
Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
2.2. Cell Culture
2.3. Induction of Cellular Senescence
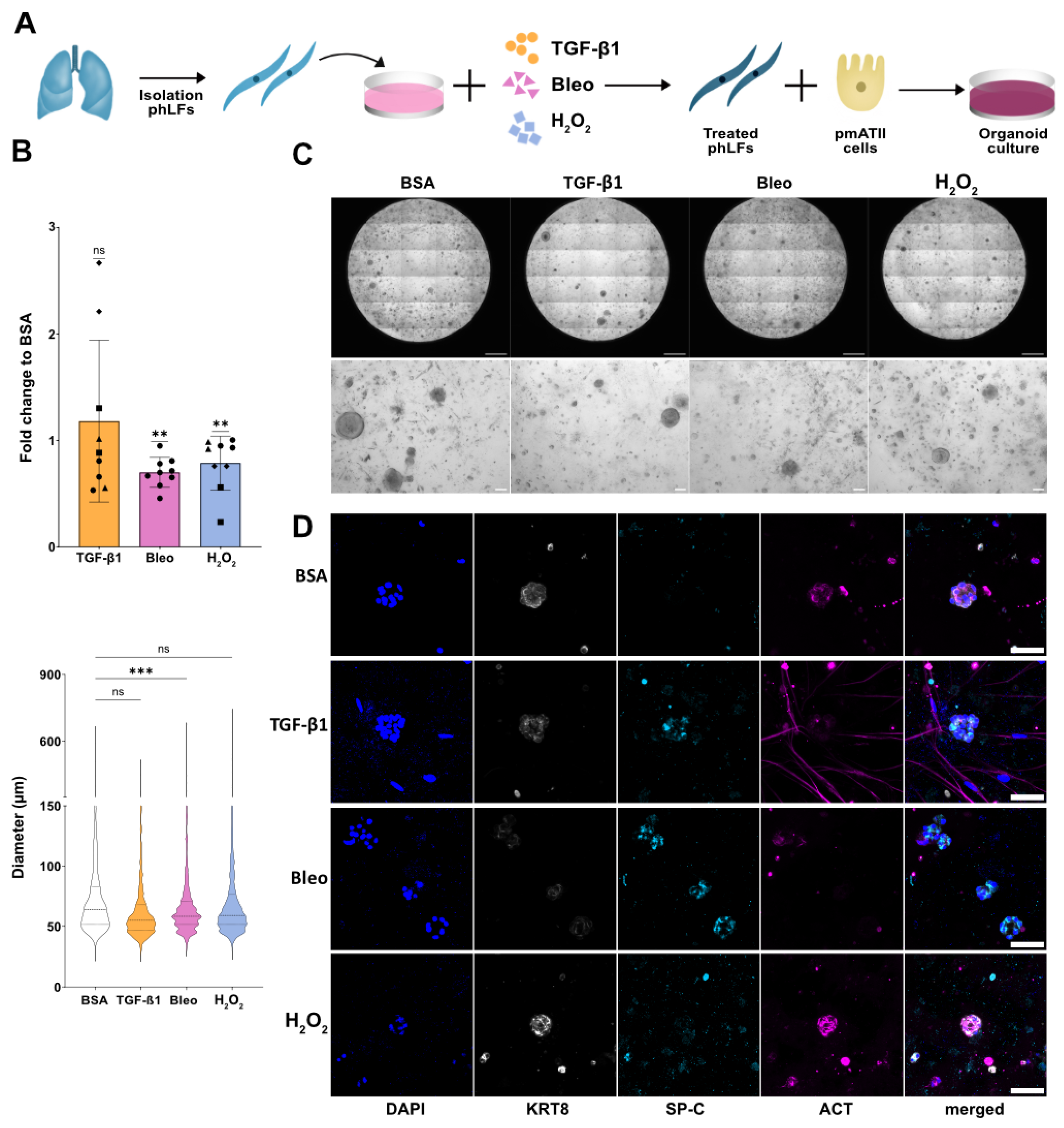
2.4. Organoid Assay
2.5. RT-qPCR
2.6. ELISA
2.7. Senescence Associated β-Galactosidase Staining
2.8. Immunofluorescence Staining
2.9. Bulk-RNA Sequencing
2.10. Analysis of Human Lung COPD Single Cell RNA Sequencing Data
2.11. Analysis of Human Lung IPF Single Cell RNA Sequencing Data
2.12. Microarray Analysis
2.13. Data Collection and Analysis
3. Results
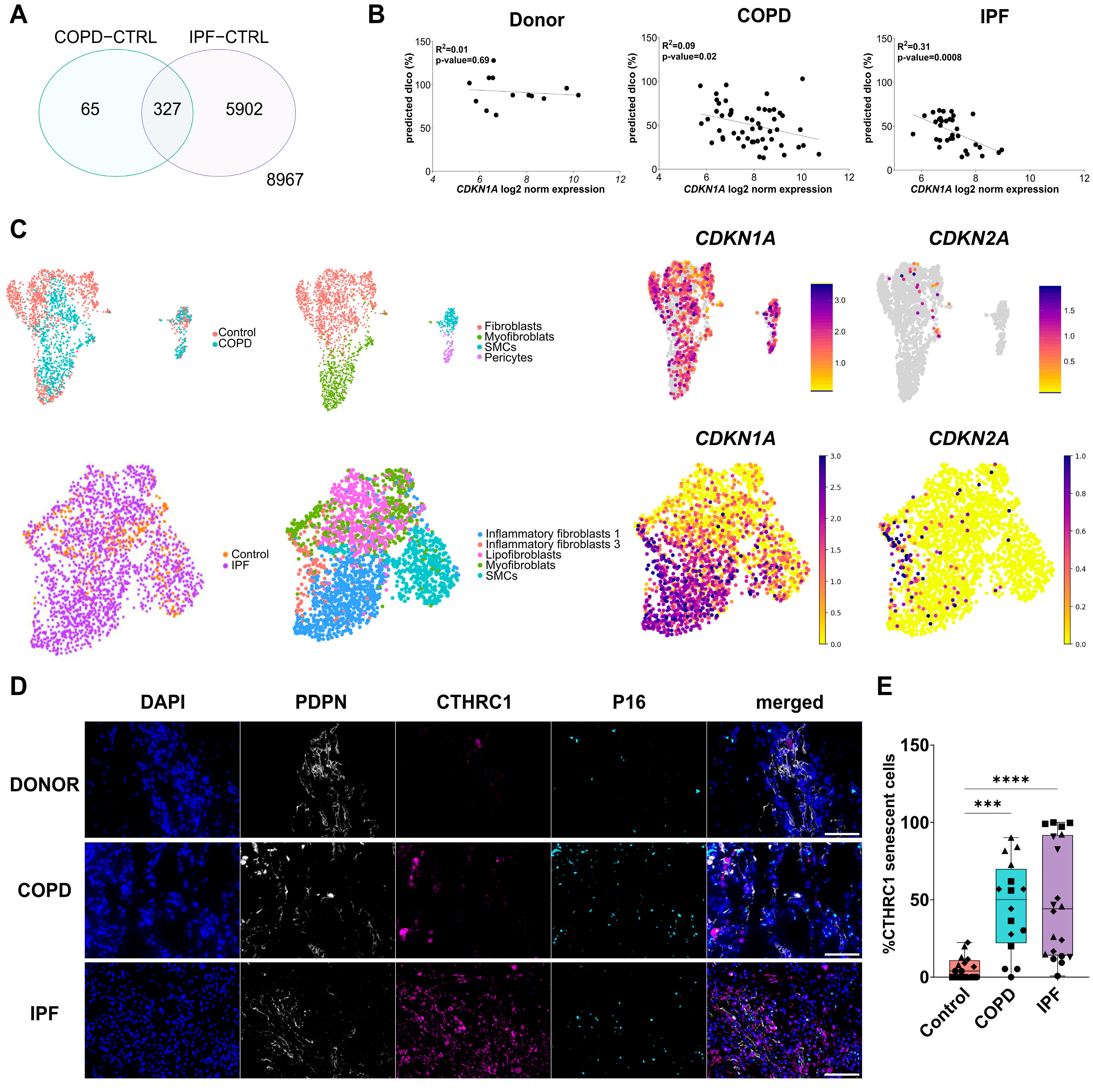
3.1. Senescent Fibroblasts Accumulate in IPF and COPD Lungs
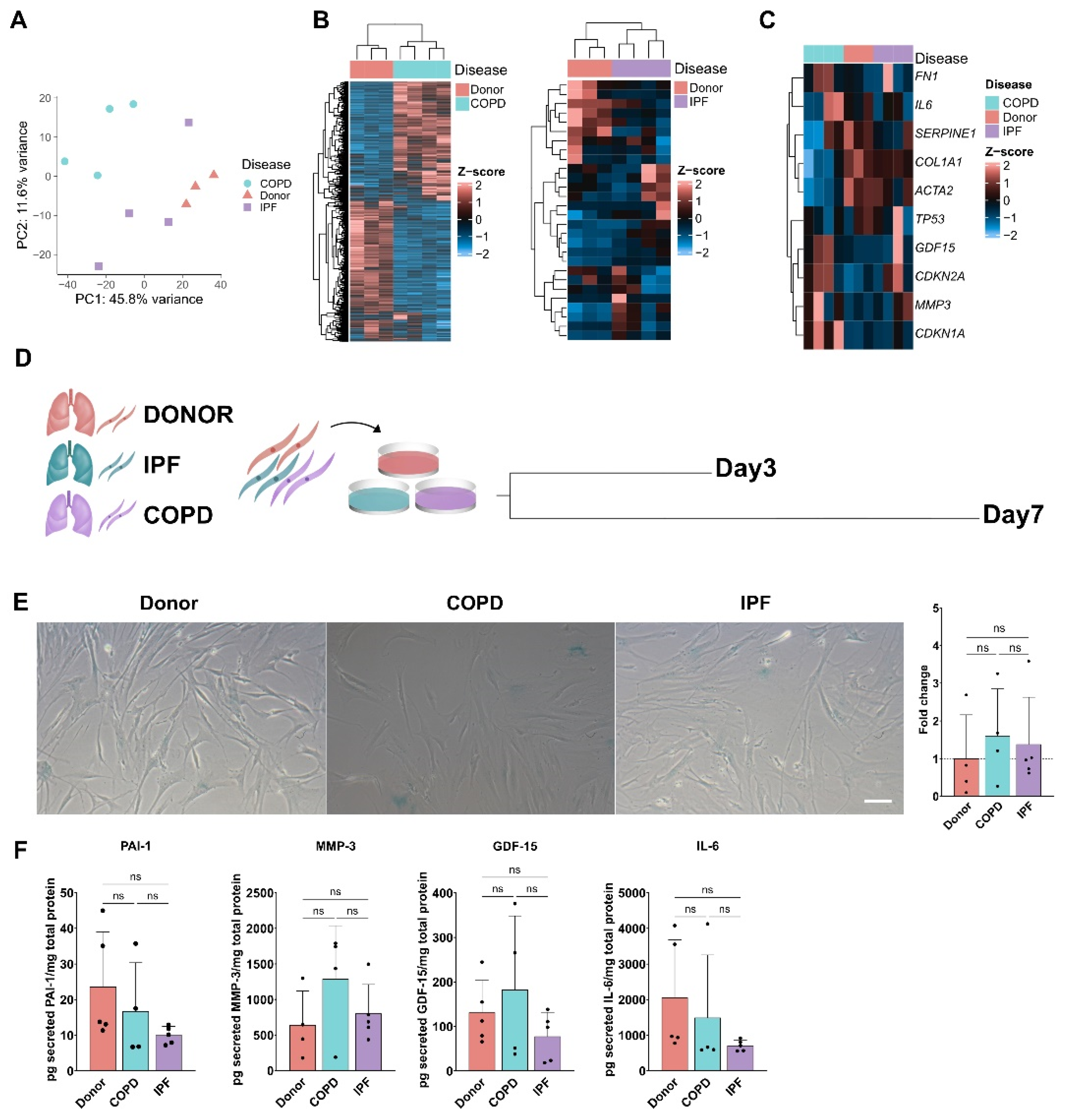
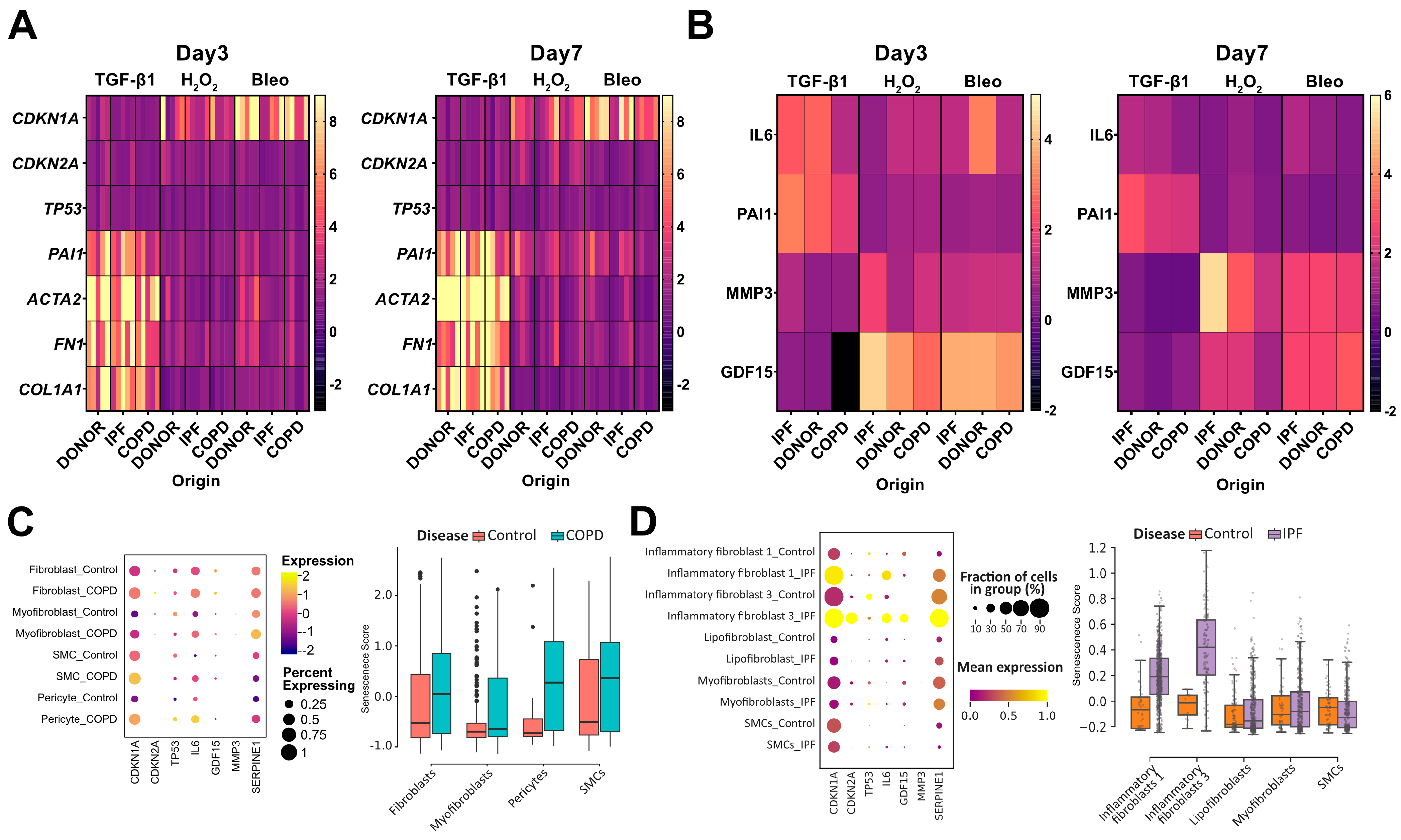
3.2. Senescence Program in phLF Derived from Different Diseases Backgrounds
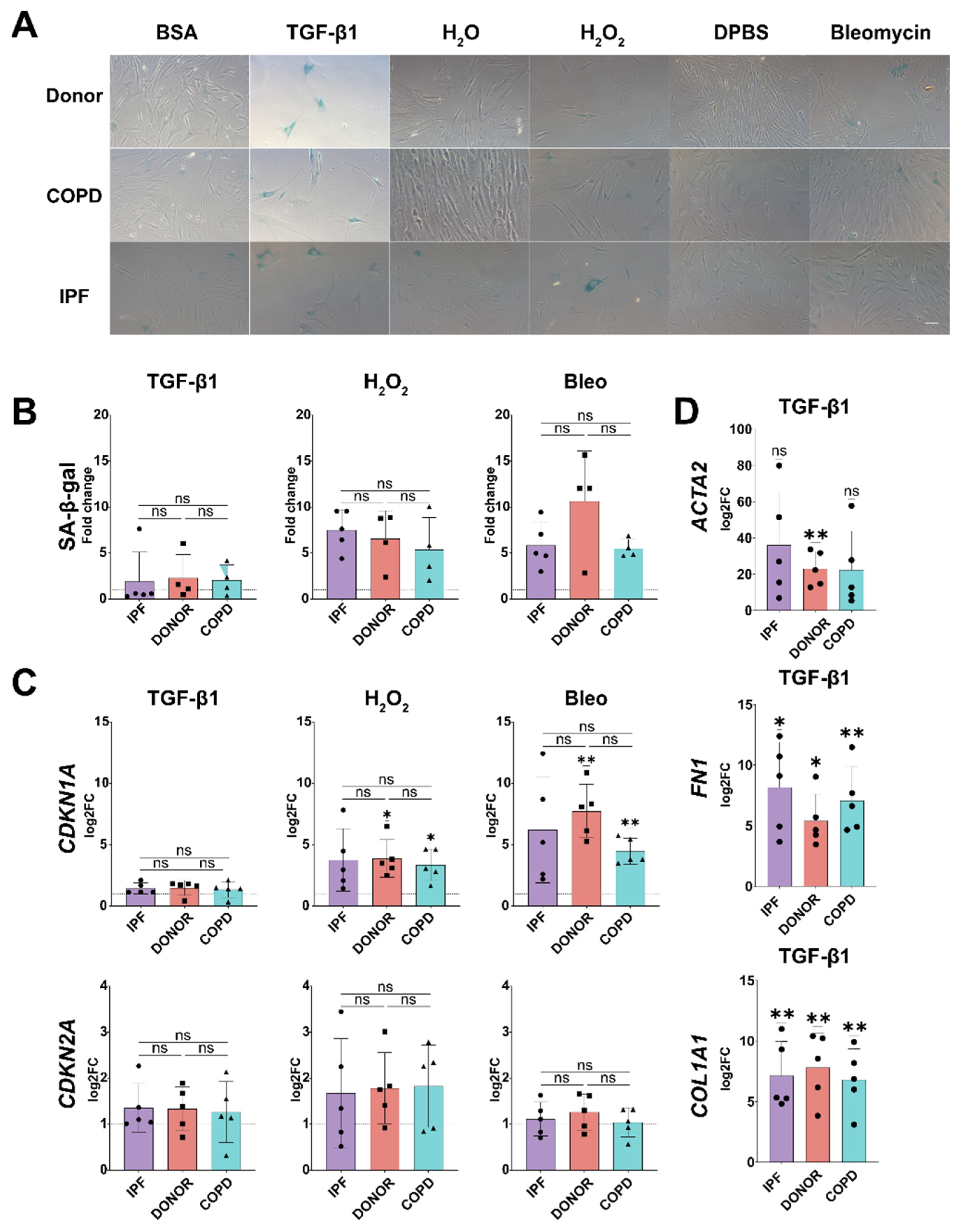
3.3. Disease Relevant Stimuli Can Induce Senescence in phLF
3.4. Senescence Induction in phLF Is Not Impacted by Disease Background
3.5. Senescence Induction in phLF Is Stimuli-Specific
3.6. Senescent Fibroblasts Disrupt Progenitor Potential of Distal Alveolar Epithelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momtazmanesh, S.; Moghaddam, S.S.; Ghamari, S.-H.; Rad, E.M.; Rezaei, N.; Shobeiri, P.; Aali, A.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abdelmasseh, M.; et al. Global Burden of Chronic Respiratory Diseases and Risk Factors, 1990–2019: An Update from the Global Burden of Disease Study 2019. eClinicalMedicine 2023, 59, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic Obstructive Pulmonary Disease. Lancet Lond. Engl. 2017, 389, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Postma, D.S.; Bush, A.; van den Berge, M. Risk Factors and Early Origins of Chronic Obstructive Pulmonary Disease. Lancet Lond. Engl. 2015, 385, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic Pulmonary Fibrosis. Nat. Rev. Dis. Primer 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K. Idiopathic Pulmonary Fibrosis: Update on Genetic Discoveries. Proc. Am. Thorac. Soc. 2011, 8, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Nureki, S.-I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of Mutant Sftpc in Murine Alveolar Epithelia Drives Spontaneous Lung Fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the Ageing Lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Carlier, F.M.; Dupasquier, S.; Ambroise, J.; Detry, B.; Lecocq, M.; Biétry–Claudet, C.; Boukala, Y.; Gala, J.-L.; Bouzin, C.; Verleden, S.E.; et al. Canonical WNT Pathway Is Activated in the Airway Epithelium in Chronic Obstructive Pulmonary Disease. eBioMedicine 2020, 61. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef]
- Barnes, P.J. Small Airway Fibrosis in COPD. Int. J. Biochem. Cell Biol. 2019, 116, 105598. [Google Scholar] [CrossRef]
- Barnes, P.J. Senescence in COPD and Its Comorbidities. Annu. Rev. Physiol. 2017, 79, 517–539. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; Heijink, I.H.; van den Berge, M.; Timens, W.; Oliver, B.G.G.; de Vries, M.; Brandsma, C.-A. COPD-Derived Fibroblasts Secrete Higher Levels of Senescence-Associated Secretory Phenotype Proteins. Thorax 2021, 76, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF Lung Fibroblasts Have a Senescent Phenotype. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-Cell RNA-Seq Reveals Ectopic and Aberrant Lung-Resident Cell Populations in Idiopathic Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A Molecular Cell Atlas of the Human Lung from Single-Cell RNA Sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Peyser, R.; MacDonnell, S.; Gao, Y.; Cheng, L.; Kim, Y.; Kaplan, T.; Ruan, Q.; Wei, Y.; Ni, M.; Adler, C.; et al. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am. J. Respir. Cell Mol. Biol. 2019, 61, 74–85. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Melo-Narváez, M.C.; Stegmayr, J.; Wagner, D.E.; Lehmann, M. Lung Regeneration: Implications of the Diseased Niche and Ageing. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2020, 29, 200222. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Hu, Q.; Hu, Y.; Hafner, K.; Costa, R.; van den Berg, A.; Königshoff, M. Chronic WNT/β-Catenin Signaling Induces Cellular Senescence in Lung Epithelial Cells. Cell. Signal. 2020, 70, 109588. [Google Scholar] [CrossRef] [PubMed]
- Kastlmeier, M.T.; Gonzalez-Rodriguez, E.; Cabanis, P.; Guenther, E.M.; König, A.-C.; Han, L.; Hauck, S.M.; See, F.; Asgharpour, S.; Bukas, C.; et al. Cytokine Signaling Converging on IL11 in ILD Fibroblasts Provokes Aberrant Epithelial Differentiation Signatures. Front. Immunol. 2023, 14, 1128239. [Google Scholar] [CrossRef] [PubMed]
- Bukas, C. HelmholtzAI-Consultants-Munich/Napari-Organoid-Counter: Latest Versions of Dependencies. 2022. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Heinzelmann, K.; Lehmann, M.; Gerckens, M.; Noskovičová, N.; Frankenberger, M.; Lindner, M.; Hatz, R.; Behr, J.; Hilgendorff, A.; Königshoff, M.; et al. Cell-Surface Phenotyping Identifies CD36 and CD97 as Novel Markers of Fibroblast Quiescence in Lung Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L682–L696. [Google Scholar] [CrossRef]
- Heinzelmann, K.; Noskovičová, N.; Merl-Pham, J.; Preissler, G.; Winter, H.; Lindner, M.; Hatz, R.; Hauck, S.M.; Behr, J.; Eickelberg, O. Surface Proteome Analysis Identifies Platelet Derived Growth Factor Receptor-Alpha as a Critical Mediator of Transforming Growth Factor-Beta-Induced Collagen Secretion. Int. J. Biochem. Cell Biol. 2016, 74, 44–59. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Core Team (2021). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Compu-ting: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 10 July 2023).
- Mayr, C.H.; Simon, L.M.; Leuschner, G.; Ansari, M.; Schniering, J.; Geyer, P.E.; Angelidis, I.; Strunz, M.; Singh, P.; Kneidinger, N.; et al. Integrative Analysis of Cell State Changes in Lung Fibrosis with Peripheral Protein Biomarkers. EMBO Mol. Med. 2021, 13, e12871. [Google Scholar] [CrossRef]
- Tsukui, T.; Sun, K.-H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-Producing Lung Cell Atlas Identifies Multiple Subsets with Distinct Localization and Relevance to Fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Cortijo, J. Pirfenidone Anti-Fibrotic Effects Are Partially Mediated by the Inhibition of MUC1 Bioactivation. Oncotarget 2020, 11, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Blokland, K.E.C.; Habibie, H.; Borghuis, T.; Teitsma, G.J.; Schuliga, M.; Melgert, B.N.; Knight, D.A.; Brandsma, C.-A.; Pouwels, S.D.; Burgess, J.K. Regulation of Cellular Senescence Is Independent from Profibrotic Fibroblast-Deposited ECM. Cells 2021, 10, 1628. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.W.; Schuliga, M.; Pathinayake, P.S.; Wei, L.; Tan, H.-Y.; Blokland, K.E.C.; Jaffar, J.; Westall, G.P.; Burgess, J.K.; Prêle, C.M.; et al. A Senescence Bystander Effect in Human Lung Fibroblasts. Biomedicines 2021, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1–PD-1 Improves Senescence Surveillance and Ageing Phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent Cells Evade Immune Clearance via HLA-E-Mediated NK and CD8+ T Cell Inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Lee, S.; Islam, M.N.; Boostanpour, K.; Aran, D.; Jin, G.; Christenson, S.; Matthay, M.A.; Eckalbar, W.L.; DePianto, D.J.; Arron, J.R.; et al. Molecular Programs of Fibrotic Change in Aging Human Lung. Nat. Commun. 2021, 12, 6309. [Google Scholar] [CrossRef]
- Hamsanathan, S.; Alder, J.K.; Sellares, J.; Rojas, M.; Gurkar, A.U.; Mora, A.L. Cellular Senescence: The Trojan Horse in Chronic Lung Diseases. Am. J. Respir. Cell Mol. Biol. 2019, 61, 21–30. [Google Scholar] [CrossRef]
- Holz, O.; Zühlke, I.; Jaksztat, E.; Müller, K.C.; Welker, L.; Nakashima, M.; Diemel, K.D.; Branscheid, D.; Magnussen, H.; Jörres, R.A. Lung Fibroblasts from Patients with Emphysema Show a Reduced Proliferation Rate in Culture. Eur. Respir. J. 2004, 24, 575–579. [Google Scholar] [CrossRef]
- Rodriguez, L.R.; Emblom-Callahan, M.; Chhina, M.; Bui, S.; Aljeburry, B.; Tran, L.H.; Novak, R.; Lemma, M.; Nathan, S.D.; Grant, G.M. Global Gene Expression Analysis in an in Vitro Fibroblast Model of Idiopathic Pulmonary Fibrosis Reveals Potential Role for CXCL14/CXCR4. Sci. Rep. 2018, 8, 3983. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; López-Diazguerrero, N.E.; Maciel-Barón, L.A.; Morales-Rosales, S.L.; Galván-Arzate, S.; Fernandez-Perrino, F.J.; Zentella, A.; Pérez, V.I.; Gomez-Quiroz, L.E.; Königsberg, M. Cell Proliferation Arrest and Redox State Status as Part of Different Stages during Senescence Establishment in Mouse Fibroblasts. Biogerontology 2014, 15, 165–176. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; de Vries, M.; Timens, W.; van den Berge, M.; Demaria, M.; Oliver, B.G.G.; Heijink, I.H.; Brandsma, C.-A. Link between Increased Cellular Senescence and Extracellular Matrix Changes in COPD. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L48–L60. [Google Scholar] [CrossRef]
- Wrench, C.; Baker, J.; Fenwick, P.; Donnelly, L.; Barnes, P. Small Airway Fibroblasts from COPD Patients Are Senescent and Pro-Fibrotic. Eur. Respir. J. 2018, 52, PA2172. [Google Scholar] [CrossRef]
- Baarsma, H.A.; Skronska-Wasek, W.; Mutze, K.; Ciolek, F.; Wagner, D.E.; John-Schuster, G.; Heinzelmann, K.; Günther, A.; Bracke, K.R.; Dagouassat, M.; et al. Noncanonical WNT-5A Signaling Impairs Endogenous Lung Repair in COPD. J. Exp. Med. 2016, 214, 143–163. [Google Scholar] [CrossRef]
- Baranyi, U.; Winter, B.; Gugerell, A.; Hegedus, B.; Brostjan, C.; Laufer, G.; Messner, B. Primary Human Fibroblasts in Culture Switch to a Myofibroblast-Like Phenotype Independently of TGF Beta. Cells 2019, 8, 721. [Google Scholar] [CrossRef]
- Wrench, C.L.; Baker, J.R.; Monkley, S.; Fenwick, P.S.; Murray, L.; Donnelly, L.E.; Barnes, P.J. Small Airway Fibroblasts from Chronic Obstructive Pulmonary Disease Patients Exhibit Cellular Senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2024, 326, L266–L279. [Google Scholar] [CrossRef]
- Mak, J.C.W.; Chan-Yeung, M.M.W.; Ho, S.P.; Chan, K.S.; Choo, K.; Yee, K.S.; Chau, C.H.; Cheung, A.H.K.; Ip, M.S.M. Elevated Plasma TGF-Β1 Levels in Patients with Chronic Obstructive Pulmonary Disease. Respir. Med. 2009, 103, 1083–1089. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Ziesche, R.; Zeische, R.; Fraifeld, V.E. Cellular Senescence-like Features of Lung Fibroblasts Derived from Idiopathic Pulmonary Fibrosis Patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef]
- DePianto, D.J.; Heiden, J.A.V.; Morshead, K.B.; Sun, K.-H.; Modrusan, Z.; Teng, G.; Wolters, P.J.; Arron, J.R. Molecular Mapping of Interstitial Lung Disease Reveals a Phenotypically Distinct Senescent Basal Epithelial Cell Population. Available online: https://insight.jci.org/articles/view/143626/figure/2 (accessed on 10 July 2023).
- Chuliá-Peris, L.; Carreres-Rey, C.; Gabasa, M.; Alcaraz, J.; Carretero, J.; Pereda, J. Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int. J. Mol. Sci. 2022, 23, 6894. [Google Scholar] [CrossRef]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C.; et al. Matrix Metalloproteinase 3 Is a Mediator of Pulmonary Fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, A.; Cottage, C.T.; Piras, A.; Overed-Sayer, C.; Sihlbom, C.; Budida, R.; Wrench, C.; Connor, J.; Monkley, S.; Hazon, P.; et al. Increased Expression and Accumulation of GDF15 in IPF Extracellular Matrix Contribute to Fibrosis. JCI Insight 2022, 7, e153058. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Liu, G.; Cai, L.; Deshane, J.; Antony, V.; Thannickal, V.J.; Liu, R.-M. Divergent Regulation of Alveolar Type 2 Cell and Fibroblast Apoptosis by Plasminogen Activator Inhibitor 1 in Lung Fibrosis. Am. J. Pathol. 2021, 191, 1227–1239. [Google Scholar] [CrossRef]
- Shioya, S.; Masuda, T.; Senoo, T.; Horimasu, Y.; Miyamoto, S.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Hattori, N. Plasminogen Activator Inhibitor-1 Serves an Important Role in Radiation-induced Pulmonary Fibrosis. Exp. Ther. Med. 2018, 16, 3070–3076. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Age | Smoker | Diagnosis |
|---|---|---|---|
| female | 72 | ex-smoker | Ctrl Donor |
| female | 67 | smoker | Ctrl Donor |
| male | 84 | ex-smoker | Ctrl Donor |
| female | 57 | smoker | Ctrl Donor |
| female | 56 | ex-smoker | IPF |
| male | 72 | n/a | IPF |
| female | 54 | never smoker | IPF |
| male | 73 | n/a | IPF |
| male | 63 | ex-smoker | IPF |
| male | 67 | ex-smoker | COPD |
| male | 73 | ex-smoker | COPD |
| female | 69 | ex-smoker | COPD |
| male | 62 | ex-smoker | COPD |
| female | n/a | n/a | COPD |
| male | 64 | smoker | COPD |
| male | 58 | smoker | COPD |
| female | 60 | smoker | COPD |
| female | 61 | smoker | COPD |
| unknown | n/a | n/a | Ctrl Donor |
| unknown | n/a | n/a | Ctrl Donor |
| unknown | n/a | n/a | Ctrl Donor |
| unknown | n/a | n/a | Ctrl Donor |
| male | 58 | ex-smoker | IPF |
| male | 63 | ex-smoker | IPF |
| male | n/a | never smoker | IPF |
| male | 61 | n/a | IPF |
| female | 29 | never smoker | Ctrl Donor |
| female | 71 | ex-smoker | Ctrl Donor |
| male | 59 | ex-smoker | Ctrl Donor |
| male | 49 | never smoker | Ctrl Donor |
| male | 62 | ex-smoker | COPD |
| male | 58 | ex-smoker | COPD |
| male | 60 | ex-smoker | COPD |
| male | 65 | ex-smoker | COPD |
| male | 57 | ex-smoker | IPF |
| female | 40 | never smoker | IPF |
| male | 52 | ex-smoker | IPF |
| male | 62 | ex-smoker | IPF |
| male | 59 | ex-smoker | IPF |
| Primer | Sequence (5′-3′) |
|---|---|
| ACTA2_fw | CGAGATCTCACTGACTACCTCATGA |
| ACTA2_rv | AGAGCTACATAACACAGTTTCTCCTTGA |
| FN-1_fw | CCGACCAGAAGTTTGGGTTCT |
| FN-1_rv | CAATGCGGTACATGACCCCT |
| COL1a1_fw | CAAGAGGAAGGCCAAGTCGAG |
| COL1a1_rv | TTGTCGCAGACGCAGATCC |
| PAI-1 fw | GACATCCTGGAACTGCCCTA |
| PAI-1 rv | GGTCATGTTGCCTTTCCAGT |
| CDKN2A_fw | ACCAGAGGCAGTAACCATGC |
| CDKN2A_rev | CCTGTAGGACCTTCGGTGAC |
| CDKN1A_fw | GTCAGTTCCTTGTGGAGCCG |
| CDKN1A_rev | TGGGTTCTGACGGACATCCC |
| TP53_fw | CGCTTCGAGATGTTCCGAGA |
| TP53_rv | CTTCAGGTGGCTGGAGTGAG |
| HPRT_fw | AAGGACCCCACGAAGTGTTG |
| HPRT_rv | GGCTTTGTATTTTGCTTTTCCA |
| Target Protein | Host | Company | Ref. No |
|---|---|---|---|
| P21 | rabbit | Abcam | ab109520 |
| Phospho-histone H2A.X | mouse | Millipore | 05–636 |
| ACT | mouse | Abcam | ab24610 |
| SP-C | rabbit | Abcam | ab3786 |
| Krt8 | rat | DSHB | TROMAI |
| Anti-rat-488 | donkey | Invitrogen | A21208 |
| Anti-rabbit-647 | donkey | Invitrogen | A31573 |
| Anti-mouse-568 | donkey | Invitrogen | A10037 |
| Anti-mouse-555 | goat | Invitrogen | A21424 |
| Anti-rabbit-488 | goat | Invitrogen | A11008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melo-Narváez, M.C.; Bramey, N.; See, F.; Heinzelmann, K.; Ballester, B.; Steinchen, C.; Jain, E.; Federl, K.; Hu, Q.; Dhakad, D.; et al. Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function. Cells 2024, 13, 1129. https://doi.org/10.3390/cells13131129
Melo-Narváez MC, Bramey N, See F, Heinzelmann K, Ballester B, Steinchen C, Jain E, Federl K, Hu Q, Dhakad D, et al. Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function. Cells. 2024; 13(13):1129. https://doi.org/10.3390/cells13131129
Chicago/Turabian StyleMelo-Narváez, Maria Camila, Nora Bramey, Fenja See, Katharina Heinzelmann, Beatriz Ballester, Carina Steinchen, Eshita Jain, Kathrin Federl, Qianjiang Hu, Deepesh Dhakad, and et al. 2024. "Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function" Cells 13, no. 13: 1129. https://doi.org/10.3390/cells13131129
APA StyleMelo-Narváez, M. C., Bramey, N., See, F., Heinzelmann, K., Ballester, B., Steinchen, C., Jain, E., Federl, K., Hu, Q., Dhakad, D., Behr, J., Eickelberg, O., Yildirim, A. Ö., Königshoff, M., & Lehmann, M. (2024). Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function. Cells, 13(13), 1129. https://doi.org/10.3390/cells13131129