
MLIP and Its Potential Influence on Key Oncogenic Pathways


 ,
,
Abstract
1. Introduction
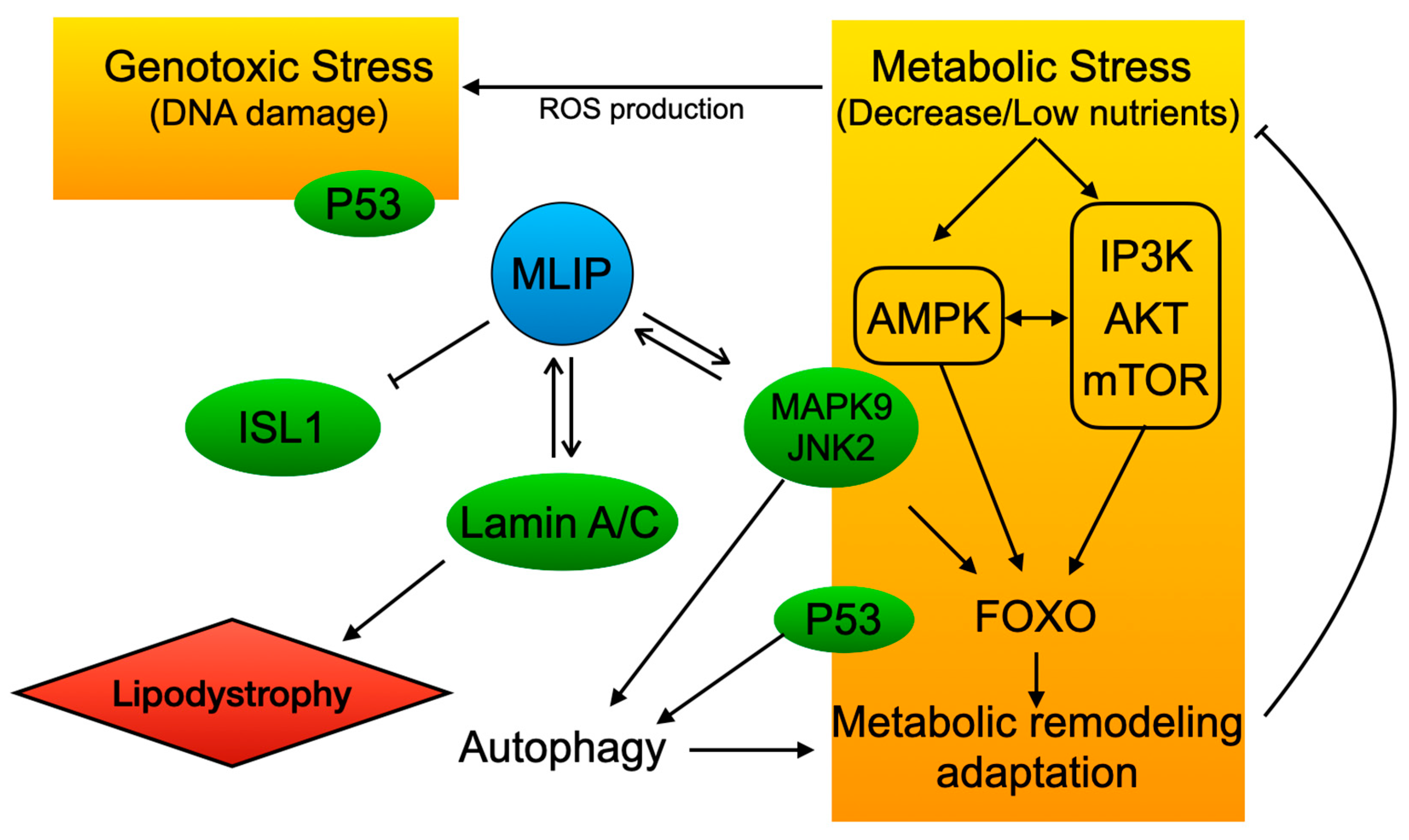
2. MLIP Discovery and Cellular Role
3. MLIP Expression in Cancer
Expression of MLIP in Different Types of Cancer
4. Molecular Relationship of MLIP with Pro-Survival/Oncogenic Pathways and Tumor Suppressors
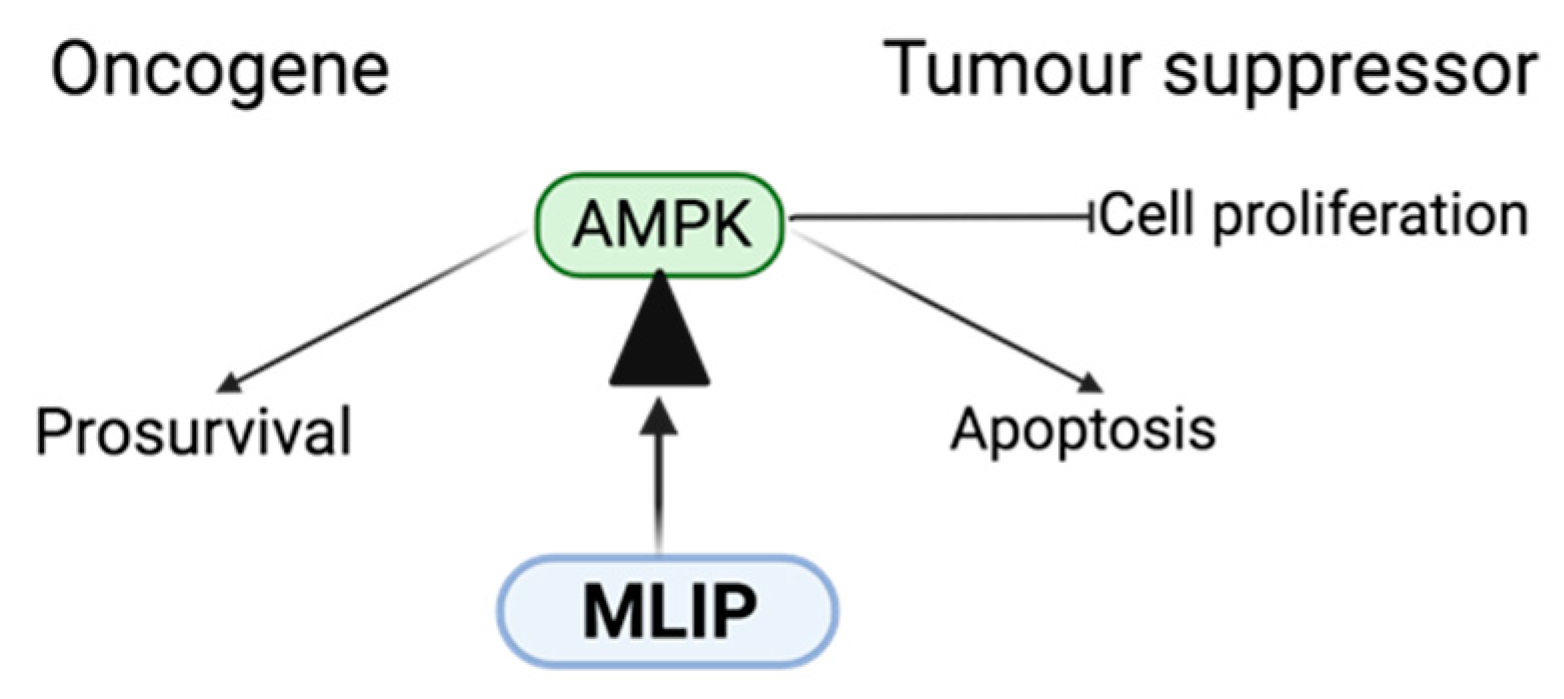
4.1. MLIP: AMPK Function and Dysfunction in Cancer
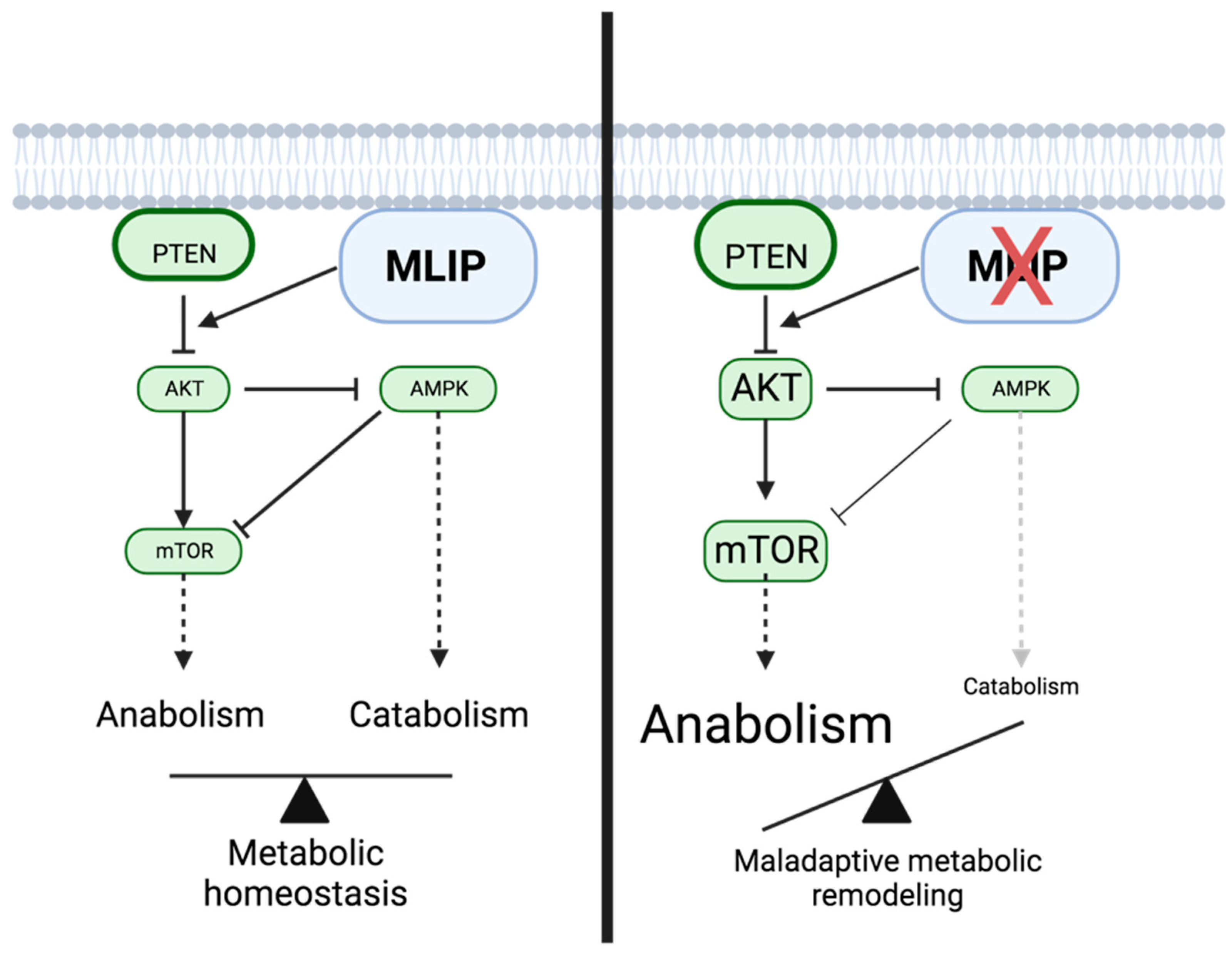
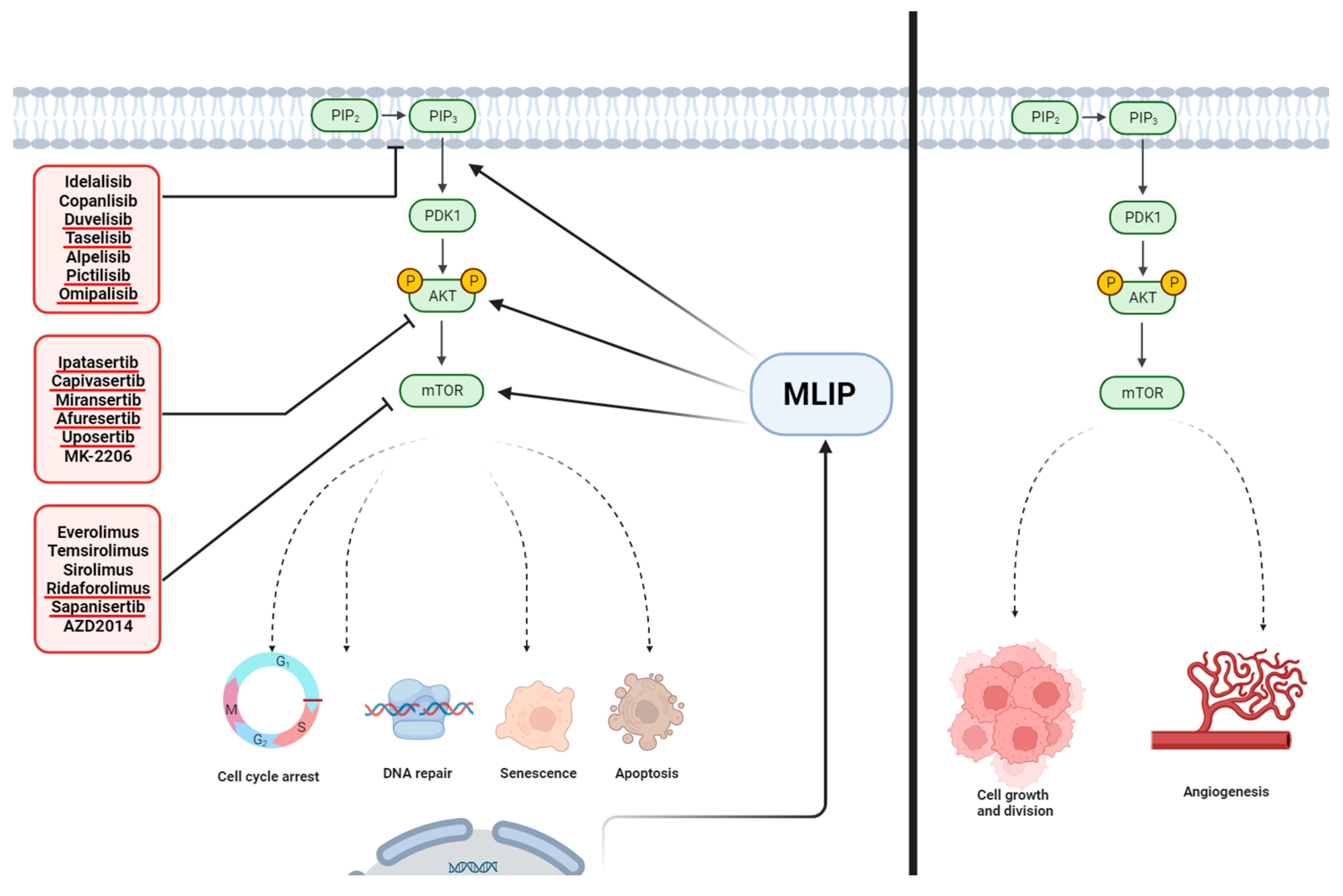
4.2. MLIP and the PI3K/Akt/mTOR Pathway
4.3. Role of MLIP in FOXO1 Signaling
4.4. MLIP and P53
4.5. MLIP and MAPK9 (Jak2)
5. Conclusions and MLIP as a Potential Therapeutic Target
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell Cycle Control in Cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. MTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/MTOR Signaling Pathway for Targeted Therapeutic Treatment in Human Cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling Integrators for Homeostasis Maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Patterson, C. The FOXO Family in Cardiac Function and Dysfunction. Annu. Rev. Physiol. 2010, 72, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. P53 and Metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Jose, E.; March-Steinman, W.; Wilson, B.A.; Shanks, L.; Paek, A.L. Temporal Coordination of the Transcription Factor Response to H2O2 Stress. Nat. Commun. 2024, 15, 3440. [Google Scholar] [CrossRef]
- Jȩśko, H.; Strosznajder, R.P. Sirtuins and Their Interactions with Transcription Factors and Poly(ADP-Ribose) Polymerases. Folia Neuropathol. 2016, 54, 212–233. [Google Scholar] [CrossRef]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor Suppressor Genes and ROS: Complex Networks of Interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of P53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Battiprolu, P.K.; Lopez-Crisosto, C.; Wang, Z.V.; Nemchenko, A.; Lavandero, S.; Hill, J.A. Diabetic Cardiomyopathy and Metabolic Remodeling of the Heart. Life Sci. 2013, 92, 609–615. [Google Scholar] [CrossRef] [PubMed]
- AbuEid, M.; McAllister, D.M.; McOlash, L.; Harwig, M.C.; Cheng, G.; Drouillard, D.; Boyle, K.A.; Hardy, M.; Zielonka, J.; Johnson, B.D.; et al. Synchronous Effects of Targeted Mitochondrial Complex I Inhibitors on Tumor and Immune Cells Abrogate Melanoma Progression. iScience 2021, 24, 102653. [Google Scholar] [CrossRef] [PubMed]
- Zečić, A.; Braeckman, B.P. DAF-16/FOXO in Caenorhabditis Elegans and Its Role in Metabolic Remodeling. Cells 2020, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Jiang, Z.; He, J.; Zhang, B.; Wang, L.; Long, C.; Zhao, B.; Yang, Y.; Du, L.; Luo, W.; Hu, J.; et al. A Potential “Anti-Warburg Effect” in Circulating Tumor Cell-Mediated Metastatic Progression? Aging Dis. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Chae, H.S.; Hong, S.T. Overview of Cancer Metabolism and Signaling Transduction. Int. J. Mol. Sci. 2023, 24, 12. [Google Scholar] [CrossRef]
- Farhan, M.; Silva, M.; Xingan, X.; Huang, Y.; Zheng, W. Role of FOXO Transcription Factors in Cancer Metabolism and Angiogenesis. Cells 2020, 9, 1586. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, Y.; Wei, W. Cell Cycle on the Crossroad of Tumorigenesis and Cancer Therapy. Trends Cell Biol. 2022, 32, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Tseng, L.M.; Lee, H.C. Role of Mitochondrial Alterations in Human Cancer Progression and Cancer Immunity. J. Biomed. Sci. 2023, 30, 61. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, E.; Blais, A.; Burgon, P.G. Muscle Enriched Lamin Interacting Protein (MLIP) Binds Chromatin and Is Required for Myoblast Differentiation. Cells 2021, 10, 615. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, E.; Deeke, S.A.; Rabaa, S.; Kouri, L.; Kenney, L.; Stewart, A.F.R.; Burgon, P.G. Identification of a Novel Muscle A-Type Lamin-Interacting Protein (MLIP). J. Biol. Chem. 2011, 286, 19702–19713. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.-E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (Muscle-Enriched A-Type Lamin-Interacting Protein) Leads to Cardiac Hyperactivation of Akt/Mammalian Target of Rapamycin (MTOR) and Impaired Cardiac Adaptation. J. Biol. Chem. 2015, 290, 26699–26714. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.-E.; Deeke, S.A.; Dick, S.A.; Verret-Borsos, Z.J.A.; Tennakoon, G.; Gupta, R.; Mak, E.; Roeske, C.L.; Weldrick, J.J.; Megeney, L.A.; et al. Expression of Murine Muscle-Enriched A-Type Lamin-Interacting Protein (MLIP) Is Regulated by Tissue-Specific Alternative Transcription Start Sites. J. Biol. Chem. 2018, 293, 19761–19770. [Google Scholar] [CrossRef] [PubMed]
- Lopes Abath Neto, O.; Medne, L.; Donkervoort, S.; Rodríguez-García, M.E.; Bolduc, V.; Hu, Y.; Guadagnin, E.; Foley, A.R.; Brandsema, J.F.; Glanzman, A.M.; et al. MLIP Causes Recessive Myopathy with Rhabdomyolysis, Myalgia and Baseline Elevated Serum Creatine Kinase. Brain 2021, 144, 2722–2731. [Google Scholar] [CrossRef]
- Huang, Z.-P.; Kataoka, M.; Chen, J.; Wu, G.; Ding, J.; Nie, M.; Lin, Z.; Liu, J.; Hu, X.; Ma, L.; et al. Cardiomyocyte-Enriched Protein CIP Protects against Pathophysiological Stresses and Regulates Cardiac Homeostasis. J. Clin. Investig. 2015, 125, 4122–4134. [Google Scholar] [CrossRef]
- Zhang, Y.; Tong, G.H.; Wei, X.X.; Chen, H.Y.; Liang, T.; Tang, H.P.; Wu, C.A.; Wen, G.M.; Yang, W.K.; Liang, L.; et al. Identification of Five Cytotoxicity-Related Genes Involved in the Progression of Triple-Negative Breast Cancer. Front. Genet. 2022, 12, 723477. [Google Scholar] [CrossRef]
- Burgon, P. Human Genome Organization—Symbolized Muscle-Enriched a-Type Lamin-Interacting Protein to Clear up Confusion. Circ. Res. 2012, 111, e252; author reply e253-4. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-P.; Young Seok, H.; Zhou, B.; Chen, J.J.-F.; Chen, J.J.-F.; Tao, Y.; Pu, W.T.; Wang, D.-Z. CIP, a Cardiac Isl1-Interacting Protein, Represses Cardiomyocyte Hypertrophy. Circ. Res. 2012, 110, 818–830. [Google Scholar] [CrossRef]
- Bullock, M. FOXO Factors and Breast Cancer: Outfoxing Endocrine Resistance. Endocr. Relat. Cancer 2016, 23, R113–R130. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, C.; Cheng, J. The Role and Molecular Mechanism of FOXO1 in Mediating Cardiac Hypertrophy. ESC Heart Fail. 2020, 7, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Helton, E.S.; Chen, X. P53 Modulation of the DNA Damage Response. J. Cell Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Shi, X.; Li, Y.; Sun, Y.; Zhao, X.; Sun, X.; Gong, T.; Liang, Z.; Ma, Y.; Zhang, X. Genome-Wide Analysis of LncRNAs, MiRNAs, and MRNAs Forming a Prognostic Scoring System in Esophageal Squamous Cell Carcinoma. PeerJ 2020, 2020, e8368. [Google Scholar] [CrossRef]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline Copy Number Variations Are Associated with Breast Cancer Risk and Prognosis. Sci. Rep. 2017, 7, 14621. [Google Scholar] [CrossRef]
- Breast Cancer Facts & Stats|Incidence, Age, Survival, & More. Available online: https://www.nationalbreastcancer.org/breast-cancer-facts/ (accessed on 26 October 2023).
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-Scale Genotyping Identifies 41 New Loci Associated with Breast Cancer Risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef]
- Fachal, L.; Dunning, A.M. From Candidate Gene Studies to GWAS and Post-GWAS Analyses in Breast Cancer. Curr. Opin. Genet. Dev. 2015, 30, 32–41. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as Well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.H.; Hardie, D.G. AMP-Activated Protein Kinase Is Activated by Low Glucose in Cell Lines Derived from Pancreatic Beta Cells, and May Regulate Insulin Release. Biochem. J. 1998, 335 Pt 3, 533–539. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef]
- Liong, S.; Lappas, M. Activation of AMPK Improves Inflammation and Insulin Resistance in Adipose Tissue and Skeletal Muscle from Pregnant Women. J. Physiol. Biochem. 2015, 71, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [PubMed]
- Keerthana, C.K.; Rayginia, T.P.; Shifana, S.C.; Anto, N.P.; Kalimuthu, K.; Isakov, N.; Anto, R.J. The Role of AMPK in Cancer Metabolism and Its Impact on the Immunomodulation of the Tumor Microenvironment. Front. Immunol. 2023, 14, 1114582. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a Metabolic Tumor Suppressor: Control of Metabolism and Cell Growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Zordoky, B.N.; Bujak, A.L.; Lally, J.S.V.; Fung, D.; Young, M.E.; Horman, S.; Miller, E.J.; Light, P.E.; Kemp, B.E.; et al. AMPK Deficiency in Cardiac Muscle Results in Dilated Cardiomyopathy in the Absence of Changes in Energy Metabolism. Cardiovasc. Res. 2015, 107, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/MTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/MTOR Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Polivka, J.; Janku, F. Molecular Targets for Cancer Therapy in the PI3K/AKT/MTOR Pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Bou Zeid, N.; Yazbeck, V. PI3k Inhibitors in NHL and CLL: An Unfulfilled Promise. Blood Lymphat. Cancer 2023, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Hassenrück, F.; Hallek, M. Copanlisib for Treatment of B-Cell Malignancies: The Development of a PI3K Inhibitor with Considerable Differences to Idelalisib. Drug Des. Devel Ther. 2018, 12, 2577–2590. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Danilov, A.V.; Pagel, J.M. Duvelisib for CLL/SLL and Follicular Non-Hodgkin Lymphoma. Blood 2019, 134, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Redman, M.W.; Wade, J.L.; Aggarwal, C.; Bradley, J.D.; Crawford, J.; Stella, P.J.; Knapp, M.H.; Miao, J.; Minichiello, K.; et al. SWOG S1400B (NCT02785913), a Phase II Study of GDC-0032 (Taselisib) for Previously Treated PI3K-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Sub-Study). J. Thorac. Oncol. 2019, 14, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Chia, S.; Kanakamedala, H.; Hsu, W.C.; Park, J.; Chandiwana, D.; Ridolfi, A.; Yu, C.L.; Zarate, J.P.; Rugo, H.S. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. Oncologist 2021, 26, e1133–e1142. [Google Scholar] [CrossRef] [PubMed]
- Bertho, M.; Patsouris, A.; Augereau, P.; Robert, M.; Frenel, J.S.; Blonz, C.; Campone, M. A Pharmacokinetic Evaluation of Alpelisib for the Treatment of HR+, HER2-Negative, PIK3CA-Mutated Advanced or Metastatic Breast Cancer. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan-Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.M.; Van Der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2016, 22, 1932–1939. [Google Scholar] [CrossRef]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Bang, Y.J.; Kang, Y.K.; Ng, M.; Chung, H.C.; Wainberg, Z.A.; Gendreau, S.; Chan, W.Y.; Xu, N.; Maslyar, D.; Meng, R.; et al. A Phase II, Randomised Study of MFOLFOX6 with or without the Akt Inhibitor Ipatasertib in Patients with Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer. Eur. J. Cancer 2019, 108, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef] [PubMed]
- Forde, K.; Resta, N.; Ranieri, C.; Rea, D.; Kubassova, O.; Hinton, M.; Andrews, K.A.; Semple, R.; Irvine, A.D.; Dvorakova, V. Clinical Experience with the AKT1 Inhibitor Miransertib in Two Children with PIK3CA-Related Overgrowth Syndrome. Orphanet J. Rare Dis. 2021, 16, 109. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I Study of the MEK Inhibitor Trametinib in Combination with the AKT Inhibitor Afuresertib in Patients with Solid Tumors and Multiple Myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Bell-McGuinn, K.M.; Burris, H.A.; Siu, L.L.; Stayner, L.A.; Wheler, J.J.; Hong, D.S.; Kurkjian, C.; Pant, S.; Santiago-Walker, A.; et al. A Phase I, Open-Label, Two-Stage Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Oral AKT Inhibitor GSK2141795 in Patients with Solid Tumors. Invest. New Drugs 2018, 36, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.P.; Konstantinopoulos, P.A.; Barry, W.T.; Luo, W.; Broaddus, R.R.; Makker, V.; Drapkin, R.; Liu, J.; Doyle, A.; Horowitz, N.S.; et al. Phase II, 2-Stage, 2-Arm, PIK3CA Mutation Stratified Trial of MK-2206 in Recurrent Endometrial Cancer. Int. J. Cancer 2020, 147, 413–422. [Google Scholar] [CrossRef]
- Besse, B.; Leighl, N.; Bennouna, J.; Papadimitrakopoulou, V.A.; Blais, N.; Traynor, A.M.; Soria, J.C.; Gogov, S.; Miller, N.; Jehl, V.; et al. Phase II Study of Everolimus-Erlotinib in Previously Treated Patients with Advanced Non-Small-Cell Lung Cancer. Ann. Oncol. 2014, 25, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Heist, R.S.; Papadmitrakopoulou, V.A.; Camidge, D.R.; Beck, J.T.; Schmid, P.; Mulatero, C.; Miller, N.; Dimitrijevic, S.; Urva, S.; et al. A Phase Ib Dose-Escalation Study of Everolimus Combined with Cisplatin and Etoposide as First-Line Therapy in Patients with Extensive-Stage Small-Cell Lung Cancer. Ann. Oncol. 2014, 25, 505–511. [Google Scholar] [CrossRef]
- Gandhi, L.; Bahleda, R.; Tolaney, S.M.; Kwak, E.L.; Cleary, J.M.; Pandya, S.S.; Hollebecque, A.; Abbas, R.; Ananthakrishnan, R.; Berkenblit, A.; et al. Phase I Study of Neratinib in Combination with Temsirolimus in Patients with Human Epidermal Growth Factor Receptor 2-Dependent and Other Solid Tumors. J. Clin. Oncol. 2014, 32, 68–75. [Google Scholar] [CrossRef]
- Moran, T.; Palmero, R.; Provencio, M.; Insa, A.; Majem, M.; Reguart, N.; Bosch-Barrera, J.; Isla, D.; Costa, E.C.; Lee, C.; et al. A Phase Ib Trial of Continuous Once-Daily Oral Afatinib plus Sirolimus in Patients with Epidermal Growth Factor Receptor Mutation-Positive Non-Small Cell Lung Cancer and/or Disease Progression Following Prior Erlotinib or Gefitinib. Lung Cancer 2017, 108, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.D.J.; Federico, S.M.; Aerts, I.; Hargrave, D.R.; DuBois, S.G.; Iannone, R.; Geschwindt, R.D.; Wang, R.; Haluska, F.G.; Trippett, T.M.; et al. A Phase 1 Study of Oral Ridaforolimus in Pediatric Patients with Advanced Solid Tumors. Oncotarget 2016, 7, 84736–84747. [Google Scholar] [CrossRef] [PubMed]
- García-Saenz, J.A.; Martínez-Jañez, N.; Cubedo, R.; Jerez, Y.; Lahuerta, A.; Gonzalez-Santiago, S.; Ferrer, N.; Ramos, M.; Alonso-Romero, J.L.; Anton, A.; et al. Sapanisertib plus Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive/HER2-Negative Advanced Breast Cancer after Progression on Aromatase Inhibitor. Clin. Cancer Res. 2022, 28, 1107–1116. [Google Scholar] [CrossRef]
- Heudel, P.; Frenel, J.S.; Dalban, C.; Bazan, F.; Joly, F.; Arnaud, A.; Abdeddaim, C.; Chevalier-Place, A.; Augereau, P.; Pautier, P.; et al. Safety and Efficacy of the MTOR Inhibitor, Vistusertib, Combined with Anastrozole in Patients with Hormone Receptor-Positive Recurrent or Metastatic Endometrial Cancer: The VICTORIA Multicenter, Open-Label, Phase 1/2 Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Wunderle, L.; Badura, S.; Schleyer, E.; Brüggemann, M.; Serve, H.; Schnittger, S.; Gökbuget, N.; Pfeifer, H.; Wagner, S.; et al. A Phase I Study of a Dual PI3-Kinase/MTOR Inhibitor BEZ235 in Adult Patients with Relapsed or Refractory Acute Leukemia. BMC Pharmacol. Toxicol. 2020, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D.; et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, versus Everolimus in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2016, 34, 1660–1668. [Google Scholar] [CrossRef]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/MTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.A.; Stayner, L.A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A Phase Ib Dose-Escalation Study of the MEK Inhibitor Trametinib in Combination with the PI3K/MTOR Inhibitor GSK2126458 in Patients with Advanced Solid Tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Alsina, M.; Soares, H.P.; Braña, I.; Britten, C.D.; Del Conte, G.; Ezeh, P.; Houk, B.; Kern, K.A.; Leong, S.; et al. A Multi-Arm Phase I Study of the PI3K/MTOR Inhibitors PF-04691502 and Gedatolisib (PF-05212384) plus Irinotecan or the MEK Inhibitor PD-0325901 in Advanced Cancer. Target. Oncol. 2017, 12, 775–785. [Google Scholar] [CrossRef]
- Morscher, R.J.; Brard, C.; Berlanga, P.; Marshall, L.V.; André, N.; Rubino, J.; Aerts, I.; De Carli, E.; Corradini, N.; Nebchi, S.; et al. First-in-Child Phase I/II Study of the Dual MTORC1/2 Inhibitor Vistusertib (AZD2014) as Monotherapy and in Combination with Topotecan-Temozolomide in Children with Advanced Malignancies: Arms E and F of the AcSé-ESMART Trial. Eur. J. Cancer 2021, 157, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Porta, C.; Suárez, C.; Hainsworth, J.; Voog, E.; Duran, I.; Reeves, J.; Czaykowski, P.; Castellano, D.; Chen, J.; et al. Randomized Phase II Trial of Sapanisertib ± TAK-117 vs. Everolimus in Patients with Advanced Renal Cell Carcinoma After VEGF-Targeted Therapy. Oncologist 2022, 27, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Starks, D.C.; Rojas-Espaillat, L.; Meissner, T.; Williams, C.B. Phase I Dose Escalation Study of Dual PI3K/MTOR Inhibition by Sapanisertib and Serabelisib in Combination with Paclitaxel in Patients with Advanced Solid Tumors. Gynecol. Oncol. 2022, 166, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, V.S.; Torres, F.F.; da Silva, D.G.H. FOXO3 and Oxidative Stress: A Multifaceted Role in Cellular Adaptation. J. Mol. Med. 2023, 101, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Tsitsipatis, D.; Klotz, L.O.; Steinbrenner, H. Multifaceted Functions of the Forkhead Box Transcription Factors FOXO1 and FOXO3 in Skin. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1057–1064. [Google Scholar] [CrossRef]
- Hosaka, T.; Biggs, W.H.; Tieu, D.; Boyer, A.D.; Varki, N.M.; Cavenee, W.K.; Arden, K.C. Disruption of Forkhead Transcription Factor (FOXO) Family Members in Mice Reveals Their Functional Diversification. Proc. Natl. Acad. Sci. USA 2004, 101, 2975–2980. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FOXO Transcription Factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Jiramongkol, Y.; Lam, E.W.F. FOXO Transcription Factor Family in Cancer and Metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Rodriguez-Colman, M.J.; Dansen, T.B.; Burgering, B.M.T. FOXO Transcription Factors as Mediators of Stress Adaptation. Nat. Rev. Mol. Cell Biol. 2023, 25, 46–64. [Google Scholar] [CrossRef]
- Freycon, C.; Lupo, P.J.; Witkowski, L.; Budd, C.; Foulkes, W.D.; Goudie, C. A Systematic Review of the Prevalence of Pathogenic or Likely Pathogenic Germline Variants in Individuals with FOXO1 Fusion-Positive Rhabdomyosarcoma. Pediatr. Blood Cancer 2023, 70, 30651. [Google Scholar] [CrossRef]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical Role of FOXO3a in Carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef]
- Habrowska-Górczyńska, D.E.; Kozieł, M.J.; Kowalska, K.; Piastowska-Ciesielska, A.W. FOXO3a and Its Regulators in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 12530. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. P53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Gnanasundram, S.V.; Bonczek, O.; Wang, L.; Chen, S.; Fahraeus, R. P53 MRNA Metabolism Links with the DNA Damage Response. Genes 2021, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, N.; Dornreiter, I.; Grosse, F.; Ella, K.; Wiesmüller, L.; Deppert, W. Maintenance of Genomic Integrity by P53: Complementary Roles for Activated and Non-Activated P53. Oncogene 1999, 18, 7706–7717. [Google Scholar] [CrossRef]
- Gatz, S.A.; Wiesmüller, L. P53 in Recombination and Repair. Cell Death Differ. 2006, 13, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting P53 for the Treatment of Cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. P53 as a Target for the Treatment of Cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Nagano, H.; Tanaka, T. The Role of Tumor Suppressor P53 in Metabolism and Energy Regulation, and Its Implication in Cancer and Lifestyle-Related Diseases. Endocr. J. 2019, 66, 485–496. [Google Scholar] [CrossRef]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic Functions of the P53 Tumor Suppressor: Regulation of Energy Metabolism and Antioxidant Defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant P53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell Biol. 2017, 37, e00328-17. [Google Scholar] [CrossRef]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type P53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef]
- Zhuang, W.; Lian, G.; Huang, B.; Du, A.; Gong, J.; Xiao, G.; Xu, C.; Wang, H.; Xie, L. CPT1 Regulates the Proliferation of Pulmonary Artery Smooth Muscle Cells through the AMPK-P53-P21 Pathway in Pulmonary Arterial Hypertension. Mol. Cell Biochem. 2019, 455, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Its Mutants in Cancer Metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A Reference Map of the Human Binary Protein Interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.S.; Meyer, S.C. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals 2022, 15, 160. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, S.A.; Girnius, N.; Vernia, S.; Davis, R.J. Role of the MAPK/CJun NH2-Terminal Kinase Signaling Pathway in Starvation-Induced Autophagy. Autophagy 2018, 14, 1586–1595. [Google Scholar] [CrossRef]
- Li, D.; You, Y.; Bi, F.F.; Zhang, T.N.; Jiao, J.; Wang, T.R.; Zhou, Y.M.; Shen, Z.Q.; Wang, X.X.; Yang, Q. Autophagy Is Activated in the Ovarian Tissue of Polycystic Ovary Syndrome. Reproduction 2018, 155, 85–92. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the Physiological Endometrium and Cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated with Patient Survival. Gastroenterology 2016, 151, 180–193.E12. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F.; Tossberg, J.T.; Fuchs, H.A.; Olsen, N.J.; Aune, T.M. Methotrexate Increases Expression of Cell Cycle Checkpoint Genes via JNK Activation. Arthritis Rheum. 2012, 64, 1780–1789. [Google Scholar] [CrossRef]
- Yan, Y.; Long, T.; Su, Q.; Wang, Y.; Chen, K.; Yang, T.; Zhao, G.; Ma, Q.; Hu, X.; Liu, C.; et al. Cardiac ISL1-Interacting Protein, a Cardioprotective Factor, Inhibits the Transition from Cardiac Hypertrophy to Heart Failure. Front. Cardiovasc. Med. 2022, 9, 857049. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Status | Reference |
|---|---|---|
| Breast cancer | 1 out of 6 genes associated with breast cancer risk and recurrence-free survival | [35] |
| Triple-negative breast cancer | Upregulated in and associated with patient survival in triple-negative breast cancer | [36] |
| Esophageal cancer | One of seven risk RNAs for esophageal cancer | [37] |
| Drug | Molecular Target | Status | Reference |
|---|---|---|---|
| Idelalisib (Zydelig) | PI3K | relapsed chronic lymphocytic leukemia (CLL), follicular lymphoma, and small lymphocytic lymphoma (SLL) | [55] |
| Copanlisib (Aliqopa) | PI3K | relapsed follicular lymphoma | [56] |
| Duvelisib (Copiktra) | PI3K | relapsed or refractory CLL, SLL, and follicular lymphoma | [57] |
| Taselisib (GDC-0032) | PI3K | investigational drug in clinical trials for various types of cancer, including breast cancer and lung cancer | [58] |
| Alpelisib (Piqray) | PI3K | HR-positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer | [59,60] |
| Pictilisib (GDC-0941) | PI3K | investigational drug in clinical trials for various types of cancer, including breast cancer and non-small cell lung cancer | [61] |
| Omipalisib (GSK2126458) | PI3K | investigational drug in clinical trials for various types of cancer, including melanoma and pancreatic cancer | [62] |
| Ipatasertib (GDC-0068) | AKT | investigational drug in clinical trials for various types of cancer, including breast cancer, prostate cancer, and ovarian cancer | [63,64] |
| Capivaserib (AZD5363) | AKT | investigational drug in clinical trials for various types of cancer, including breast cancer, prostate cancer, and non-small cell lung cancer | [65] |
| Miransertib (ARQ092) | AKT | investigational drug in clinical trials for various types of cancer, including endometrial cancer, solid tumors, and proteus syndrome | [66] |
| Afuresertib (GSK2110183) | AKT | investigational drug in clinical trials for multiple myeloma and other hematologic malignancies | [67] |
| Uprosertib (GSK2141795) | AKT | investigational drug in clinical trials for various types of cancer, including solid tumors and lymphomas | [68] |
| MK-2206 | AKT | investigational drug in clinical trials for various types of cancer, including breast cancer, colorectal cancer, and non-small cell lung cancer | [69] |
| Everolimus (Afinitor, Zortress) | mTOR | advanced renal cell carcinoma (RCC), progressive neuroendocrine tumors of pancreatic origin (PNET), advanced hormone receptor-positive, HER2-negative breast cancer, renal angiomyolipoma with tuberous sclerosis complex (TSC), and subependymal giant cell astrocytoma (SEGA) associated with TSC | [70,71] |
| Temsirolimus (Torisel) | mTOR | advanced renal cell carcinoma (RCC) | [72] |
| Sirolimus (Rapamune) | mTOR | potential anti-cancer properties in certain cancers, such as TSC-associated lymphangioleiomyomatosis (LAM) | [73] |
| Ridaforolimus (AP23573, MK-8669) | mTOR | investigational drug in clinical trials for various types of cancer, including sarcomas, endometrial cancer, and other solid tumors | [74] |
| Sapanisertib (INK128, TAK-228) | mTOR | investigational drug in clinical trials for various types of cancer, including breast cancer, renal cell carcinoma, and non-Hodgkin’s lymphoma | [75] |
| AZD2014 (Vistusertib) | mTOR | investigational drug in clinical trials for various types of cancer, including endometrial cancer, breast cancer, and non-small cell lung cancer | [76] |
| Dactolisib (BEZ235) | dual PI3K/mTOR | preclinical and early-phase clinical trials for various types of solid tumors, including breast, prostate, and renal cell carcinoma | [77] |
| Apitolisib (GDC-0980) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors, including colorectal, breast, and prostate cancer | [78,79] |
| Bimiralisib (PQR309) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and lymphomas | [80] |
| Omipalisib (GSK2126458) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies | [62,81] |
| Gedatolisib (PF-05212384) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies | [82] |
| Vistusertib (AZD2014) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies | [83] |
| Serabelisib (INK1117, MLN0128, TAK-228) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies | [84,85] |
| FOXO Type | Role in Cell Biology | Role in Cancer | Types of Tumors | References |
|---|---|---|---|---|
| FOXO1 | Regulation of gluconeogenesis, cell proliferation, apoptosis, metabolism, inflammation, differentiation, and stress resistance. Global deletion causes embryonic cell death due to incomplete vascular development. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Lymphoma, soft tissue sarcoma, acute myeloid leukemia (AML), breast cancer | [90] |
| FOXO2 | Involved in multiple important biological processes, such as cell cycle arrest, DNA repair, apoptosis, glucose metabolism, aging, and autophagy. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Not specified | [90] |
| FOXO3 | Affects lymph proliferation, widespread organ inflammation. Expression found in most tissues, including lymphocytes and myeloid cells. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Neuroblastoma, breast cancer, colorectal cancer, glioblastoma, pancreatic ductal adenocarcinoma | [90] |
| FOXO4 | Required for stem cell function in multiple tissues, including the maintenance of hematopoietic, neural, and muscle stem cell pools. | Tumor suppressor, regulation of cell cycle arrest and apoptosis | Not specified | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamwi, M.N.; Elsayed, E.; Dabash, H.; Abuawad, A.; Aweer, N.A.; Al Zeir, F.; Pedersen, S.; Al-Mansoori, L.; Burgon, P.G. MLIP and Its Potential Influence on Key Oncogenic Pathways. Cells 2024, 13, 1109. https://doi.org/10.3390/cells13131109
Hamwi MN, Elsayed E, Dabash H, Abuawad A, Aweer NA, Al Zeir F, Pedersen S, Al-Mansoori L, Burgon PG. MLIP and Its Potential Influence on Key Oncogenic Pathways. Cells. 2024; 13(13):1109. https://doi.org/10.3390/cells13131109
Chicago/Turabian StyleHamwi, Mahmoud N., Engy Elsayed, Hanan Dabash, Amani Abuawad, Noor A. Aweer, Faissal Al Zeir, Shona Pedersen, Layla Al-Mansoori, and Patrick G. Burgon. 2024. "MLIP and Its Potential Influence on Key Oncogenic Pathways" Cells 13, no. 13: 1109. https://doi.org/10.3390/cells13131109
APA StyleHamwi, M. N., Elsayed, E., Dabash, H., Abuawad, A., Aweer, N. A., Al Zeir, F., Pedersen, S., Al-Mansoori, L., & Burgon, P. G. (2024). MLIP and Its Potential Influence on Key Oncogenic Pathways. Cells, 13(13), 1109. https://doi.org/10.3390/cells13131109