
Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy?

.png)
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of Actin Polymerization
3. Actin Polymerization in the Nucleus
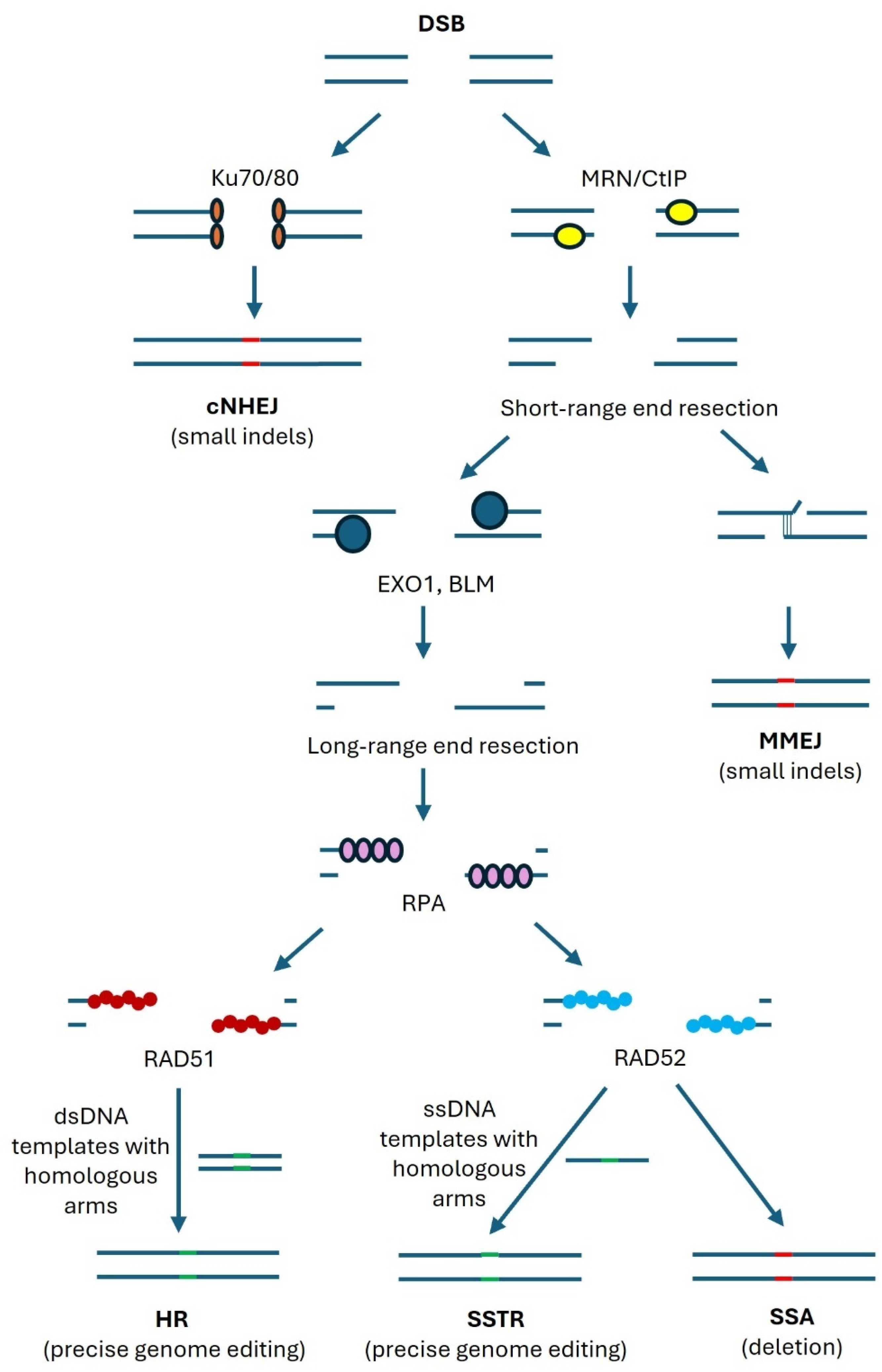
4. Repair Mechanisms of Double-Strand DNA Breaks
5. Methodological Challenges
6. Nuclear Actin Polymerization Promotes DNA Repair by HR
7. DSB Movement and Repair
8. Is Nuclear Actin Polymerization Required for Genome Maintenance?
9. Can Facilitated Actin Polymerization Promote Genome Editing by HR?
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tambuyzer, E.; Vandendriessche, B.; Austin, C.P.; Brooks, P.J.; Larsson, K.; Miller Needleman, K.I.; Valentine, J.; Davies, K.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapeutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef] [PubMed]
- Pacesa, M.; Pelea, O.; Jinek, M. Past, present, and future of CRISPR genome editing technologies. Cell 2024, 187, 1076–1100. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed]
- Picco, A.; Kukulski, W.; Manenschijn, H.E.; Specht, T.; Briggs, J.A.G.; Kaksonen, M. The contributions of the actin machinery to endocytic membrane bending and vesicle formation. Mol. Biol. Cell 2018, 29, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Ulferts, S.; Prajapati, B.; Grosse, R.; Vartiainen, M.K. Emerging Properties and Functions of Actin and Actin Filaments Inside the Nucleus. Cold Spring Harb. Perspect. Biol. 2021, 13, a040121. [Google Scholar] [CrossRef] [PubMed]
- Hurst, V.; Shimada, K.; Gasser, S.M. Nuclear Actin and Actin-Binding Proteins in DNA Repair. Trends Cell Biol. 2019, 29, 462–476. [Google Scholar] [CrossRef]
- Caridi, C.P.; Plessner, M.; Grosse, R.; Chiolo, I. Nuclear actin filaments in DNA repair dynamics. Nat. Cell Biol. 2019, 21, 1068–1077. [Google Scholar] [CrossRef]
- Le, S.; Yu, M.; Bershadsky, A.; Yan, J. Mechanical regulation of formin-dependent actin polymerization. Semin. Cell Dev. Biol. 2020, 102, 73–80. [Google Scholar] [CrossRef]
- Ding, B.; Narvaez-Ortiz, H.Y.; Singh, Y.; Hocky, G.M.; Chowdhury, S.; Nolen, B.J. Structure of Arp2/3 complex at a branched actin filament junction resolved by single-particle cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2022, 119, e2202723119. [Google Scholar] [CrossRef]
- Wang, J.; Nakamura, F. Identification of Filamin A Mechanobinding Partner II: Fimbacin Is a Novel Actin Cross-Linking and Filamin A Binding Protein. Biochemistry 2019, 58, 4737–4743. [Google Scholar] [CrossRef] [PubMed]
- Senger, F.; Pitaval, A.; Ennomani, H.; Kurzawa, L.; Blanchoin, L.; Théry, M. Spatial integration of mechanical forces by α-actinin establishes actin network symmetry. J. Cell Sci. 2019, 132, jcs236604. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Kudryashov, D.S.; Reisler, E. Actin Bundles Dynamics and Architecture. Biomolecules 2023, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Leech, G.; Rust, M.J.; Das, M.; McGorty, R.J.; Ross, J.L.; Robertson-Anderson, R.M. Myosin-driven actin-microtubule networks exhibit self-organized contractile dynamics. Sci. Adv. 2021, 7, eabe4334. [Google Scholar] [CrossRef]
- Dopie, J.; Skarp, K.P.; Rajakylä, E.K.; Tanhuanpää, K.; Vartiainen, M.K. Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E544–E552. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef]
- Bernard, O. Lim kinases, regulators of actin dynamics. Int. J. Biochem. Cell Biol. 2007, 39, 1071–1076. [Google Scholar] [CrossRef]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Baarlink, C.; Plessner, M.; Sherrard, A.; Morita, K.; Misu, S.; Virant, D.; Kleinschnitz, E.M.; Harniman, R.; Alibhai, D.; Baumeister, S.; et al. A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 2017, 19, 1389–1399. [Google Scholar] [CrossRef]
- Dopie, J.; Rajakylä, E.K.; Joensuu, M.S.; Huet, G.; Ferrantelli, E.; Xie, T.; Jäälinoja, H.; Jokitalo, E.; Vartiainen, M.K. Genome-wide RNAi screen for nuclear actin reveals a network of cofilin regulators. J. Cell Sci. 2015, 128, 2388–2400. [Google Scholar] [CrossRef]
- Stüven, T.; Hartmann, E.; Görlich, D. Exportin 6: A novel nuclear export receptor that is specific for profilin.actin complexes. EMBO J. 2003, 22, 5928–5940. [Google Scholar] [CrossRef]
- Belin, B.J.; Lee, T.; Mullins, R.D. Correction: DNA damage induces nuclear actin filament assembly by Formin-2 and Spire-1/2 that promotes efficient DNA repair. eLife 2015, 4, e11935. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Stüven, T.; Kuhn, C.; Cordes, V.C.; Görlich, D. A selective block of nuclear actin export stabilizes the giant nuclei of Xenopus oocytes. Nat. Cell Biol. 2006, 8, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef]
- Ceppi, I.; Howard, S.M.; Kasaciunaite, K.; Pinto, C.; Anand, R.; Seidel, R.; Cejka, P. CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. Proc. Natl. Acad. Sci. USA 2020, 117, 8859–8869. [Google Scholar] [CrossRef]
- Kamp, J.A.; Lemmens, B.; Romeijn, R.J.; Changoer, S.C.; van Schendel, R.; Tijsterman, M. Helicase Q promotes homology-driven DNA double-strand break repair and prevents tandem duplications. Nat. Commun. 2021, 12, 7126. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef]
- Volkova, N.V.; Meier, B.; González-Huici, V.; Bertolini, S.; Gonzalez, S.; Vöhringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 2169. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, N.; Wimberger, S.; Kühn, R.; Laugesen, A.; Turan, V.; Larsen, B.D.; Sørensen, C.S.; Helin, K.; Bennett, E.P.; Maresca, M.; et al. The methylation inhibitor 3DZNep promotes HDR pathway choice during CRISPR-Cas9 genome editing. Gene Genome Ed. 2023, 5, 100023. [Google Scholar] [CrossRef]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef]
- Melak, M.; Plessner, M.; Grosse, R. Actin visualization at a glance. J. Cell Sci. 2017, 130, 525–530. [Google Scholar] [CrossRef]
- Du, J.; Fan, Y.L.; Chen, T.L.; Feng, X.Q. Lifeact and Utr230 induce distinct actin assemblies in cell nuclei. Cytoskeleton 2015, 72, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.M.; De Silva, S.A.; Hayward, R.; Sek, K.; Ulferts, S.; Grosse, R.; Shanahan, C.M. Filamentous nuclear actin regulation of PML NBs during the DNA damage response is deregulated by prelamin A. Cell Death Dis. 2022, 13, 1042. [Google Scholar] [CrossRef]
- Palumbieri, M.D.; Merigliano, C.; González-Acosta, D.; Kuster, D.; Krietsch, J.; Stoy, H.; von Känel, T.; Ulferts, S.; Welter, B.; Frey, J.; et al. Nuclear actin polymerization rapidly mediates replication fork remodeling upon stress by limiting PrimPol activity. Nat. Commun. 2023, 14, 7819. [Google Scholar] [CrossRef] [PubMed]
- Torii, T.; Sugimoto, W.; Itoh, K.; Kinoshita, N.; Gessho, M.; Goto, T.; Uehara, I.; Nakajima, W.; Budirahardja, Y.; Miyoshi, D.; et al. Loss of p53 function promotes DNA damage-induced formation of nuclear actin filaments. Cell Death Dis. 2023, 14, 766. [Google Scholar] [CrossRef]
- Miné-Hattab, J.; Liu, S.; Taddei, A. Repair Foci as Liquid Phase Separation: Evidence and Limitations. Genes 2022, 13, 1846. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.J.; Ahn, Y.T.; Kim, Y.; Kim, J.M.; An, W.G. Actin disruption agents induce phosphorylation of histone H2AX in human breast adenocarcinoma MCF-7 cells. Oncol. Rep. 2011, 25, 1313–1319. [Google Scholar] [CrossRef]
- Andrin, C.; McDonald, D.; Attwood, K.M.; Rodrigue, A.; Ghosh, S.; Mirzayans, R.; Masson, J.Y.; Dellaire, G.; Hendzel, M.J. A requirement for polymerized actin in DNA double-strand break repair. Nucleus 2012, 3, 384–395. [Google Scholar] [CrossRef]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 2013, 340, 864–867. [Google Scholar] [CrossRef]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Lamm, N.; Read, M.N.; Nobis, M.; Van Ly, D.; Page, S.G.; Masamsetti, V.P.; Timpson, P.; Biro, M.; Cesare, A.J. Nuclear F-actin counteracts nuclear deformation and promotes fork repair during replication stress. Nat. Cell Biol. 2020, 22, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Wen, K.K.; García-Rubio, M.L.; Wold, M.S.; Aguilera, A.; Niedzwiedz, W.; Vyas, Y.M. WASp modulates RPA function on single-stranded DNA in response to replication stress and DNA damage. Nat. Commun. 2022, 13, 3743. [Google Scholar] [CrossRef] [PubMed]
- Nieminuszczy, J.; Martin, P.R.; Broderick, R.; Krwawicz, J.; Kanellou, A.; Mocanu, C.; Bousgouni, V.; Smith, C.; Wen, K.K.; Woodward, B.L.; et al. Actin nucleators safeguard replication forks by limiting nascent strand degradation. Nucleic Acids Res. 2023, 51, 6337–6354. [Google Scholar] [CrossRef]
- Mouilleron, S.; Guettler, S.; Langer, C.A.; Treisman, R.; McDonald, N.Q. Molecular basis for G-actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J. 2008, 27, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Marco, S.; Neilson, M.; Moore, M.; Perez-Garcia, A.; Hall, H.; Mitchell, L.; Lilla, S.; Blanco, G.R.; Hedley, A.; Zanivan, S.; et al. Nuclear-capture of endosomes depletes nuclear G-actin to promote SRF/MRTF activation and cancer cell invasion. Nat. Commun. 2021, 12, 6829. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Chen, M.; Winkler, D.D.; Luger, K.; Shen, X. Evidence for monomeric actin function in INO80 chromatin remodeling. Nat. Struct. Mol. Biol. 2013, 20, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.A.; Stojiljkovic, L.; Fuchsova, B.; Vargas, G.M.; Mavrommatis, E.; Philimonenko, V.; Kysela, K.; Goodrich, J.A.; Lessard, J.L.; Hope, T.J.; et al. Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 2004, 6, 1094–1101. [Google Scholar] [CrossRef]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef]
- Fréchard, A.; Faux, C.; Hexnerova, R.; Crucifix, C.; Papai, G.; Smirnova, E.; McKeon, C.; Ping, F.L.Y.; Helmlinger, D.; Schultz, P.; et al. The structure of the NuA4-Tip60 complex reveals the mechanism and importance of long-range chromatin modification. Nat. Struct. Mol. Biol. 2023, 30, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Lademann, C.A.; Renkawitz, J.; Pfander, B.; Jentsch, S. The INO80 Complex Removes H2A.Z to Promote Presynaptic Filament Formation during Homologous Recombination. Cell Rep. 2017, 19, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Lee, G.; Mullins, A.; Mody, P.; Watanabe, S.; Tan, D. DNA-translocation-independent role of INO80 remodeler in DNA damage repairs. J. Biol. Chem. 2023, 299, 105245. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Vaissiere, T.; Krastev, D.B.; Legube, G.; Anachkova, B.; Herceg, Z. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol. Cell. Biol. 2011, 31, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Lawrimore, J.; Lin, H.; Sanaullah, S.; Seitz, C.; Segall, D.; Kefer, P.; Salvador Moreno, N.; Lietz, B.; Anderson, R.; et al. DNA damage reduces heterogeneity and coherence of chromatin motions. Proc. Natl. Acad. Sci. USA 2022, 119, e2205166119. [Google Scholar] [CrossRef] [PubMed]
- García Fernández, F.; Fabre, E. The Dynamic Behavior of Chromatin in Response to DNA Double-Strand Breaks. Genes 2022, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Lawrimore, J.; Barry, T.M.; Barry, R.M.; York, A.C.; Friedman, B.; Cook, D.M.; Akialis, K.; Tyler, J.; Vasquez, P.; Yeh, E.; et al. Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol. Biol. Cell 2017, 28, 1701–1711. [Google Scholar] [CrossRef]
- Strecker, J.; Gupta, G.D.; Zhang, W.; Bashkurov, M.; Landry, M.C.; Pelletier, L.; Durocher, D. DNA damage signalling targets the kinetochore to promote chromatin mobility. Nat. Cell Biol. 2016, 18, 281–290. [Google Scholar] [CrossRef]
- Amaral, N.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef]
- García Fernández, F.; Almayrac, E.; Carré Simon, À.; Batrin, R.; Khalil, Y.; Boissac, M.; Fabre, E. Global chromatin mobility induced by a DSB is dictated by chromosomal conformation and defines the HR outcome. eLife 2022, 11, e78015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Vidi, P.A.; Lelièvre, S.A.; Irudayaraj, J.M. Nanoscale histone localization in live cells reveals reduced chromatin mobility in response to DNA damage. J. Cell Sci. 2015, 128, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef]
- Piazza, A.; Wright, W.D.; Heyer, W.D. Multi-invasions Are Recombination Byproducts that Induce Chromosomal Rearrangements. Cell 2017, 170, 760–773.e15. [Google Scholar] [CrossRef]
- Zagelbaum, J.; Schooley, A.; Zhao, J.; Schrank, B.R.; Callen, E.; Zha, S.; Gottesman, M.E.; Nussenzweig, A.; Rabadan, R.; Dekker, J.; et al. Author Correction: Multiscale reorganization of the genome following DNA damage facilitates chromosome translocations via nuclear actin polymerization. Nat. Struct. Mol. Biol. 2023, 30, 1048. [Google Scholar] [CrossRef]
- Li, H.; McCord, R.P. Actin up: Shifting chromosomes toward repair, but also translocations. Nat. Struct. Mol. Biol. 2023, 30, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef]
- Candotti, F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 2018, 38, 13–27. [Google Scholar] [CrossRef]
- Dubash, A.D.; Guilluy, C.; Srougi, M.C.; Boulter, E.; Burridge, K.; García-Mata, R. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS ONE 2011, 6, e17380. [Google Scholar] [CrossRef]
- Magalhaes, Y.T.; Boell, V.K.; Cardella, G.D.; Forti, F.L. Downregulation of the Rho GTPase pathway abrogates resistance to ionizing radiation in wild-type p53 glioblastoma by suppressing DNA repair mechanisms. Cell Death Dis. 2023, 14, 283. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Brakebusch, C. Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy? Cells 2024, 13, 1093. https://doi.org/10.3390/cells13131093
He X, Brakebusch C. Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy? Cells. 2024; 13(13):1093. https://doi.org/10.3390/cells13131093
Chicago/Turabian StyleHe, Xiubin, and Cord Brakebusch. 2024. "Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy?" Cells 13, no. 13: 1093. https://doi.org/10.3390/cells13131093
APA StyleHe, X., & Brakebusch, C. (2024). Regulation of Precise DNA Repair by Nuclear Actin Polymerization: A Chance for Improving Gene Therapy? Cells, 13(13), 1093. https://doi.org/10.3390/cells13131093