
Hypoxia Promotes Invadosome Formation by Lung Fibroblasts

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Experimental Mouse Model
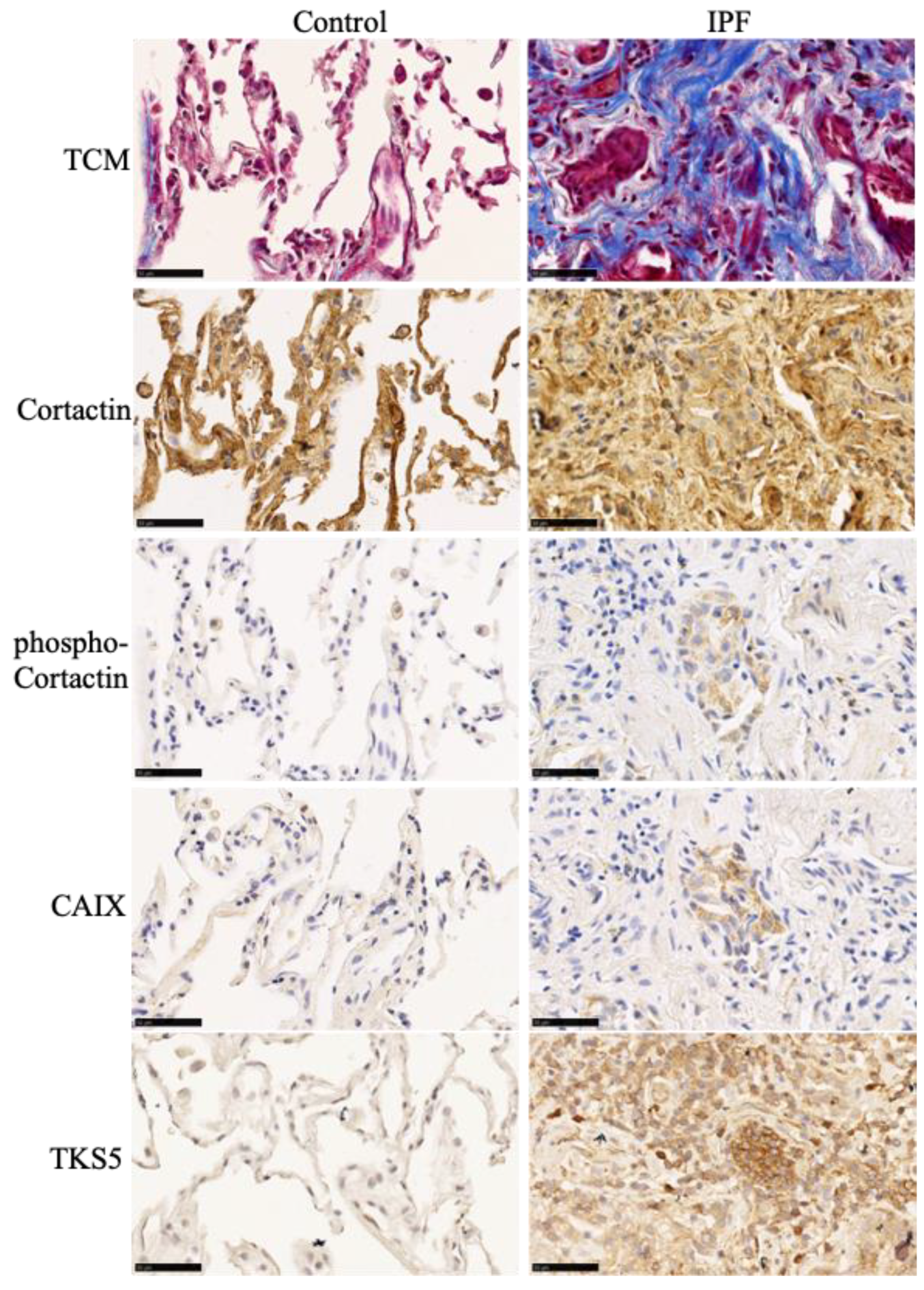
2.3. Immunohistochemistry
2.4. Isolation and Culture of Lung Fibroblasts
2.5. Invadosome Formation Assessment
2.6. mRNA Expression Analysis
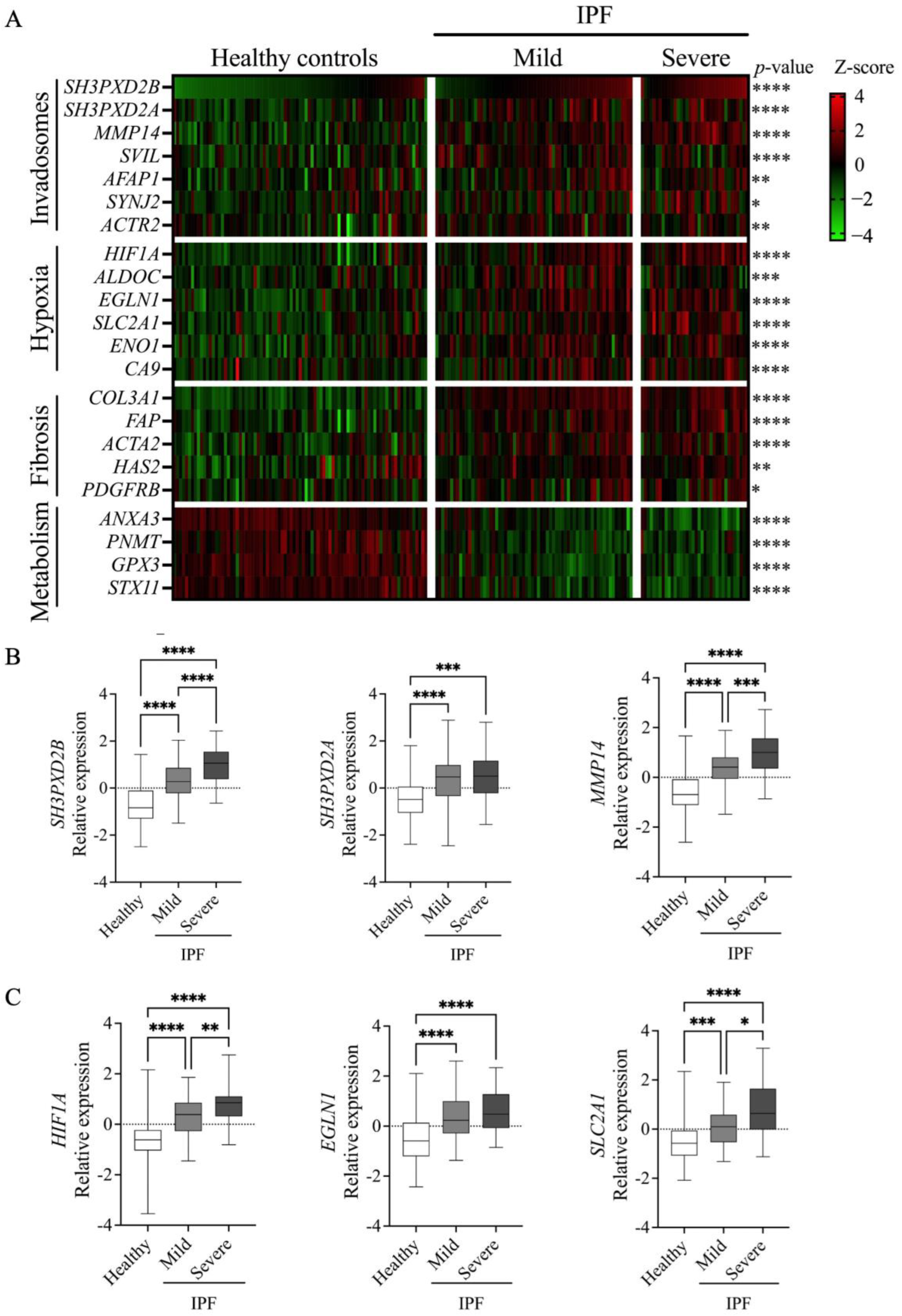
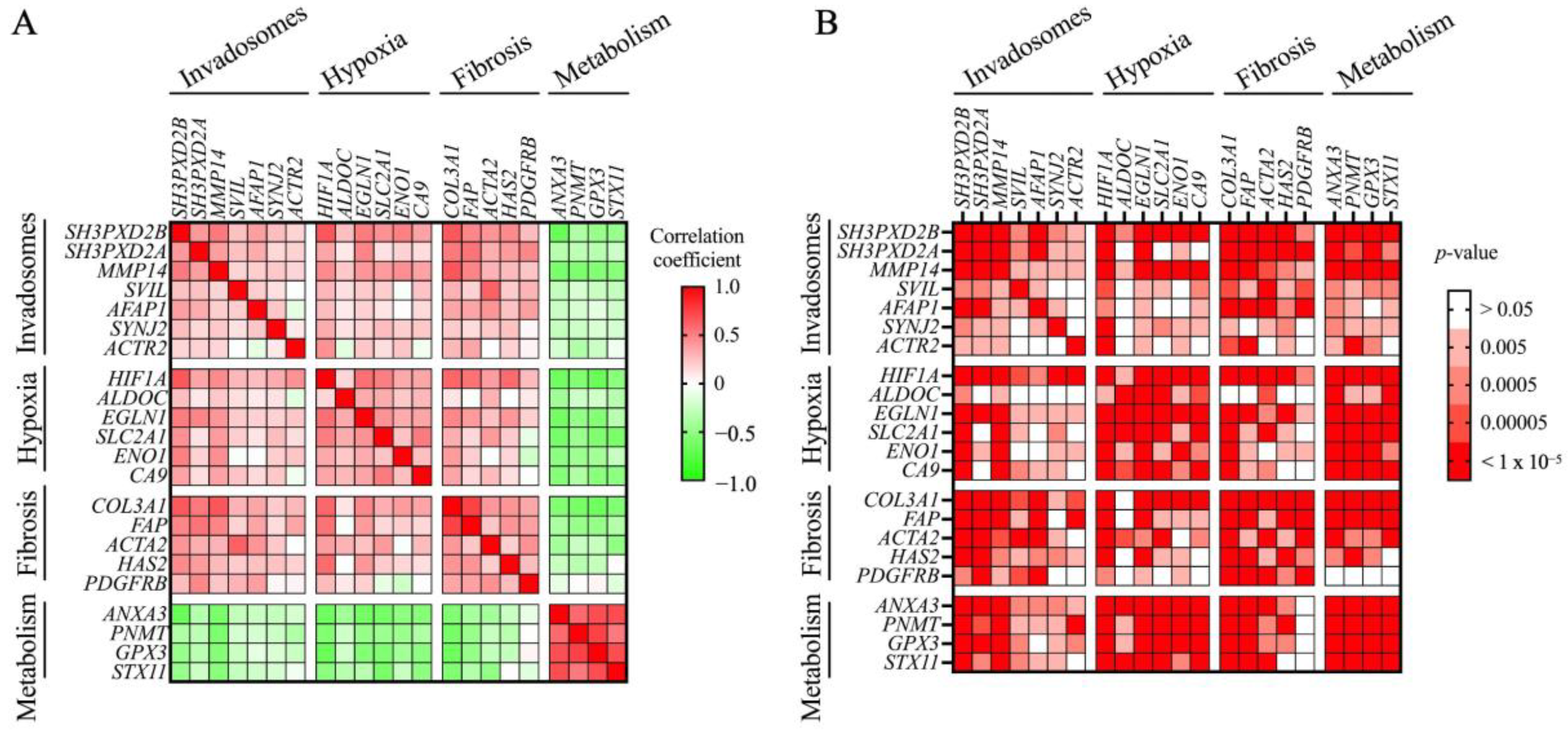
2.7. RNAseq Data Analysis of Lung Tissue
2.8. Immunoblotting and Immunoprecipitation
2.9. Autotaxin Measurement by ELISA
2.10. Statistical Analyses
3. Results
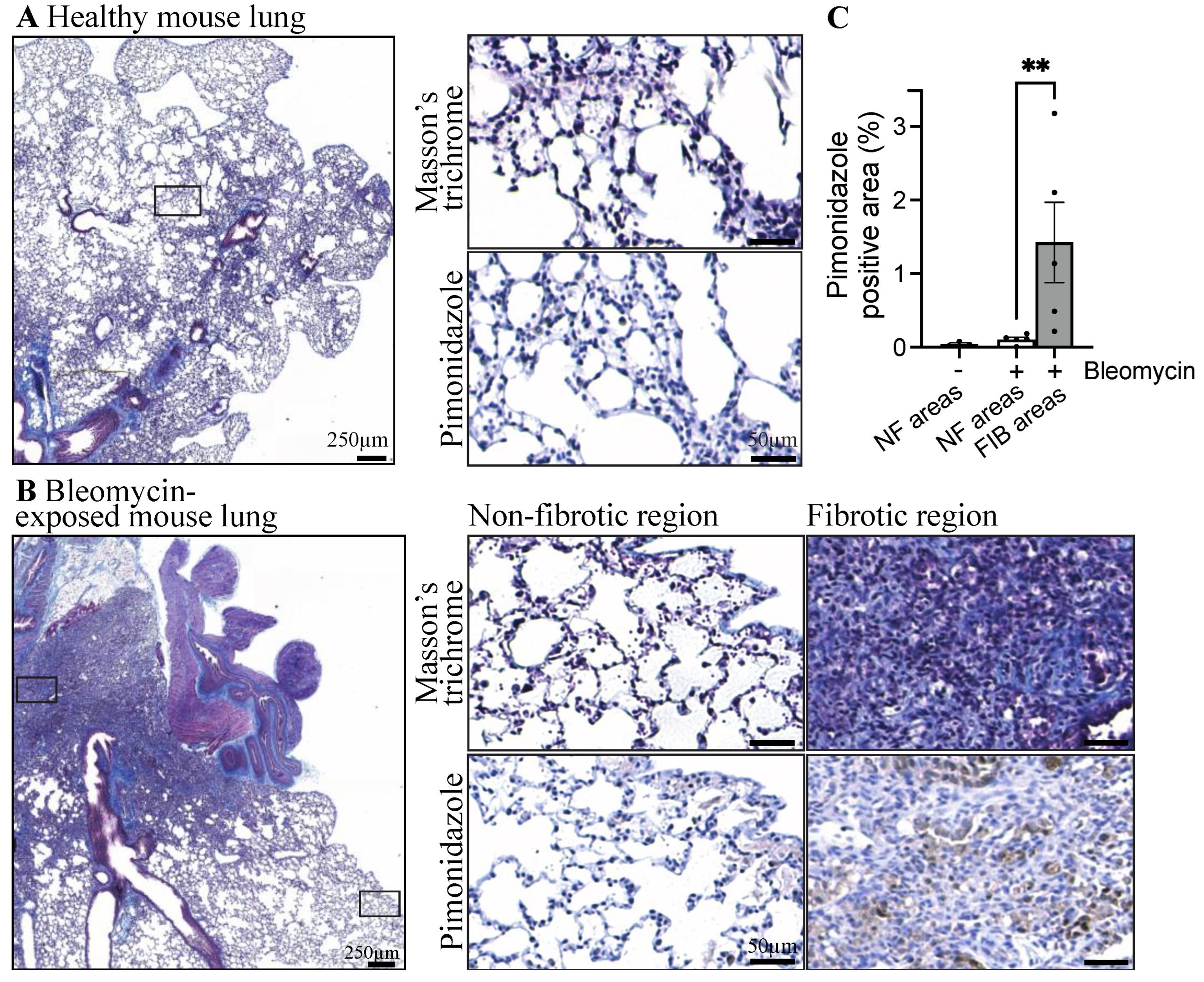
3.1. Fibrotic Lungs of Mice Exposed to Bleomycin Exhibit Hypoxic Areas
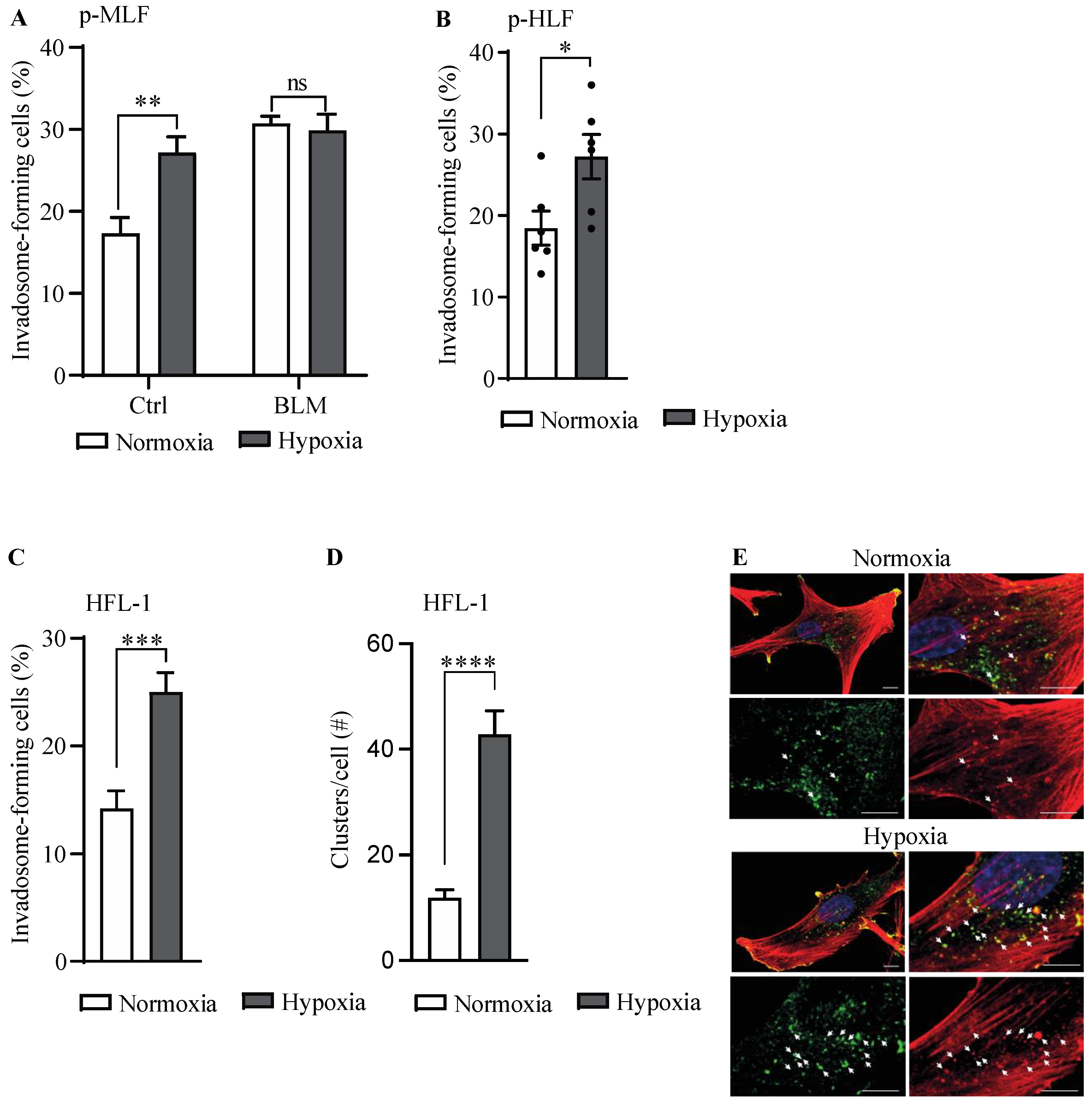
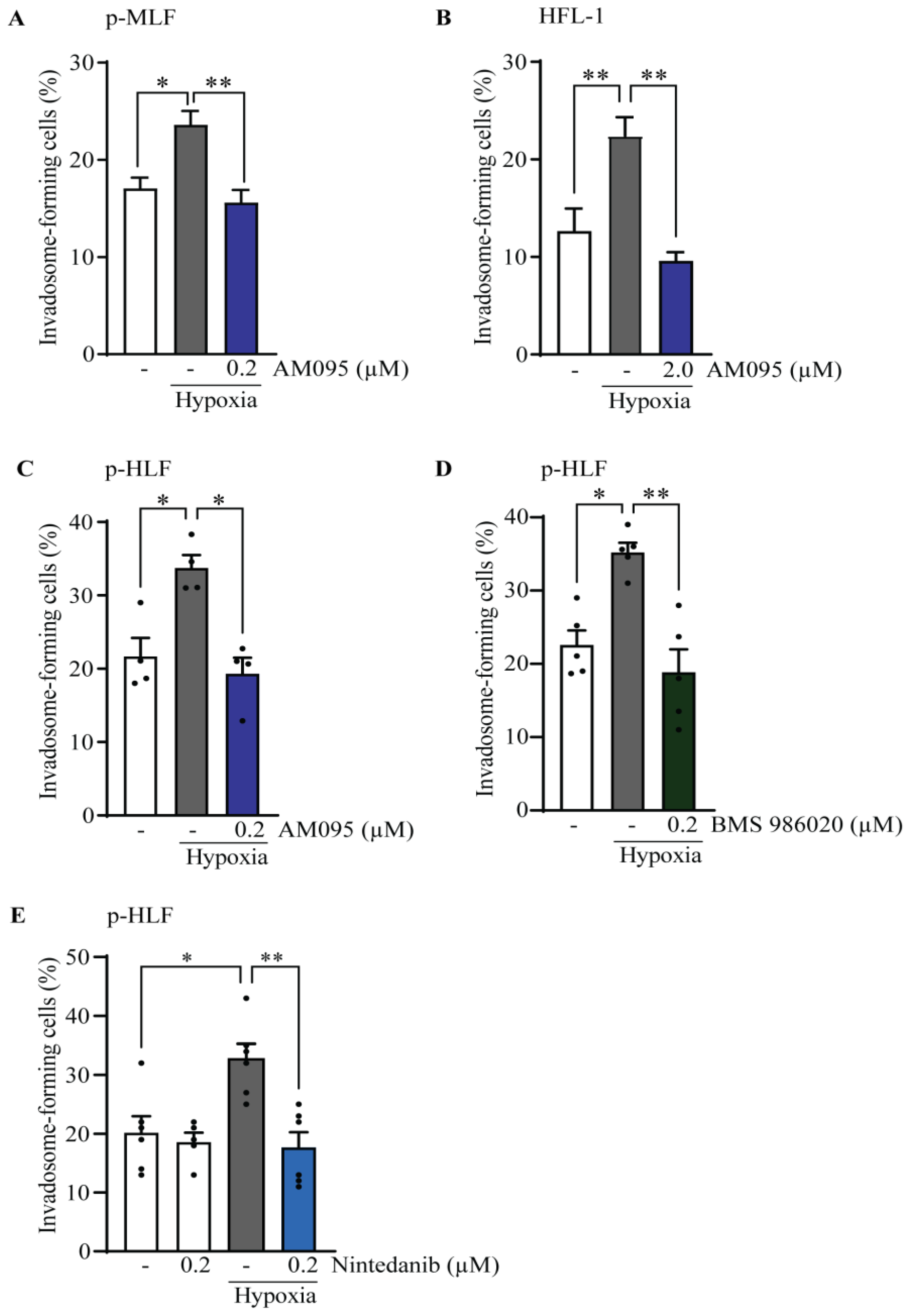
3.2. Hypoxia Stimulates Invadosome Formation in Lung Fibroblasts
3.3. The Effect of Lysophosphatidic Acid Receptor 1 (LPA1) Activation on Invadosome Formation in Lung Fibroblasts
3.4. Hypoxia Implicates LPA1 and Receptor Tyrosine Kinases (RTK) for Invadosome Formation in Lung Fibroblasts
3.5. LPA1 Is Involved in Hypoxic Stimulation of PDGFRβ Phosphorylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic Pulmonary Fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. The Pathway for Oxygen: Structure and Function in the Mammalian Respiratory System, 1st ed.; Harvard University Press: Cambridge, MA, USA, 1984. [Google Scholar]
- Ito, Y.; Ahmad, A.; Kewley, E.; Mason, R.J. Hypoxia-Inducible Factor Regulates Expression of Surfactant Protein in Alveolar Type II Cells in Vitro. Am. J. Respir. Cell Mol. Biol. 2011, 45, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of Reduced Mucus Oxygen Concentration in Airway Pseudomonas Infections of Cystic Fibrosis Patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kouthouridis, S.; Goepp, J.; Martini, C.; Matthes, E.; Hanrahan, J.W.; Moraes, C. Oxygenation as a Driving Factor in Epithelial Differentiation at the Air-Liquid Interface. Integr. Biol. 2021, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Grubb, B.R.; Rogers, T.D.; Dang, H.; Asakura, T.; Kota, P.; Gilmore, R.C.; Okuda, K.; Morton, L.C.; Sun, L.; et al. Chronic Airway Epithelial Hypoxia Exacerbates Injury in Muco-Obstructive Lung Disease through Mucus Hyperconcentration. Sci. Transl. Med. 2023, 15, eabo7728. [Google Scholar] [CrossRef] [PubMed]
- Petersson, J.; Glenny, R.W. Gas Exchange and Ventilation-Perfusion Relationships in the Lung. Eur. Respir. J. 2014, 44, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. What Makes a Good Lung? Swiss Med. Wkly. 2009, 139, 375–386. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative Expression Profiling in Pulmonary Fibrosis Suggests a Role of Hypoxia-Inducible Factor-1alpha in Disease Pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic Acid Is Elevated in Idiopathic Pulmonary Fibrosis and Induces Myofibroblast Differentiation via pH-Dependent Activation of Transforming Growth Factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef]
- Tanguy, J.; Goirand, F.; Bouchard, A.; Frenay, J.; Moreau, M.; Mothes, C.; Oudot, A.; Helbling, A.; Guillemin, M.; Bonniaud, P.; et al. [18F]FMISO PET/CT Imaging of Hypoxia as a Non-Invasive Biomarker of Disease Progression and Therapy Efficacy in a Preclinical Model of Pulmonary Fibrosis: Comparison with the [18F]FDG PET/CT Approach. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3058–3074. [Google Scholar] [CrossRef]
- Goodwin, J.; Choi, H.; Hsieh, M.-H.; Neugent, M.L.; Ahn, J.-M.; Hayenga, H.N.; Singh, P.K.; Shackelford, D.B.; Lee, I.-K.; Shulaev, V.; et al. Targeting Hypoxia-Inducible Factor-1α/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, T.A.; Knudsen, L.; Leitz, D.H.W.; Duerr, J.; Beers, M.F.; Mall, M.A.; Ochs, M. Linking Fibrotic Remodeling and Ultrastructural Alterations of Alveolar Epithelial Cells after Deletion of Nedd4-2. Int. J. Mol. Sci. 2021, 22, 7607. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Bodempudi, V.; Hergert, P.; Smith, K.; Xia, H.; Herrera, J.; Peterson, M.; Khalil, W.; Kahm, J.; Bitterman, P.B.; Henke, C.A. miR-210 Promotes IPF Fibroblast Proliferation in Response to Hypoxia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L283–L294. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Rice, C.; Prasad, J.; Bone, H.; Farkas, L.; Rojas, M.; Horowitz, J.C. Persistent Hypoxia Promotes Myofibroblast Differentiation via GPR-81 and Differential Regulation of LDH Isoenzymes in Normal and Idiopathic Pulmonary Fibrosis Fibroblasts. Physiol. Rep. 2023, 11, e15759. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.; Brochu-Gaudreau, K.; Saucier, C.; Dubois, C.M. Hypoxia Downregulates LPP3 and Promotes the Spatial Segregation of ATX and LPP1 During Cancer Cell Invasion. Cancers 2019, 11, 1403. [Google Scholar] [CrossRef] [PubMed]
- Takai, M.; Takauchi, M.; Kuribayashi, M.; Tsujiuchi, T. LPA Receptor-Mediated Signaling Regulates Cell Motility and Survival to Anticancer Drug of Pancreatic Cancer Cells under Glucose-Deprived and Hypoxic Conditions. Biochem. Biophys. Res. Commun. 2023, 661, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Tager, A.M. Role of the Lysophospholipid Mediators Lysophosphatidic Acid and Sphingosine 1-Phosphate in Lung Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The Lysophosphatidic Acid Receptor LPA1 Promotes Epithelial Cell Apoptosis after Lung Injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef]
- Sinclair, K.A.; Yerkovich, S.T.; Hopkins, P.M.-A.; Fieuw, A.M.; Ford, P.; Powell, J.E.; O’Sullivan, B.; Chambers, D.C. The Autotaxin-Lysophosphatidic Acid Pathway Mediates Mesenchymal Cell Recruitment and Fibrotic Contraction in Lung Transplant Fibrosis. J. Heart Lung Transplant. 2021, 40, 12–23. [Google Scholar] [CrossRef]
- Harper, K.; Lavoie, R.R.; Charbonneau, M.; Brochu-Gaudreau, K.; Dubois, C.M. The Hypoxic Tumor Microenvironment Promotes Invadopodia Formation and Metastasis through LPA1 Receptor and EGFR Cooperation. Mol. Cancer Res. 2018, 16, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Cliche, D.O.; Charbonneau, M.; Adam, D.; Brochiero, E.; Dubois, C.M.; Cantin, A.M. Invadosome Formation by Lung Fibroblasts in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 24, 499. [Google Scholar] [CrossRef] [PubMed]
- Barbayianni, I.; Kanellopoulou, P.; Fanidis, D.; Nastos, D.; Ntouskou, E.-D.; Galaris, A.; Harokopos, V.; Hatzis, P.; Tsitoura, E.; Homer, R.; et al. SRC and TKS5 Mediated Podosome Formation in Fibroblasts Promotes Extracellular Matrix Invasion and Pulmonary Fibrosis. Nat. Commun. 2023, 14, 5882. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.-A.; Morrison, A.; Schwock, J.; Aviel-Ronen, S.; Iakovlev, V.; Tsao, M.-S.; Ho, J.; Hedley, D.W. Quantitative Image Analysis of Immunohistochemical Stains Using a CMYK Color Model. Diagn. Pathol. 2007, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Ye, I.C.; Fertig, E.J.; DiGiacomo, J.W.; Considine, M.; Godet, I.; Gilkes, D.M. Molecular Portrait of Hypoxia in Breast Cancer: A Prognostic Signature and Novel HIF-Regulated Genes. Mol. Cancer Res. 2018, 16, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Courtneidge, S.A. Tks Adaptor Proteins at a Glance. J. Cell Sci. 2018, 131, jcs203661. [Google Scholar] [CrossRef] [PubMed]
- Ninio, L.; Nissani, A.; Meirson, T.; Domovitz, T.; Genna, A.; Twafra, S.; Srikanth, K.D.; Dabour, R.; Avraham, E.; Davidovich, A.; et al. Hepatitis C Virus Enhances the Invasiveness of Hepatocellular Carcinoma via EGFR-Mediated Invadopodia Formation and Activation. Cells 2019, 8, 1395. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The “ins” and “Outs” of Podosomes and Invadopodia: Characteristics, Formation and Function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef]
- Leiro, J.A.; Heinonen, M.H.; Kokko, K. XPS Spectra and the Electronic Structure of CaCu and CaAg Alloys. Phys. Rev. B Condens. Matter 1994, 49, 14153–14159. [Google Scholar] [CrossRef]
- Cui, Y.; Ji, J.; Hou, J.; Tan, Y.; Han, X. Identification of Key Candidate Genes Involved in the Progression of Idiopathic Pulmonary Fibrosis. Molecules 2021, 26, 1123. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Gálvez, A.; González-Ávila, G.; Jiménez-Sánchez, L.L.; Maldonado-Martínez, H.A.; Cisneros, J.; Toscano-Marquez, F.; Castillejos-López, M.; Torres-Espíndola, L.M.; Velázquez-Cruz, R.; Rodríguez, V.H.O.; et al. Dysregulated Expression of Hypoxia-Inducible Factors Augments Myofibroblasts Differentiation in Idiopathic Pulmonary Fibrosis. Respir. Res. 2019, 20, 130. [Google Scholar] [CrossRef] [PubMed]
- Bowden, E.T.; Onikoyi, E.; Slack, R.; Myoui, A.; Yoneda, T.; Yamada, K.M.; Mueller, S.C. Co-Localization of Cortactin and Phosphotyrosine Identifies Active Invadopodia in Human Breast Cancer Cells. Exp. Cell Res. 2006, 312, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, P.; Besson, A. Cortactin Function in Invadopodia. Small GTPases 2020, 11, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Broadhead, A.R.; Bain, G.; Santini, A.M.; Darlington, J.; King, C.D.; Baccei, C.S.; et al. Pharmacokinetic and Pharmacodynamic Characterization of an Oral Lysophosphatidic Acid Type 1 Receptor-Selective Antagonist. J. Pharmacol. Exp. Ther. 2011, 336, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A Novel, Orally Active LPA(1) Receptor Antagonist Inhibits Lung Fibrosis in the Mouse Bleomycin Model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Burman, A.; Kropski, J.A.; Calvi, C.L.; Serezani, A.P.; Pascoalino, B.D.; Han, W.; Sherrill, T.; Gleaves, L.; Lawson, W.E.; Young, L.R.; et al. Localized Hypoxia Links ER Stress to Lung Fibrosis through Induction of C/EBP Homologous Protein. JCI Insight 2018, 3, e99543. [Google Scholar] [CrossRef] [PubMed]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1α Triggers ER Stress and CHOP-Mediated Apoptosis in Alveolar Epithelial Cells, a Key Event in Pulmonary Fibrosis. Sci. Rep. 2018, 8, 17939. [Google Scholar] [CrossRef]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia Are Required for Cancer Cell Extravasation and Are a Therapeutic Target for Metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef]
- Brochu-Gaudreau, K.; Charbonneau, M.; Harper, K.; Dubois, C.M. Hypoxia Selectively Increases a SMAD3 Signaling Axis to Promote Cancer Cell Invasion. Cancers 2022, 14, 2751. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Brochu-Gaudreau, K.; Arsenault, D.; Harper, K.; Dubois, C.M. Hypoxia-Induced Invadopodia Formation Involves Activation of NHE-1 by the P90 Ribosomal S6 Kinase (p90RSK). PLoS ONE 2011, 6, e28851. [Google Scholar] [CrossRef]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-Induced Invadopodia Formation Promotes Tumor Metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Sengupta, S.; Berk, M.; Kwak, Y.-G.; Escobar, P.F.; Belinson, J.; Mok, S.C.; Xu, Y. Hypoxia Enhances Lysophosphatidic Acid Responsiveness in Ovarian Cancer Cells and Lysophosphatidic Acid Induces Ovarian Tumor Metastasis in Vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The Lysophosphatidic Acid Receptor LPA1 Links Pulmonary Fibrosis to Lung Injury by Mediating Fibroblast Recruitment and Vascular Leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef] [PubMed]
- Herr, K.J.; Herr, D.R.; Lee, C.-W.; Noguchi, K.; Chun, J. Stereotyped Fetal Brain Disorganization Is Induced by Hypoxia and Requires Lysophosphatidic Acid Receptor 1 (LPA1) Signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15444–15449. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.-H.; Park, S.G.; Choi, J.-S.; Xia, Y.; Sung, J.-H. The Pivotal Role of Reactive Oxygen Species Generation in the Hypoxia-Induced Stimulation of Adipose-Derived Stem Cells. Stem Cells Dev. 2011, 20, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.K.; Yoon, C.; Yi, B.C.; Tap, W.D.; Simon, M.C.; Yoon, S.S. Platelet-Derived Growth Factor Receptor-α and -β Promote Cancer Stem Cell Phenotypes in Sarcomas. Oncogenesis 2018, 7, 47. [Google Scholar] [CrossRef]
- Baxter, R.M.; Secrist, J.P.; Vaillancourt, R.R.; Kazlauskas, A. Full Activation of the Platelet-Derived Growth Factor Beta-Receptor Kinase Involves Multiple Events. J. Biol. Chem. 1998, 273, 17050–17055. [Google Scholar] [CrossRef]
- Kazlauskas, A.; Cooper, J.A. Phosphorylation of the PDGF Receptor Beta Subunit Creates a Tight Binding Site for Phosphatidylinositol 3 Kinase. EMBO J. 1990, 9, 3279–3286. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Primer Sequences |
|---|---|
| SH3PXD2A | Forward: 5′-TGCCAAGAAGGAGATCAGCC-3′ Reverse: 5′-TGGAGGTCTTGTCCGTAGGT-3′ |
| RPL13 | Forward: 5′-CTCAAGGTCGTGCGTCTG-3′ Reverse: 5′-TGGCTTTCTCTTTCCTCTTCTC-3′ |
| CAIX | Forward: 5′-CCTCCATAGCGCCAATGACT-3′ Reverse: 5′-CCTCAAGAACCCCAGATTATTGC-3′ |
| ENPP2 | Forward: 5′-GGGAGACCACGGATTTGATAA-3′ Reverse: 5′-ATCCCAGGAGATCACACATAAC-3′ |
| LPAR1 | Forward: 5′-AATCGAGAGGCACATTACGG-3′ Reverse: 5′-TGTGGACAGCACACGTCTAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebel, M.; Cliche, D.O.; Charbonneau, M.; Brochu-Gaudreau, K.; Adam, D.; Brochiero, E.; Dubois, C.M.; Cantin, A.M. Hypoxia Promotes Invadosome Formation by Lung Fibroblasts. Cells 2024, 13, 1152. https://doi.org/10.3390/cells13131152
Lebel M, Cliche DO, Charbonneau M, Brochu-Gaudreau K, Adam D, Brochiero E, Dubois CM, Cantin AM. Hypoxia Promotes Invadosome Formation by Lung Fibroblasts. Cells. 2024; 13(13):1152. https://doi.org/10.3390/cells13131152
Chicago/Turabian StyleLebel, Mégane, Dominic O. Cliche, Martine Charbonneau, Karine Brochu-Gaudreau, Damien Adam, Emmanuelle Brochiero, Claire M. Dubois, and André M. Cantin. 2024. "Hypoxia Promotes Invadosome Formation by Lung Fibroblasts" Cells 13, no. 13: 1152. https://doi.org/10.3390/cells13131152
APA StyleLebel, M., Cliche, D. O., Charbonneau, M., Brochu-Gaudreau, K., Adam, D., Brochiero, E., Dubois, C. M., & Cantin, A. M. (2024). Hypoxia Promotes Invadosome Formation by Lung Fibroblasts. Cells, 13(13), 1152. https://doi.org/10.3390/cells13131152