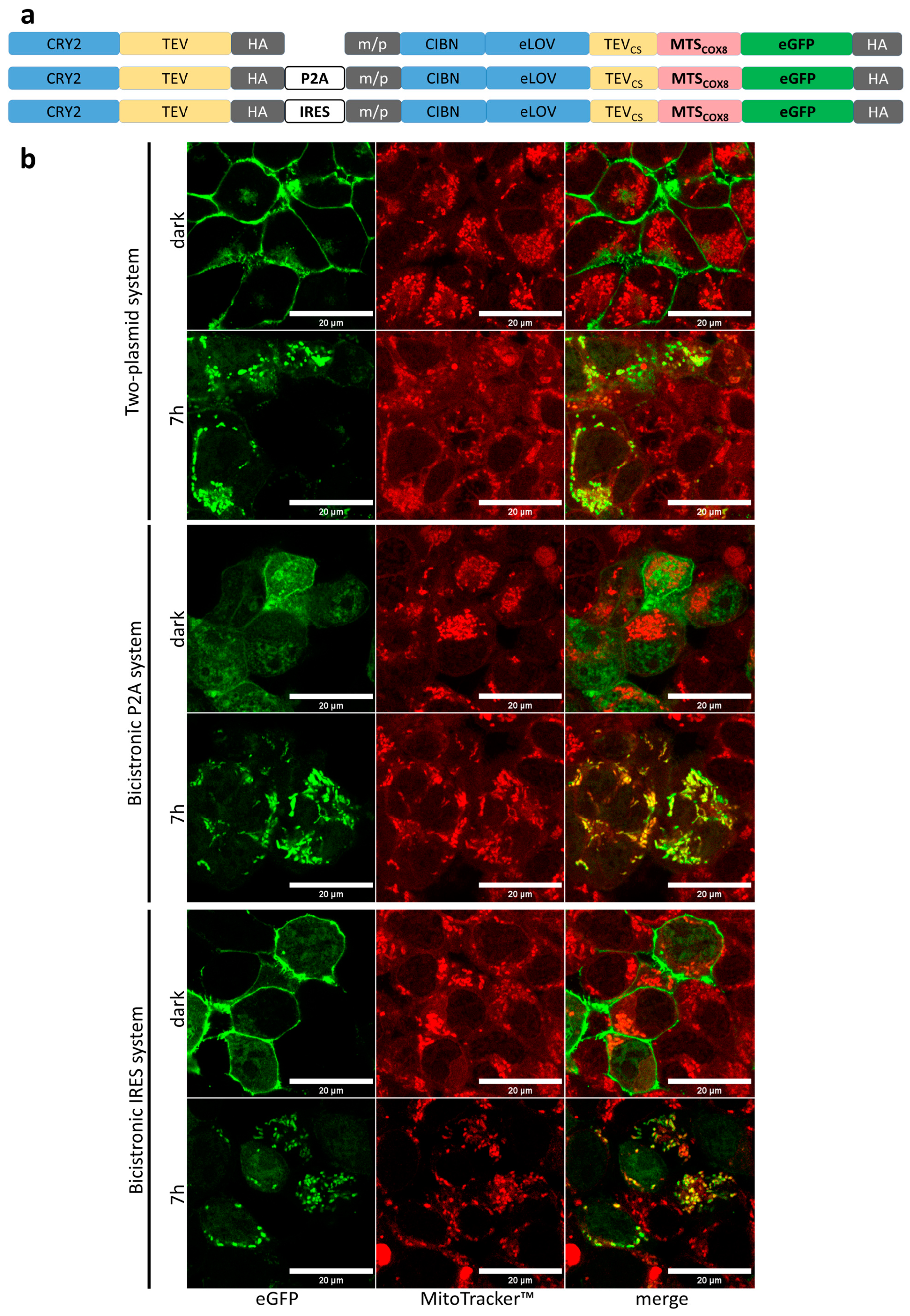
3.3. Light-Induced Mitochondrial Import Based on Bicistronic Constructs
3.4. Light-Induced Mitochondrial Import of MTS Fusion Proteins Depends on the Membrane Potential across the Inner Mitochondrial Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, M.; Peikert, C.D.; Lübbert, P.; Suppanz, I.; Klemm, C.; Alka, O.; Steiert, C.; Naumenko, N.; Schendzielorz, A.; Melchionda, L.; et al. Quantitative High-Confidence Human Mitochondrial Proteome and Its Dynamics in Cellular Context. Cell Metab. 2021, 33, 2464–2483.e18. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An Updated Mitochondrial Proteome Now with Sub-Organelle Localization and Pathway Annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.N.; Forny, P.; Pagliarini, D.J. Mitochondrial Proteome Research: The Road Ahead. Nat. Rev. Mol. Cell Biol. 2024, 25, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.-W. The Mitochondrial Genome: Structure, Transcription, Translation and Replication. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Busch, J.D.; Fielden, L.F.; Pfanner, N.; Wiedemann, N. Mitochondrial Protein Transport: Versatility of Translocases and Mechanisms. Mol. Cell 2023, 83, 890–910. [Google Scholar] [CrossRef]
- Haastrup, M.O.; Vikramdeo, K.S.; Singh, S.; Singh, A.P.; Dasgupta, S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. Int. J. Mol. Sci. 2023, 24, 2479. [Google Scholar] [CrossRef]
- den Brave, F.; Pfanner, N.; Becker, T. Mitochondrial Entry Gate as Regulatory Hub. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2024, 1871, 119529. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.P.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global Analysis of the Mitochondrial N-Proteome Identifies a Processing Peptidase Critical for Protein Stability. Cell 2009, 139, 428–439. [Google Scholar] [CrossRef]
- Calvo, S.E.; Julien, O.; Clauser, K.R.; Shen, H.; Kamer, K.J.; Wells, J.A.; Mootha, V.K. Comparative Analysis of Mitochondrial N-Termini from Mouse, Human, and Yeast. Mol. Cell. Proteom. 2017, 16, 512–523. [Google Scholar] [CrossRef]
- Moulin, C.; Caumont-Sarcos, A.; Ieva, R. Mitochondrial Presequence Import: Multiple Regulatory Knobs Fine-Tune Mitochondrial Biogenesis and Homeostasis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.-H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular Architecture of the Active Mitochondrial Protein Gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.I.; Chen, Y.; Lynch, D.L.; Gumbart, J.C.; Park, E. Structural Basis of Mitochondrial Protein Import by the TIM23 Complex. Nature 2023, 621, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Hutu, D.P.; Guiard, B.; Chacinska, A.; Becker, D.; Pfanner, N.; Rehling, P.; van der Laan, M. Mitochondrial Protein Import Motor: Differential Role of Tim44 in the Recruitment of Pam17 and J-Complex to the Presequence Translocase. MBoC 2008, 19, 2642–2649. [Google Scholar] [CrossRef]
- Backes, S.; Herrmann, J.M. Protein Translocation into the Intermembrane Space and Matrix of Mitochondria: Mechanisms and Driving Forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef]
- Kücükköse, C.; Taskin, A.A.; Marada, A.; Brummer, T.; Dennerlein, S.; Vögtle, F.-N. Functional Coupling of Presequence Processing and Degradation in Human Mitochondria. FEBS J. 2021, 288, 600–613. [Google Scholar] [CrossRef]
- Gomez-Fabra Gala, M.; Vögtle, F.-N. Mitochondrial Proteases in Human Diseases. FEBS Lett. 2021, 595, 1205–1222. [Google Scholar] [CrossRef]
- Yogev, O.; Naamati, A.; Pines, O. Fumarase: A Paradigm of Dual Targeting and Dual Localized Functions. FEBS J. 2011, 278, 4230–4242. [Google Scholar] [CrossRef]
- Pines, O.; Horwitz, M.; Herrmann, J.M. Privileged Proteins with a Second Residence: Dual Targeting and Conditional Re-Routing of Mitochondrial Proteins. FEBS J. 2024. [Google Scholar] [CrossRef]
- den Brave, F.; Schulte, U.; Fakler, B.; Pfanner, N.; Becker, T. Mitochondrial Complexome and Import Network. Trends Cell Biol. 2024, 34, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Kolar, K.; Knobloch, C.; Stork, H.; Žnidarič, M.; Weber, W. OptoBase: A Web Platform for Molecular Optogenetics. ACS Synth. Biol. 2018, 7, 1825–1828. [Google Scholar] [CrossRef] [PubMed]
- Goglia, A.G.; Toettcher, J.E. A Bright Future: Optogenetics to Dissect the Spatiotemporal Control of Cell Behavior. Curr. Opin. Chem. Biol. 2019, 48, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.M.; Lataster, L.; Weber, W.; Radziwill, G. Optogenetic Approaches for the Spatiotemporal Control of Signal Transduction Pathways. Int. J. Mol. Sci. 2021, 22, 5300. [Google Scholar] [CrossRef]
- Shin, Y.; Berry, J.; Pannucci, N.; Haataja, M.P.; Toettcher, J.E.; Brangwynne, C.P. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 2017, 168, 159–171.e14. [Google Scholar] [CrossRef]
- Mühlhäuser, W.W.D.; Weber, W.; Radziwill, G. OpEn-Tag—A Customizable Optogenetic Toolbox To Dissect Subcellular Signaling. ACS Synth. Biol. 2019, 8, 1679–1684. [Google Scholar] [CrossRef]
- Kramer, M.M.; Mühlhäuser, W.W.D.; Weber, W.; Radziwill, G. Multichromatic Control of Signaling Pathways in Mammalian Cells. Adv. Biol. 2021, 5, 2000196. [Google Scholar] [CrossRef]
- Lataster, L.; Huber, H.M.; Böttcher, C.; Föller, S.; Takors, R.; Radziwill, G. Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein P21. Biology 2023, 12, 1194. [Google Scholar] [CrossRef]
- Zhou, Y.; Kong, D.; Wang, X.; Yu, G.; Wu, X.; Guan, N.; Weber, W.; Ye, H. A Small and Highly Sensitive Red/Far-Red Optogenetic Switch for Applications in Mammals. Nat. Biotechnol. 2022, 40, 262–272. [Google Scholar] [CrossRef]
- Armbruster, A.; Mohamed, A.M.; Phan, H.T.; Weber, W. Lighting the Way: Recent Developments and Applications in Molecular Optogenetics. Curr. Opin. Biotechnol. 2024, 87, 103126. [Google Scholar] [CrossRef]
- Sanchez, M.I.; Ting, A.Y. Directed Evolution Improves the Catalytic Efficiency of TEV Protease. Nat. Methods 2020, 17, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid Blue-Light–Mediated Induction of Protein Interactions in Living Cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Bugaj, L.J.; Lim, W.A. High-Throughput Multicolor Optogenetics in Microwell Plates. Nat. Protoc. 2019, 14, 2205–2228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, O.S.; Hörner, M.; Weber, W. A Graphical User Interface to Design High-Throughput Optogenetic Experiments with the optoPlate-96. Nat. Protoc. 2020, 15, 2785–2787. [Google Scholar] [CrossRef] [PubMed]
- De Michele, R.; Carimi, F.; Frommer, W.B. Mitochondrial Biosensors. Int. J. Biochem. Cell Biol. 2014, 48, 39–44. [Google Scholar] [CrossRef]
- Kang, Y.C.; Son, M.; Kang, S.; Im, S.; Piao, Y.; Lim, K.S.; Song, M.-Y.; Park, K.-S.; Kim, Y.-H.; Pak, Y.K. Cell-Penetrating Artificial Mitochondria-Targeting Peptide-Conjugated Metallothionein 1A Alleviates Mitochondrial Damage in Parkinson’s Disease Models. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed Evolution of O6-Alkylguanine-DNA Alkyltransferase for Efficient Labeling of Fusion Proteins with Small Molecules In Vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, R.; Zhao, P.; Xia, Q. 2A Self-Cleaving Peptide-Based Multi-Gene Expression System in the Silkworm Bombyx Mori. Sci. Rep. 2015, 5, 16273. [Google Scholar] [CrossRef]
- Jang, S.K.; Kräusslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A Segment of the 5′ Nontranslated Region of Encephalomyocarditis Virus RNA Directs Internal Entry of Ribosomes during In Vitro Translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Internal Initiation of Translation of Eukaryotic mRNA Directed by a Sequence Derived from Poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Palmenberg, A.C. Translational Efficiency of EMCV IRES in Bicistronic Vectors Is Dependent upon IRES Sequence and Gene Location. BioTechniques 2006, 41, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Martin, F.; Guiard, B.; Pfanner, N.; Voos, W. The Mitochondrial Hsp70-dependent Import System Actively Unfolds Preproteins and Shortens the Lag Phase of Translocation. EMBO J. 2001, 20, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Jamdar, S.N.; Goyal, V.D.; Kumar, A.; Ghosh, B.; Makde, R.D. Structure of the Human Aminopeptidase XPNPEP3 and Comparison of Its in Vitro Activity with Icp55 Orthologs: Insights into Diverse Cellular Processes. J. Biol. Chem. 2017, 292, 10035–10047. [Google Scholar] [CrossRef] [PubMed]
- Bogorodskiy, A.; Okhrimenko, I.; Maslov, I.; Maliar, N.; Burkatovskii, D.; von Ameln, F.; Schulga, A.; Jakobs, P.; Altschmied, J.; Haendeler, J.; et al. Accessing Mitochondrial Protein Import in Living Cells by Protein Microinjection. Front. Cell Dev. Biol. 2021, 9, 698658. [Google Scholar] [CrossRef]
- Ford, H.C.; Allen, W.J.; Pereira, G.C.; Liu, X.; Dillingham, M.S.; Collinson, I. Towards a Molecular Mechanism Underlying Mitochondrial Protein Import through the TOM and TIM23 Complexes. eLife 2022, 11, e75426. [Google Scholar] [CrossRef]
- Schäfer, J.A.; Bozkurt, S.; Michaelis, J.B.; Klann, K.; Münch, C. Global Mitochondrial Protein Import Proteomics Reveal Distinct Regulation by Translation and Translocation Machinery. Mol. Cell 2022, 82, 435–446.e7. [Google Scholar] [CrossRef]
- Berry, B.J.; Wojtovich, A.P. Mitochondrial Light Switches: Optogenetic Approaches to Control Metabolism. FEBS J. 2020, 287, 4544–4556. [Google Scholar] [CrossRef]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef]
- Benej, M.; Papandreou, I.; Denko, N.C. Hypoxic Adaptation of Mitochondria and Its Impact on Tumor Cell Function. Semin. Cancer Biol. 2024, 100, 28–38. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Althoff, L.F.J.; Kramer, M.M.; Bührer, B.; Gaspar, D.; Radziwill, G. Optogenetic Control of the Mitochondrial Protein Import in Mammalian Cells. Cells 2024, 13, 1671. https://doi.org/10.3390/cells13191671
Althoff LFJ, Kramer MM, Bührer B, Gaspar D, Radziwill G. Optogenetic Control of the Mitochondrial Protein Import in Mammalian Cells. Cells. 2024; 13(19):1671. https://doi.org/10.3390/cells13191671
Chicago/Turabian StyleAlthoff, Lukas F. J., Markus M. Kramer, Benjamin Bührer, Denise Gaspar, and Gerald Radziwill. 2024. "Optogenetic Control of the Mitochondrial Protein Import in Mammalian Cells" Cells 13, no. 19: 1671. https://doi.org/10.3390/cells13191671
APA StyleAlthoff, L. F. J., Kramer, M. M., Bührer, B., Gaspar, D., & Radziwill, G. (2024). Optogenetic Control of the Mitochondrial Protein Import in Mammalian Cells. Cells, 13(19), 1671. https://doi.org/10.3390/cells13191671