
Chronological and Biological Aging in Amyotrophic Lateral Sclerosis and the Potential of Senolytic Therapies

Abstract
1. Introduction
2. Chronological and Biological Aging in Disease and Neurodegeneration
3. Understanding ALS Incidence Rates in the Context of Aging and Neurological Decline

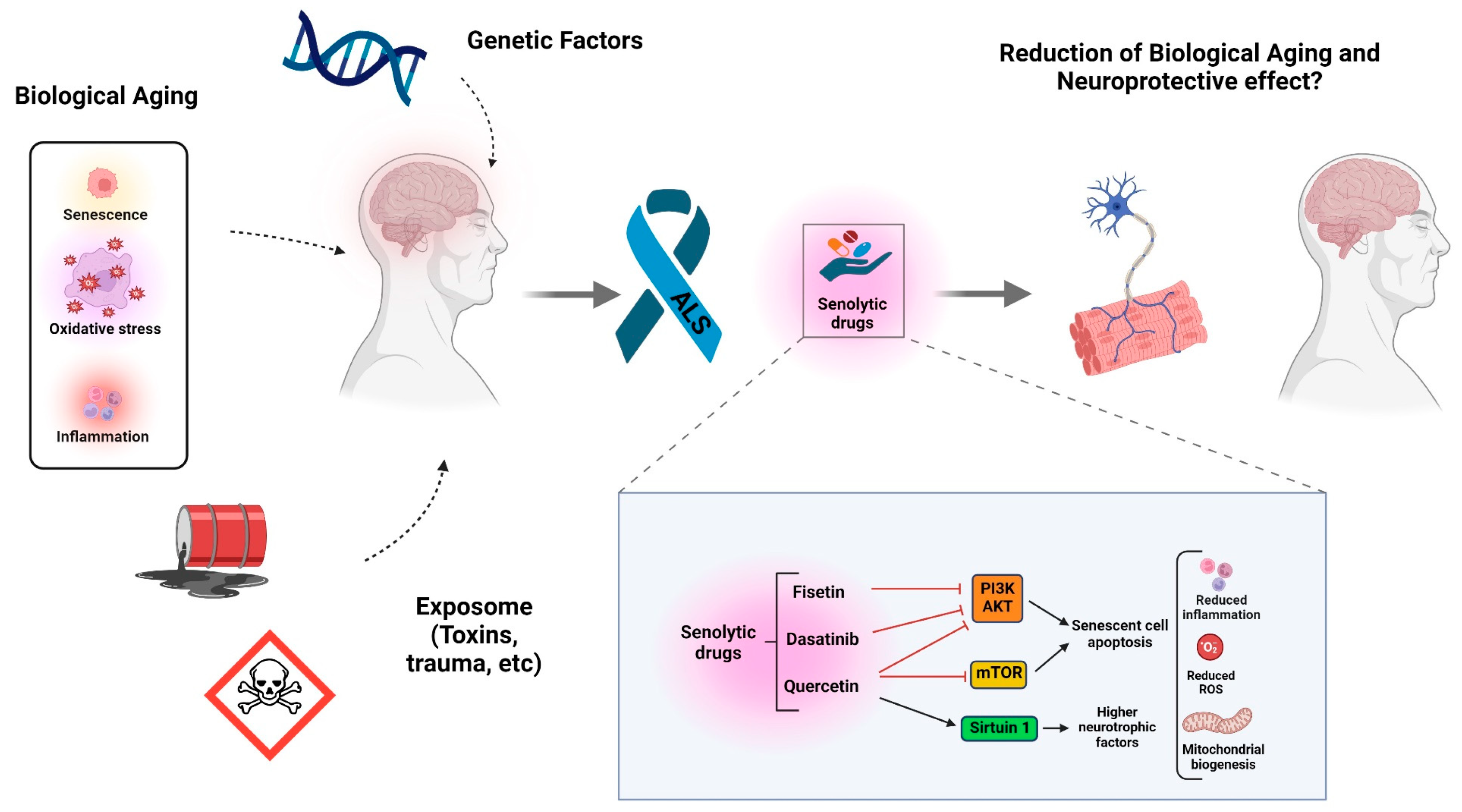
4. Hallmarks of Biological Aging and ALS

{kind=link}
{kind=link}
| Gene | Hallmarks of Aging | Evidence in ALS | References |
|---|---|---|---|
| C9orf72 TDP43 FUS SOD1 | Genome Instability | Patient, iPSC-derived motor neuron Patient, iPSC-derived motor neuron, SH-SY5Y cells Patients, iPSC-derived motor neuron, mouse model Patients, cell models and mouse models sALS patients | [53,54,74,75,76,77,78] [79,80,81,82] [83,84,85,86] [12,87,88,89,90,91,92,93] [81,93,94] |
| C9orf72 TDP43 FUS SOD1 | Epigenetic Alterations | Patient, iPSC-derived motor neuron Patient, SHSY5Y cell model Patients, EPSC- and iPSC-derived motor neuron, mouse, and yeast model sALS patients | [95,96,97] [57,98,99] [100,101,102] [97,103,104,105] [15,16,106,107,108,109] |
| C9orf72 SOD1 TDP43 | Inflammation | Patients, mouse, and cell model Patients, mouse, rat, and cell models Mouse model sALS patients, iPSC-derived astrocyte from sALS patients | [110,111,112,113] [66,111,113,114,115,116,117,118] [78] [119] |
| SOD1 C9orf72 TDP-43 FUS | Mitochondrial Dysfunction | Patients, mouse, and rat model Patients, iPSC-derived motor neurons PSC-derived motor neurons, fibroblast from patients, mouse model fALS tissue, primary neuron | [62,63,64,120,121,122,123] [75,124] [64,66,75] [125,126,127] |
| C9orf72 TDP43 FUS SOD1 | Loss of Proteostasis | Primary neurons, NSC-34 cells, mouse model Patients, mouse model, Zebrafish, Caenorhabditis elegans, Neuro2 cell model Patients, mouse model Patients, mouse model sALS | [128,129,130,131] [128,131,132,133,134,135] [128,131,136] [128,131,137] [138] |
| C9orf72 SOD1 | Cellular Senescence | Patients, iPSC-derived astrocyte from patients Patients, mouse, and rat models Myoblast from sALS patients sALS patients | [73,113] [139] [72] [71,73] |
5. Senescence-Associated Secretory Phenotype (SASP) and Senolytic Treatments
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, D.R.; Aslinia, F.; Yale, S.H.; Mazza, J.J. Jean-Martin Charcot: The father of neurology. Clin. Med. Res. 2011, 9, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Sohn, A.L.; Ping, L.; Glass, J.D.; Seyfried, N.T.; Hector, E.C.; Muddiman, D.C. Interrogating the Metabolomic Profile of Amyotrophic Lateral Sclerosis in the Post-Mortem Human Brain by Infrared Matrix-Assisted Laser Desorption Electrospray Ionization (IR-MALDESI) Mass Spectrometry Imaging (MSI). Metabolites 2022, 12, 1096. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.; Appel, S.; Baloh, R.H.; Barohn, R.; Brooks, B.R.; Elman, L.; Floeter, M.K.; Henderson, C.; Lomen-Hoerth, C.; Macklis, J.D.; et al. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14 (Suppl. S1), 5–18. [Google Scholar] [CrossRef] [PubMed]
- Schymick, J.C.; Talbot, K.; Traynor, B.J. Genetics of sporadic amyotrophic lateral sclerosis. Human. Mol. Genet. 2007, 16, R233–R242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, L.; Deng, M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front. Neurosci. 2023, 17, 1170996. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. Als genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 497022. [Google Scholar] [CrossRef]
- Pereira, G.R.C.; Vieira, B.A.A.; De Mesquita, J.F. Comprehensive in silico analysis and molecular dynamics of the superoxide dismutase 1 (SOD1) variants related to amyotrophic lateral sclerosis. PLoS ONE 2021, 16, e0247841. [Google Scholar] [CrossRef] [PubMed]
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 103. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Burley, S.K.; Zheng, X.F.S. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants 2022, 11, 427. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.; Atkin, J.D. DNA Damage, Defective DNA Repair, and Neurodegeneration in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 786420. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.V.; Singatulina, A.S.; Pastré, D.; Lavrik, O.I. FUSed in Sarcoma (FUS) in DNA Repair: Tango with Poly(ADP-ribose) Polymerase 1 and Compartmentalisation of Damaged DNA. Int. J. Mol. Sci. 2020, 21, 7020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xu, R. Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis. Front. Neurosci. 2023, 17, 1189470. [Google Scholar] [CrossRef] [PubMed]
- Hogden, A.; Foley, G.; Henderson, R.D.; James, N.; Aoun, S.M. Amyotrophic lateral sclerosis: Improving care with a multidisciplinary approach. J. Multidiscip. Healthc. 2017, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Tzeplaeff, L.; Wilfling, S.; Requardt, M.V.; Herdick, M. Current State and Future Directions in the Therapy of ALS. Cells 2023, 12, 1523. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Stringer, R.N.; Weiss, N. Pathophysiology of ion channels in amyotrophic lateral sclerosis. Mol. Brain 2023, 16, 82. [Google Scholar] [CrossRef]
- Pattee, G.L.; Genge, A.; Couratier, P.; Lunetta, C.; Sobue, G.; Aoki, M.; Yoshino, H.; Jackson, C.E.; Wymer, J.; Salah, A.; et al. Oral Edaravone—Introducing a Flexible Treatment Option for Amyotrophic Lateral Sclerosis. Expert. Rev. Neurother. 2023, 23, 859–866. [Google Scholar] [CrossRef]
- Castrillo-Viguera, C.; Grasso, D.L.; Simpson, E.; Shefner, J.; Cudkowicz, M.E. Clinical significance in the change of decline in ALSFRS-R. Amyotroph. Lateral Scler. 2010, 11, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Berry, J.D.; Ciepielewska, M.; Liu, Y.; Zambrano, G.S.; Zhang, J.; Hagan, M. Intravenous edaravone treatment in ALS and survival: An exploratory, retrospective, administrative claims analysis. EClinicalMedicine 2022, 52, 101590. [Google Scholar] [CrossRef]
- Dorst, J.; Genge, A. Clinical studies in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2022, 35, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef]
- NCOA. Get the Facts on Healthy Aging. Available online: https://www.ncoa.org/article/get-the-facts-on-healthy-aging (accessed on 23 May 2024).
- Xiang, D.; Hu, S.; Mai, T.; Zhang, X.; Zhang, L.; Wang, S.; Jin, K.; Huang, J. Worldwide cancer statistics of adults over 75 years old in 2019: A systematic analysis of the global burden of disease study 2019. BMC Public Health 2022, 22, 1979. [Google Scholar] [CrossRef]
- Pandey, A.; Kitzman, D.; Reeves, G. Frailty Is Intertwined with Heart Failure: Mechanisms, Prevalence, Prognosis, Assessment, and Management. JACC Heart Fail. 2019, 7, 1001–1011. [Google Scholar] [CrossRef]
- Laiteerapong, N.; Huang, E.S. Diabetes in Older Adults. In Diabetes in America; Cowie, C.C., Casagrande, S.S., Menke, A., Cissell, M.A., Eberhardt, M.S., Meigs, J.B., Gregg, E.W., Knowler, W.C., Barrett-Connor, E., Becker, D.J., et al., Eds.; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2018. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Tian, Y.E.; Cropley, V.; Maier, A.B.; Lautenschlager, N.T.; Breakspear, M.; Zalesky, A. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 2023, 29, 1221–1231. [Google Scholar] [CrossRef]
- Jazwinski, S.M.; Kim, S. Examination of the Dimensions of Biological Age. Front. Genet. 2019, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, L.; Sinclair, A.; Selman, C. Lifespan modulation in mice and the confounding effects of genetic background. J. Genet. Genom. 2014, 41, 497–503. [Google Scholar] [CrossRef]
- Willows, J.W.; Alshahal, Z.; Story, N.M.; Alves, M.J.; Vidal, P.; Harris, H.; Rodrigo, R.; Stanford, K.I.; Peng, J.; Reifsnyder, P.C.; et al. Contributions of mouse genetic strain background to age-related phenotypes in physically active HET3 mice. Neurobiol. Aging 2024, 136, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.; Verjovski-Almeida, S.; Zatz, M. Phenotypic heterogeneity in amyotrophic lateral sclerosis type 8 and modifying mechanisms of neurodegeneration. Neural Regen. Res. 2021, 16, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.A.; Arthur, K.C.; Tienari, P.J.; Houlden, H.; Chiò, A.; Traynor, B.J. Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 2017, 7, 2116. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.C. Neuromuscular Changes with Aging and Sarcopenia. J. Frailty Aging 2019, 8, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.C.; Carson, R.G. Sarcopenia and Neuroscience: Learning to Communicate. J. Gerontol. Ser. A 2021, 76, 1882–1890. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Bohannon, R.W.; Li, X.; Sindhu, B.; Kapellusch, J. Hand-Grip Strength: Normative Reference Values and Equations for Individuals 18 to 85 Years of Age Residing in the United States. J. Orthop. Sports Phys. Ther. 2018, 48, 685–693. [Google Scholar] [CrossRef]
- Mehta, P.; Raymond, J.; Punjani, R.; Larson, T.; Han, M.; Bove, F.; Horton, D.K. Incidence of amyotrophic lateral sclerosis in the United States, 2014–2016. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 378–382. [Google Scholar] [CrossRef]
- Doherty, T.J.; Vandervoort, A.A.; Brown, W.F. Effects of ageing on the motor unit: A brief review. Can. J. Appl. Physiol. 1993, 18, 331–358. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; McComas, A.J.; Petito, F. Physiological changes in ageing muscles. J. Neurol. Neurosurg. Psychiatry 1973, 36, 174–182. [Google Scholar] [CrossRef]
- Vucic, S.; van den Bos, M.; Menon, P.; Howells, J.; Dharmadasa, T.; Kiernan, M.C. Utility of threshold tracking transcranial magnetic stimulation in ALS. Clin. Neurophysiol. Pract. 2018, 3, 164–172. [Google Scholar] [CrossRef]
- Oliviero, A.; Profice, P.; Tonali, P.A.; Pilato, F.; Saturno, E.; Dileone, M.; Ranieri, F.; Di Lazzaro, V. Effects of aging on motor cortex excitability. Neurosci. Res. 2006, 55, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.C.; Taylor, J.L. Age-related changes in motor cortical properties and voluntary activation of skeletal muscle. Curr. Aging Sci. 2011, 4, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Kukull, W.A.; Higdon, R.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Schellenberg, G.D.; van Belle, G.; Jolley, L.; Larson, E.B. Dementia and Alzheimer disease incidence: A prospective cohort study. Arch. Neurol. 2002, 59, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Manfellotto, F.; D’Alessandro, R.; Marino, R.; Borra, M.; Biffali, E. Telomerase expression in amyotrophic lateral sclerosis (ALS) patients. J. Hum. Genet. 2014, 59, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Linkus, B.; Wiesner, D.; Meßner, M.; Karabatsiakis, A.; Scheffold, A.; Rudolph, K.L.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Danzer, K.M. Telomere shortening leads to earlier age of onset in ALS mice. Aging 2016, 8, 382–393. [Google Scholar] [CrossRef]
- Masala, A.; Sanna, S.; Esposito, S.; Rassu, M.; Galioto, M.; Zinellu, A.; Carru, C.; Carrì, M.T.; Iaccarino, C.; Crosio, C. Epigenetic Changes Associated with the Expression of Amyotrophic Lateral Sclerosis (ALS) Causing Genes. Neuroscience 2018, 390, 1–11. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Hegde, M.L. A Commentary on TDP-43 and DNA Damage Response in Amyotrophic Lateral Sclerosis. J. Exp. Neurosci. 2019, 13, 1179069519880166. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, W.; Perry, G.; Zhu, X.; Wang, X. Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.M.; Yen, Y.H.; Yuan, F.; Zhang, S.C.; Chong, C.M. Neuronal Senescence in the Aged Brain. Aging Dis. 2023, 14, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Neault, M.; Couteau, F.; Bonneau, É.; De Guire, V.; Mallette, F.A. Molecular Regulation of Cellular Senescence by MicroRNAs: Implications in Cancer and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2017, 334, 27–98. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, O.; Schroth, J.; Tree, T.; Turner, M.R.; Shaw, P.J.; Henson, S.M.; Malaspina, A. Senescent-like Blood Lymphocytes and Disease Progression in Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200042. [Google Scholar] [CrossRef]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombes, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar] [CrossRef]
- Vazquez-Villasenor, I.; Garwood, C.J.; Heath, P.R.; Simpson, J.E.; Ince, P.G.; Wharton, S.B. Expression of p16 and p21 in the frontal association cortex of ALS/MND brains suggests neuronal cell cycle dysregulation and astrocyte senescence in early stages of the disease. Neuropathol. Appl. Neurobiol. 2020, 46, 171–185. [Google Scholar] [CrossRef]
- Konopka, A.; Atkin, J.D. The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 3137. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Talbot, K. The Role of Mitochondrial Dysfunction and ER Stress in TDP-43 and C9ORF72 ALS. Front. Cell Neurosci. 2021, 15, 653688. [Google Scholar] [CrossRef]
- Walker, C.; Herranz-Martin, S.; Karyka, E.; Liao, C.; Lewis, K.; Elsayed, W.; Lukashchuk, V.; Chiang, S.C.; Ray, S.; Mulcahy, P.J.; et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 2017, 20, 1225–1235. [Google Scholar] [CrossRef]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.M.; Ke, Y.D. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 2021, 22, 7781. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, M.S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 2020, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, E.N.; Guerrero, E.N.; Mitra, J.; Wang, H.; Rangaswamy, S.; Hegde, P.M.; Basu, P.; Rao, K.S.J.; Hegde, M.L.; Hegde, M.L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 8, 2459–2476. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Castanedo-Vázquez, D.; Gil-Bea, F.; Tapia, O.; Arozamena, J.; Durán-Vían, C.; Sedano, M.J.; Berciano, M.T.; Lopez de Munain, A.; Lafarga, M. ALS-derived fibroblasts exhibit reduced proliferation rate, cytoplasmic TDP-43 aggregation and a higher susceptibility to DNA damage. J. Neurol. 2020, 267, 1291–1299. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Higelin, J.; Demestre, M.; Putz, S.; Delling, J.P.; Jacob, C.; Lutz, A.K.; Bausinger, J.; Huber, A.K.; Klingenstein, M.; Barbi, G.; et al. FUS Mislocalization and Vulnerability to DNA Damage in ALS Patients Derived hiPSCs and Aging Motoneurons. Front. Cell Neurosci. 2016, 10, 290. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Wang, X.D.; Zhu, M.W.; Shan, D.; Wang, S.Y.; Yin, X.; Yang, Y.Q.; Wang, T.H.; Zhang, C.T.; Wang, Y.; Liang, W.W.; et al. Spy1, a unique cell cycle regulator, alters viability in ALS motor neurons and cell lines in response to mutant SOD1-induced DNA damage. DNA Repair 2019, 74, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, Y.; Liang, W.; Wang, T.; Wang, S.; Wang, X.; Wang, Y.; Jiang, H.; Feng, H. Neuroprotection by urate on the mutant hSOD1-related cellular and Drosophila models of amyotrophic lateral sclerosis: Implication for GSH synthesis via activating Akt/GSK3β/Nrf2/GCLC pathways. Brain Res. Bull. 2019, 146, 287–301. [Google Scholar] [CrossRef]
- Warita, H.; Hayashi, T.; Murakami, T.; Manabe, Y.; Abe, K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res. Mol. Brain Res. 2001, 89, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, N.; Beal, M.F.; Matson, W.R.; Bogdanov, M.B. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic. Res. 2005, 39, 383–388. [Google Scholar] [CrossRef]
- Li, J.; Song, M.; Moh, S.; Kim, H.; Kim, D.H. Cytoplasmic Restriction of Mutated SOD1 Impairs the DNA Repair Process in Spinal Cord Neurons. Cells 2019, 8, 1502. [Google Scholar] [CrossRef]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef]
- Kok, J.R.; Palminha, N.M.; Dos Santos Souza, C.; El-Khamisy, S.F.; Ferraiuolo, L. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell Mol. Life Sci. 2021, 78, 5707–5729. [Google Scholar] [CrossRef] [PubMed]
- Wald-Altman, S.; Pichinuk, E.; Kakhlon, O.; Weil, M. A differential autophagy-dependent response to DNA double-strand breaks in bone marrow mesenchymal stem cells from sporadic ALS patients. Dis. Model. Mech. 2017, 10, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaiswal, M.K.; Chien, J.-F.; Kozlenkov, A.; Jung, J.; Zhou, P.; Gardashli, M.; Pregent, L.J.; Engelberg-Cook, E.; Dickson, D.W.; et al. Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat. Commun. 2023, 14, 5714. [Google Scholar] [CrossRef] [PubMed]
- Yazar, V.; Kühlwein, J.K.; Knehr, A.; Grozdanov, V.; Ekici, A.B.; Ludolph, A.C.; Danzer, K.M. Impaired ATF3 signaling involves SNAP25 in SOD1 mutant ALS patients. Sci. Rep. 2023, 13, 12019. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Sugai, A.; Hara, N.; Ito, J.; Yokoseki, A.; Ishihara, T.; Yamagishi, T.; Tsuboguchi, S.; Tada, M.; Ikeuchi, T.; et al. Age-related demethylation of the TDP-43 autoregulatory region in the human motor cortex. Commun. Biol. 2021, 4, 1107. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, J.; Spalloni, A.; Cappelli, S.; Ciaiola, F.; Orlando, V.; Buratti, E.; Longone, P. TDP-43 Epigenetic Facets and Their Neurodegenerative Implications. Int. J. Mol. Sci. 2023, 24, 13807. [Google Scholar] [CrossRef] [PubMed]
- Capauto, D.; Colantoni, A.; Lu, L.; Santini, T.; Peruzzi, G.; Biscarini, S.; Morlando, M.; Shneider, N.A.; Caffarelli, E.; Laneve, P.; et al. A Regulatory Circuitry Between Gria2, miR-409, and miR-495 Is Affected by ALS FUS Mutation in ESC-Derived Motor Neurons. Mol. Neurobiol. 2018, 55, 7635–7651. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef]
- Nguyen, S.; Meletis, K.; Fu, D.; Jhaveri, S.; Jaenisch, R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: A role in the regulation of NFL mRNA levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.; Schmalbach, S.; Boeselt, S.; Sarlette, A.; Dengler, R.; Petri, S. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 573–581. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef] [PubMed]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, J.M.; Smit-Oistad, I.M.; Macrander, C.; Krakora, D.; Meyer, M.G.; Suzuki, M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp. Neurol. 2016, 277, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.P.; Guarnieri, P.; Friedman, B.A.; Carrasco, M.A.; Muratet, M.; O’Keeffe, S.; Nwakeze, C.; Pauli-Behn, F.; Newberry, K.M.; Meadows, S.K.; et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA 2013, 110, E756–E765. [Google Scholar] [CrossRef] [PubMed]
- Migliarini, S.; Scaricamazza, S.; Valle, C.; Ferri, A.; Pasqualetti, M.; Ferraro, E. Microglia Morphological Changes in the Motor Cortex of hSOD1(G93A) Transgenic ALS Mice. Brain Sci. 2021, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Graber, D.J.; Hickey, W.F.; Harris, B.T. Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2010, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Trolese, M.C.; Scarpa, C.; Melfi, V.; Fabbrizio, P.; Sironi, F.; Rossi, M.; Bendotti, C.; Nardo, G. Boosting the peripheral immune response in the skeletal muscles improved motor function in ALS transgenic mice. Mol. Ther. 2022, 30, 2760–2784. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Amponsah, A.E.; Guo, R.; Liu, X.; Zhang, J.; Du, X.; Zhou, Z.; He, J.; Ma, J.; Cui, H. Autophagy-Mediated Inflammatory Cytokine Secretion in Sporadic ALS Patient iPSC-Derived Astrocytes. Oxid. Med. Cell Longev. 2022, 2022, 6483582. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Li, Q.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.-Q.; Chun, S.-J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes. Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [PubMed]
- Salam, S.; Tacconelli, S.; Smith, B.N.; Mitchell, J.C.; Glennon, E.; Nikolaou, N.; Houart, C.; Vance, C. Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 2021, 11, 13613. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Shaw, P.J.; De Vos, K.J. Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front. Mol. Neurosci. 2017, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.; Cabral-Miranda, F.; Gonzalez-Teuber, V.; Hetz, C. Proteostasis deregulation as a driver of C9ORF72 pathogenesis. J. Neurochem. 2021, 159, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Diab, R.; Pilotto, F.; Saxena, S. Autophagy and neurodegeneration: Unraveling the role of C9ORF72 in the regulation of autophagy and its relationship to ALS-FTD pathology. Front. Cell Neurosci. 2023, 17, 1086895. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 2006, 17, 4606–4618. [Google Scholar] [CrossRef]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef]
- Kato, T.; Katagiri, T.; Hirano, A.; Kawanami, T.; Sasaki, H. Lewy body-like hyaline inclusions in sporadic motor neuron disease are ubiquitinated. Acta Neuropathol. 1989, 77, 391–396. [Google Scholar] [CrossRef]
- Trias, E.; Beilby, P.R.; Kovacs, M.; Ibarburu, S.; Varela, V.; Barreto-Nunez, R.; Bradford, S.C.; Beckman, J.S.; Barbeito, L. Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS. Front. Aging Neurosci. 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes. Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-κB pathway activators as potential ageing biomarkers: Targets for new therapeutic strategies. Immun. Ageing 2013, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Das, M.M.; Svendsen, C.N. Astrocytes show reduced support of motor neurons with aging that is accelerated in a rodent model of ALS. Neurobiol. Aging 2015, 36, 1130–1139. [Google Scholar] [CrossRef]
- Torres, P.; Pamplona, R.; Portero-Otin, M. Cell senescence, loss of splicing, and lipid metabolism in TDP-43-related neurodegenerative processes. Neural. Regen. Res. 2023, 18, 1725–1726. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front. Cell. Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- McCombe, P.A.; Henderson, R.D. The Role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Schade, A.E.; Schieven, G.L.; Townsend, R.; Jankowska, A.M.; Susulic, V.; Zhang, R.; Szpurka, H.; Maciejewski, J.P. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008, 111, 1366–1377. [Google Scholar] [CrossRef]
- Bruning, A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anticancer Agents Med. Chem. 2013, 13, 1025–1031. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Smith, J.J.; Revetta, F.; Washington, M.K.; Merchant, N.B. Targeted inhibition of SRC kinase signaling attenuates pancreatic tumorigenesis. Mol. Cancer Ther. 2010, 9, 2322–2332. [Google Scholar] [CrossRef]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: Possible combinations in solid tumors. Clin. Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef]
- Zhu, M.; Meng, P.; Ling, X.; Zhou, L. Advancements in therapeutic drugs targeting of senescence. Ther. Adv. Chronic Dis. 2020, 11, 2040622320964125. [Google Scholar] [CrossRef]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1071–R1077. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Liu, Z.; Wang, S.; Sun, S.; Ma, S.; Liu, X.; Chan, P.; Sun, L.; Song, M.; Zhang, W.; et al. Low-dose quercetin positively regulates mouse healthspan. Protein Cell 2019, 10, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, F.J.; Yang, W.L.; Qiao, H.Z.; Zhang, S.J. Quercetin improves cognitive disorder in aging mice by inhibiting NLRP3 inflammasome activation. Food Funct. 2021, 12, 717–725. [Google Scholar] [CrossRef]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Cruz, M.P. Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. Pharm. Ther. 2018, 43, 25–28. [Google Scholar]
- Sala, G.; Arosio, A.; Conti, E.; Beretta, S.; Lunetta, C.; Riva, N.; Ferrarese, C.; Tremolizzo, L. Riluzole Selective Antioxidant Effects in Cell Models Expressing Amyotrophic Lateral Sclerosis Endophenotypes. Clin. Psychopharmacol. Neurosci. 2019, 17, 438–442. [Google Scholar] [CrossRef]
- Gonzales, M.M.; Garbarino, V.R.; Kautz, T.F.; Palavicini, J.P.; Lopez-Cruzan, M.; Dehkordi, S.K.; Mathews, J.J.; Zare, H.; Xu, P.; Zhang, B.; et al. Senolytic therapy in mild Alzheimer’s disease: A phase 1 feasibility trial. Nat. Med. 2023, 29, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Grosso, G.; Nieves, J.W.; Zanghì, A.; Factor-Litvak, P.; Mitsumoto, H. Metabolic Abnormalities, Dietary Risk Factors and Nutritional Management in Amyotrophic Lateral Sclerosis. Nutrients 2021, 13, 2273. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, I.; Shukitt-Hale, B.; Joseph, J.A. Neurobehavioral aspects of antioxidants in aging. Int. J. Dev. Neurosci. 2000, 18, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, A.; Ziemichód, W.; Herbet, M.; Piątkowska-Chmiel, I. The Role of Diet as a Modulator of the Inflammatory Process in the Neurological Diseases. Nutrients 2023, 15, 1436. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.P.; Hamadeh, M.J. Nutritional and exercise-based interventions in the treatment of amyotrophic lateral sclerosis. Clin. Nutr. 2009, 28, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, Y.; Xuan, R.; Huang, J.; István, B.; Fekete, G.; Gu, Y. Mixed Comparison of Different Exercise Interventions for Function, Respiratory, Fatigue, and Quality of Life in Adults with Amyotrophic Lateral Sclerosis: Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2022, 14, 919059. [Google Scholar] [CrossRef] [PubMed]
- Polsky, L.R.; Rentscher, K.E.; Carroll, J.E. Stress-induced biological aging: A review and guide for research priorities. Brain Behav. Immun. 2022, 104, 97–109. [Google Scholar] [CrossRef]
- Su, F.C.; Goutman, S.A.; Chernyak, S.; Mukherjee, B.; Callaghan, B.C.; Batterman, S.; Feldman, E.L. Association of Environmental Toxins with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 803–811. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dashtmian, A.R.; Darvishi, F.B.; Arnold, W.D. Chronological and Biological Aging in Amyotrophic Lateral Sclerosis and the Potential of Senolytic Therapies. Cells 2024, 13, 928. https://doi.org/10.3390/cells13110928
Dashtmian AR, Darvishi FB, Arnold WD. Chronological and Biological Aging in Amyotrophic Lateral Sclerosis and the Potential of Senolytic Therapies. Cells. 2024; 13(11):928. https://doi.org/10.3390/cells13110928
Chicago/Turabian StyleDashtmian, Anna Roshani, Fereshteh B. Darvishi, and William David Arnold. 2024. "Chronological and Biological Aging in Amyotrophic Lateral Sclerosis and the Potential of Senolytic Therapies" Cells 13, no. 11: 928. https://doi.org/10.3390/cells13110928
APA StyleDashtmian, A. R., Darvishi, F. B., & Arnold, W. D. (2024). Chronological and Biological Aging in Amyotrophic Lateral Sclerosis and the Potential of Senolytic Therapies. Cells, 13(11), 928. https://doi.org/10.3390/cells13110928