

The Barrier Disruption and Pyroptosis of Intestinal Epithelial Cells Caused by Perfringolysin O (PFO) from Clostridium perfringens

 ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Antibodies, Reagents, and Plasmids
2.3. Expression and Purification of PFO
2.4. Animal Experiments and Sample Collection
2.5. Transepithelial Electrical Resistance Determination
2.6. The Impact of PFO on Cell Permeability Assessed by FD-4 Infiltration
2.7. CCK-8 Assay
2.8. Mitochondrial Membrane Potential Monitoring
2.9. LDH Assay
2.10. Cell Membrane Staining
2.11. Reactive Oxygen Species (ROS) Detection
2.12. RNA Extraction and RT-qPCR
2.13. Western Blot
2.14. Immunofluorescent Staining
2.15. Statistical Analysis
3. Results
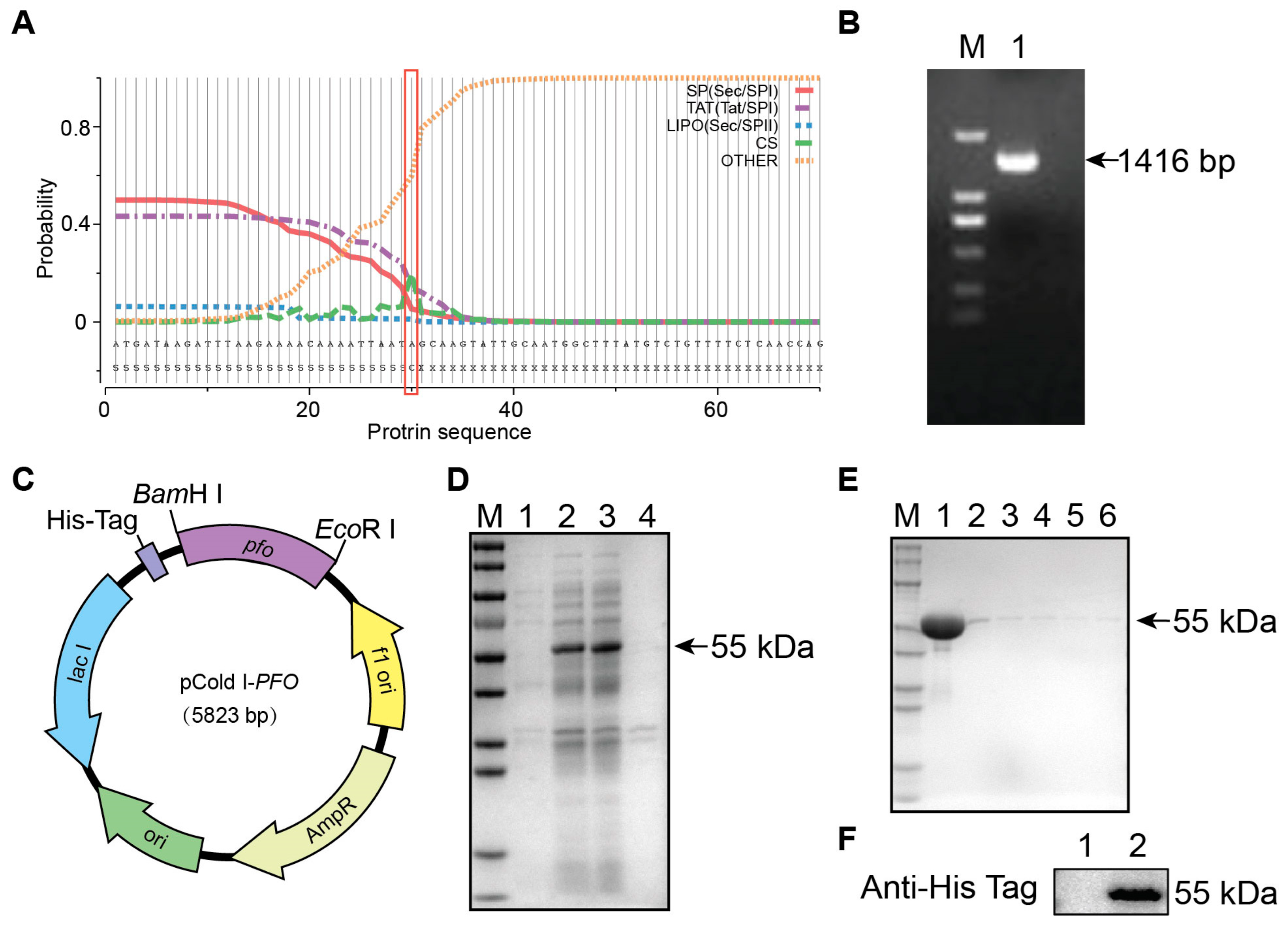
3.1. Expression and Purification of Theta Toxin (PFO)
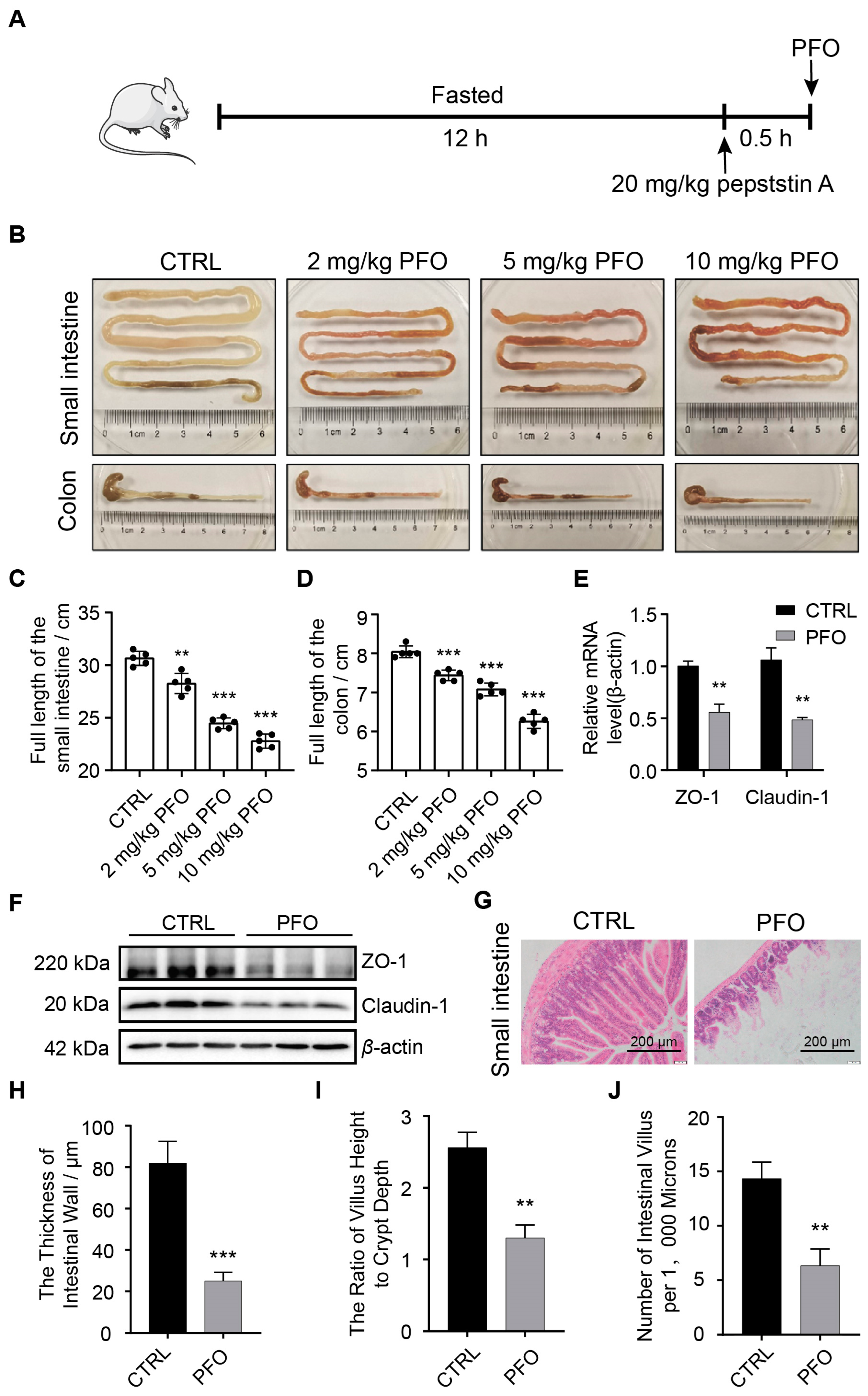
3.2. PFO Exposure Induces Intestinal Epithelial Barrier Damage In Vivo
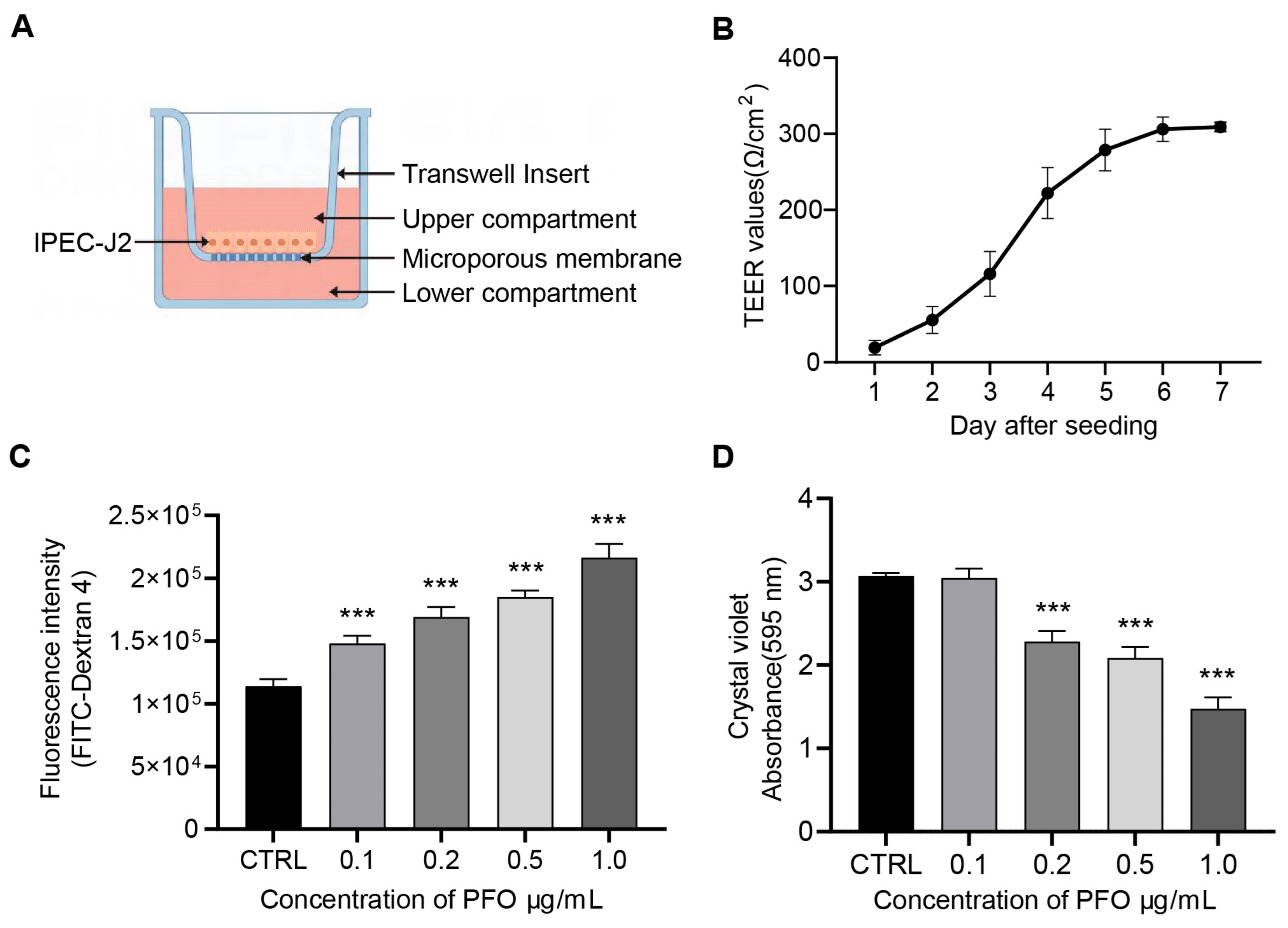
3.3. PFO Exposure Induces Damage to the Intestinal Epithelial Barrier Model In Vitro
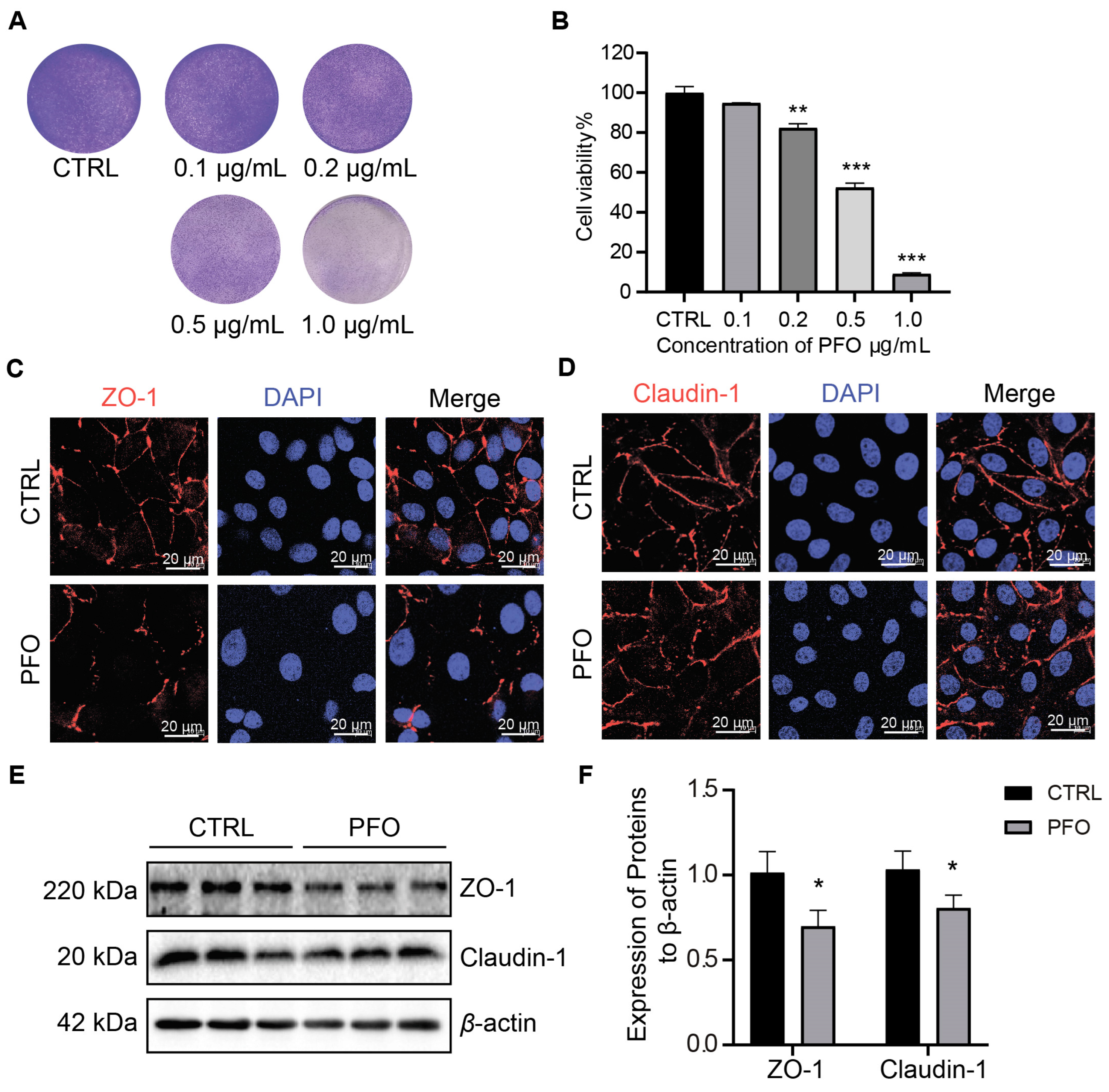
3.4. PFO Exposure Influences IPEC-J2 Cell Activity and Intercellular Tight Junction Impairment
3.5. PFO Causes Mitochondrial Damage and Increased ROS Content
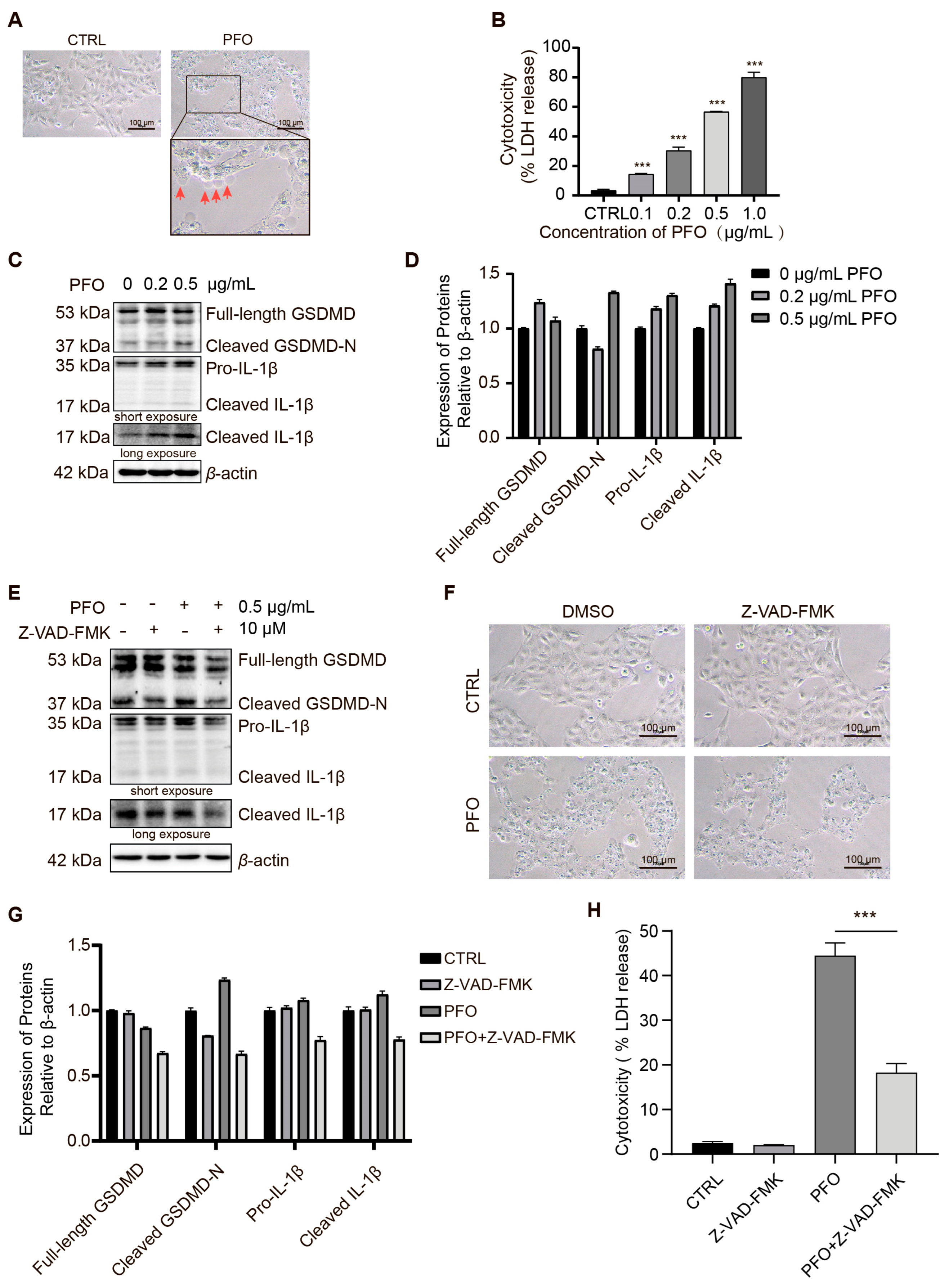
3.6. PFO Induces Pyroptosis in IPEC-J2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef]
- Huang, S.; Tian, Y.; Wang, Y.; García, P.; Liu, B.; Lu, R.; Wu, L.; Bao, H.; Pang, M.; Zhou, Y.; et al. The Broad Host Range Phage vB_CpeS_BG3P Is Able to Inhibit Clostridium perfringens Growth. Viruses 2022, 14, 676. [Google Scholar] [CrossRef]
- Marlow, M.A.; Luna-Gierke, R.E.; Griffin, P.M.; Vieira, A.R. Foodborne Disease Outbreaks in Correctional Institutions-United States, 1998–2014. Am. J. Public Health 2017, 107, 1150–1156. [Google Scholar] [CrossRef]
- Camargo, A.; Guerrero-Araya, E.; Castañeda, S.; Vega, L.; Cardenas-Alvarez, M.X.; Rodríguez, C.; Paredes-Sabja, D.; Ramírez, J.D.; Muñoz, M. Intra-species diversity of Clostridium perfringens: A diverse genetic repertoire reveals its pathogenic potential. Front. Microbiol. 2022, 13, 952081. [Google Scholar] [CrossRef]
- Kheravii, S.K.; Swick, R.A.; Choct, M.; Wu, S.B. Effect of oat hulls as a free choice feeding on broiler performance, short chain fatty acids and microflora under a mild necrotic enteritis challenge. Anim. Nutr. 2018, 4, 65–72. [Google Scholar] [CrossRef]
- Udhayavel, S.; Thippichettypalayam Ramasamy, G.; Gowthaman, V.; Malmarugan, S.; Senthilvel, K. Occurrence of Clostridium perfringens contamination in poultry feed ingredients: Isolation, identification and its antibiotic sensitivity pattern. Anim. Nutr. 2017, 3, 309–312. [Google Scholar] [CrossRef]
- Bendary, M.M.; Abd El-Hamid, M.I.; El-Tarabili, R.M.; Hefny, A.A.; Algendy, R.M.; Elzohairy, N.A.; Ghoneim, M.M.; Al-Sanea, M.M.; Nahari, M.H.; Moustafa, W.H. Clostridium perfringens Associated with Foodborne Infections of Animal Origins: Insights into Prevalence, Antimicrobial Resistance, Toxin Genes Profiles, and Toxinotypes. Biology 2022, 11, 551. [Google Scholar] [CrossRef]
- Monteiro, C.; do Carmo, M.S.; Melo, B.O.; Alves, M.S.; Dos Santos, C.I.; Monteiro, S.G.; Bomfim, M.R.Q.; Fernandes, E.S.; Monteiro-Neto, V. In Vitro Antimicrobial Activity and Probiotic Potential of Bifidobacterium and Lactobacillus against Species of Clostridium. Nutrients 2019, 11, 448. [Google Scholar] [CrossRef]
- Posthaus, H.; Kittl, S.; Tarek, B.; Bruggisser, J. Clostridium perfringens type C necrotic enteritis in pigs: Diagnosis, pathogenesis, and prevention. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc. 2020, 32, 203–212. [Google Scholar] [CrossRef]
- Ding, G.; Bai, J.; Feng, B.; Wang, L.; Qiao, X.; Zhou, H.; Jiang, Y.; Cui, W.; Tang, L.; Li, Y.; et al. An EGFP-marked recombinant lactobacillus oral tetravalent vaccine constitutively expressing α, ε, β1, and β2 toxoids for Clostridium perfringens elicits effective anti-toxins protective immunity. Virulence 2019, 10, 754–767. [Google Scholar] [CrossRef]
- Lin, Y.H.; Luck, H.; Khan, S.; Schneeberger, P.H.H.; Tsai, S.; Clemente-Casares, X.; Lei, H.; Leu, Y.L.; Chan, Y.T.; Chen, H.Y.; et al. Aryl hydrocarbon receptor agonist indigo protects against obesity-related insulin resistance through modulation of intestinal and metabolic tissue immunity. Int. J. Obes. 2019, 43, 2407–2421. [Google Scholar] [CrossRef]
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Yu, H.; Zhang, L.; Ruan, Z. Dietary Vegetable Powders Modulate Immune Homeostasis and Intestinal Microbiota in Mice. Foods 2021, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.S.; Leng, Y.X.; Yang, N.; Li, Z.Q.; Zong, Y.N.; Han, D.Y.; Li, Y.; He, J.D.; Mi, X.N.; Cong, Z.K.; et al. Inhibition of visfatin alleviates sepsis-induced intestinal damage by inhibiting Hippo signaling pathway. Inflamm. Res. 2022, 71, 911–922. [Google Scholar] [CrossRef]
- Song, X.; Pi, S.; Gao, Y.; Zhou, F.; Yan, S.; Chen, Y.; Qiao, L.; Dou, X.; Shao, D.; Xu, C. The Role of Vasoactive Intestinal Peptide and Mast Cells in the Regulatory Effect of Lactobacillus casei ATCC 393 on Intestinal Mucosal Immune Barrier. Front. Immunol. 2021, 12, 723173. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Mu, S.; Han, Y.; Chen, Y.; Kuang, Z.; Wu, X.; Luo, Y.; Tong, C.; Zhang, Y.; Yang, Y.; et al. Gpr174 Knockout Alleviates DSS-Induced Colitis via Regulating the Immune Function of Dendritic Cells. Front. Immunol. 2022, 13, 841254. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, L.; Chen, H.; Tsim, K.W.K.; Shen, X.; Li, X.; Li, X.; Lei, H. Herbicide propisochlor exposure induces intestinal barrier impairment, microbiota dysbiosis and gut pyroptosis. Ecotoxicol. Environ. Saf. 2023, 262, 115154. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Q.; Wang, J.; Sun, J.; Xiang, Y.; Jin, X. Aflatoxin B1 disrupts the intestinal barrier integrity by reducing junction protein and promoting apoptosis in pigs and mice. Ecotoxicol. Environ. Saf. 2022, 247, 114250. [Google Scholar] [CrossRef]
- Li, L.; Lv, X.; He, J.; Zhang, L.; Li, B.; Zhang, X.; Liu, S.; Zhang, Y. Chronic exposure to polystyrene nanoplastics induces intestinal mechanical and immune barrier dysfunction in mice. Ecotoxicol. Environ. Saf. 2024, 269, 115749. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.; Wang, J.; Shan, A.; Xu, L. Changes in intestinal barrier functions and gut microbiota in rats exposed to zearalenone. Ecotoxicol. Environ. Saf. 2020, 204, 111072. [Google Scholar] [CrossRef]
- Balzan, S.; de Almeida Quadros, C.; de Cleva, R.; Zilberstein, B.; Cecconello, I. Bacterial translocation: Overview of mechanisms and clinical impact. J. Gastroenterol. Hepatol. 2007, 22, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Terao, Y.; Sakurai, J.; Nagahama, M. Membrane-Binding Mechanism of Clostridium perfringens Alpha-Toxin. Toxins 2015, 7, 5268–5275. [Google Scholar] [CrossRef] [PubMed]
- Theoret, J.R.; Uzal, F.A.; McClane, B.A. Identification and characterization of Clostridium perfringens beta toxin variants with differing trypsin sensitivity and in vitro cytotoxicity activity. Infect. Immun. 2015, 83, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; McClane, B.A. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell Microbiol. 2005, 7, 129–146. [Google Scholar] [CrossRef]
- Chakrabarti, G.; Zhou, X.; McClane, B.A. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 2003, 71, 4260–4270. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Bryant, A.E. The role of clostridial toxins in the pathogenesis of gas gangrene. Clin. Infect. Dis. 2002, 35, S93–S100. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, X.; Zhang, H.; Zhang, J.; Zhou, Y.; Wang, T.; Hu, N.; Deng, X.; Bai, X.; Wang, J. Amentoflavone Attenuates Clostridium perfringens Gas Gangrene by Targeting Alpha-Toxin and Perfringolysin O. Front. Pharmacol. 2020, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Kay, C.; Xue, Y.; Pandey, A.; Lee, J.; Jing, W.; Enosi Tuipulotu, D.; Lo Pilato, J.; Feng, S.; Ngo, C.; et al. Clostridium perfringens virulence factors are nonredundant activators of the NLRP3 inflammasome. EMBO Rep. 2023, 24, e54600. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, S.; Li, C.; Ai, Z.; Shen, W.; Ren, W.; Yang, X. Discovery of a novel ferroptosis inducer-talaroconvolutin A-killing colorectal cancer cells in vitro and in vivo. Cell Death Dis. 2020, 11, 988. [Google Scholar] [CrossRef]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef]
- Xiang, X.; Zhang, F.; Fu, R.; Yan, S.; Zhou, L. Significant Differences in Bacterial and Potentially Pathogenic Communities Between Sympatric Hooded Crane and Greater White-Fronted Goose. Front. Microbiol. 2019, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. mBio 2015, 6, e02513. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh Gohari, I.; Li, J.; Navarro, M.; Uzal, F.; McClane, B. Effects of Claudin-1 on the Action of Clostridium perfringens Enterotoxin in Caco-2 Cells. Toxins 2019, 11, 582. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Troyer, B.E.; Merrick, D.T.; Mitten, J.E.; Olson, R.D. Lethal effects and cardiovascular effects of purified alpha- and theta-toxins from Clostridium perfringens. J. Infect. Dis. 1988, 157, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Dorca-Arévalo, J.; Not, A.; Cases, M.; Gómez de Aranda, I.; Martínez-Yélamos, A.; Martínez-Yélamos, S.; Solsona, C.; Blasi, J. The Cytotoxicity of Epsilon Toxin from Clostridium perfringens on Lymphocytes Is Mediated by MAL Protein Expression. Mol. Cell Biol. 2018, 38, e00086-18. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, K.; Ashida, H.; Okano, T.; Kinoshita-Daitoku, R.; Suzuki, S.; Ohtani, K.; Hamagaki, M.; Ikeda, T.; Suzuki, T. Inflammasome Activation Induced by Perfringolysin O of Clostridium perfringens and Its Involvement in the Progression of Gas Gangrene. Front. Microbiol. 2019, 10, 2406. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- McGuinness, C.; Walsh, J.C.; Bayly-Jones, C.; Dunstone, M.A.; Christie, M.P.; Morton, C.J.; Parker, M.W.; Böcking, T. Single-molecule analysis of the entire perfringolysin O pore formation pathway. eLife 2022, 11, e74901. [Google Scholar] [CrossRef]
- Su, Z.; Burchfield, J.G.; Yang, P.; Humphrey, S.J.; Yang, G.; Francis, D.; Yasmin, S.; Shin, S.Y.; Norris, D.M.; Kearney, A.L.; et al. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 2019, 10, 5486. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Xue, Q.; Kang, R.; Klionsky, D.J.; Tang, D.; Liu, J.; Chen, X. Copper metabolism in cell death and autophagy. Autophagy 2023, 19, 2175–2195. [Google Scholar] [CrossRef]
- Devant, P.; Boršić, E.; Ngwa, E.M.; Xiao, H.; Chouchani, E.T.; Thiagarajah, J.R.; Hafner-Bratkovič, I.; Evavold, C.L.; Kagan, J.C. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep. 2023, 42, 112008. [Google Scholar] [CrossRef]
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.C.; Chu, H.; Han, S.H.; Yun, C.H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95. [Google Scholar] [CrossRef]
- Peng, X.; Wang, R.; Hu, L.; Zhou, Q.; Liu, Y.; Yang, M.; Fang, Z.; Lin, Y.; Xu, S.; Feng, B.; et al. Enterococcus faecium NCIMB 10415 administration improves the intestinal health and immunity in neonatal piglets infected by enterotoxigenic Escherichia coli K88. J. Anim. Sci. Biotechnol. 2019, 10, 72. [Google Scholar] [CrossRef]
- Roos, S.; Wyder, M.; Candi, A.; Regenscheit, N.; Nathues, C.; van Immerseel, F.; Posthaus, H. Binding studies on isolated porcine small intestinal mucosa and in vitro toxicity studies reveal lack of effect of C. perfringens beta-toxin on the porcine intestinal epithelium. Toxins 2015, 7, 1235–1252. [Google Scholar] [CrossRef]
- Xie, K.; Yan, Z.; Yang, Q.; Huang, X.; Wang, P.; Gao, X.; Li, J.; Gun, S. lnc001776 Affects CPB2 Toxin-Induced Excessive Injury of Porcine Intestinal Epithelial Cells via Activating JNK/NF-kB Pathway through ssc-let-7i-5p/IL-6 Axis. Cells 2023, 12, 1036. [Google Scholar] [CrossRef]
- Adler, D.; Linden, J.R.; Shetty, S.V.; Ma, Y.; Bokori-Brown, M.; Titball, R.W.; Vartanian, T. Clostridium perfringens Epsilon Toxin Compromises the Blood-Brain Barrier in a Humanized Zebrafish Model. iScience 2019, 15, 39–54. [Google Scholar] [CrossRef]
- Verherstraeten, S.; Goossens, E.; Valgaeren, B.; Pardon, B.; Timbermont, L.; Vermeulen, K.; Schauvliege, S.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.; et al. The synergistic necrohemorrhagic action of Clostridium perfringens perfringolysin and alpha toxin in the bovine intestine and against bovine endothelial cells. Vet. Res. 2013, 44, 45. [Google Scholar] [CrossRef]
- Maekawa, M.; Yang, Y.; Fairn, G.D. Perfringolysin O Theta Toxin as a Tool to Monitor the Distribution and Inhomogeneity of Cholesterol in Cellular Membranes. Toxins 2016, 8, 67. [Google Scholar] [CrossRef]
- Chen, J.; Hu, S.; Ji, D.; Gao, Z.; Wang, H.; Yang, Y.; Chen, Y.; Gu, J. Hemolysin BL from novel Bacillus toyonensis BV-17 induces antitumor activity both in vitro and in vivo. Gut Microbes 2020, 12, 1782158. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′ to 3′) | Reverse Primer Sequence (5′ to 3′) |
|---|---|---|
| ZO-1 | ACCCGAAACTGATGCTGTGGATAG | AAATGGCCGGGCAGAACTTGTGTA |
| Claudin-1 | AGCTGCCTGTTCCATGTACT | CTCCCATTTGTCTGCTGCTC |
| β-actin | AAATCGTGCGTGACATCAAA | ATGCCACAGGATTCCATACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Mou, S.; Li, L.; Chen, Q.; Yang, R.; Guo, S.; Jin, Y.; Liu, L.; Li, T.; Chen, H.; et al. The Barrier Disruption and Pyroptosis of Intestinal Epithelial Cells Caused by Perfringolysin O (PFO) from Clostridium perfringens. Cells 2024, 13, 1140. https://doi.org/10.3390/cells13131140
Liu Z, Mou S, Li L, Chen Q, Yang R, Guo S, Jin Y, Liu L, Li T, Chen H, et al. The Barrier Disruption and Pyroptosis of Intestinal Epithelial Cells Caused by Perfringolysin O (PFO) from Clostridium perfringens. Cells. 2024; 13(13):1140. https://doi.org/10.3390/cells13131140
Chicago/Turabian StyleLiu, Zhankui, Shuang Mou, Liang Li, Qichao Chen, Ruicheng Yang, Shibang Guo, Yancheng Jin, Lixinjie Liu, Tianzhi Li, Huanchun Chen, and et al. 2024. "The Barrier Disruption and Pyroptosis of Intestinal Epithelial Cells Caused by Perfringolysin O (PFO) from Clostridium perfringens" Cells 13, no. 13: 1140. https://doi.org/10.3390/cells13131140
APA StyleLiu, Z., Mou, S., Li, L., Chen, Q., Yang, R., Guo, S., Jin, Y., Liu, L., Li, T., Chen, H., & Wang, X. (2024). The Barrier Disruption and Pyroptosis of Intestinal Epithelial Cells Caused by Perfringolysin O (PFO) from Clostridium perfringens. Cells, 13(13), 1140. https://doi.org/10.3390/cells13131140