
Reprogramming Glioblastoma Cells into Non-Cancerous Neuronal Cells as a Novel Anti-Cancer Strategy


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods and Materials
2.1. GBM Cell Lines
2.2. GBM Cell Cultures
2.3. Viral Transduction of GBM Cell Line Cultures and Generation of AAV Vector Packages
2.4. Western Blotting Analysis
2.5. MTT Assay
2.6. Wound-Healing Migration Assay
2.7. Immunoflourescent Staining In Vitro
2.8. Immunoflourescent Staining Ex Vivo
2.9. TUNEL Assay
2.10. Orthotopical GBM Mouse Model
2.11. H&E Staining of Ex Vivo Histology
2.12. Statistical Analysis
3. Results
3.1. Expression of ND1 in GBM Cells and Neuronal Reprogramming
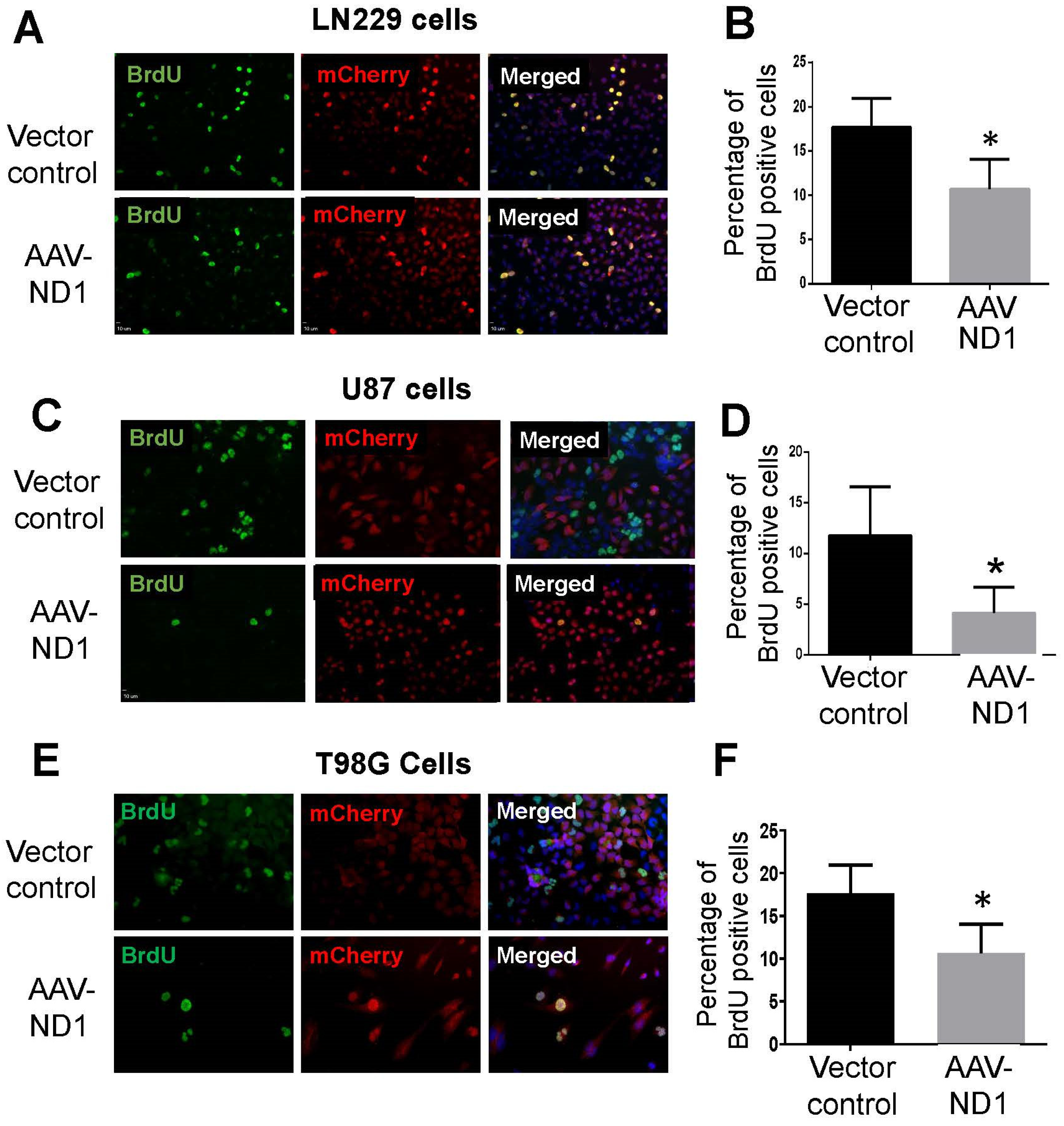
3.2. Decreased Proliferation of GBM Cells after ND1 Reprogramming
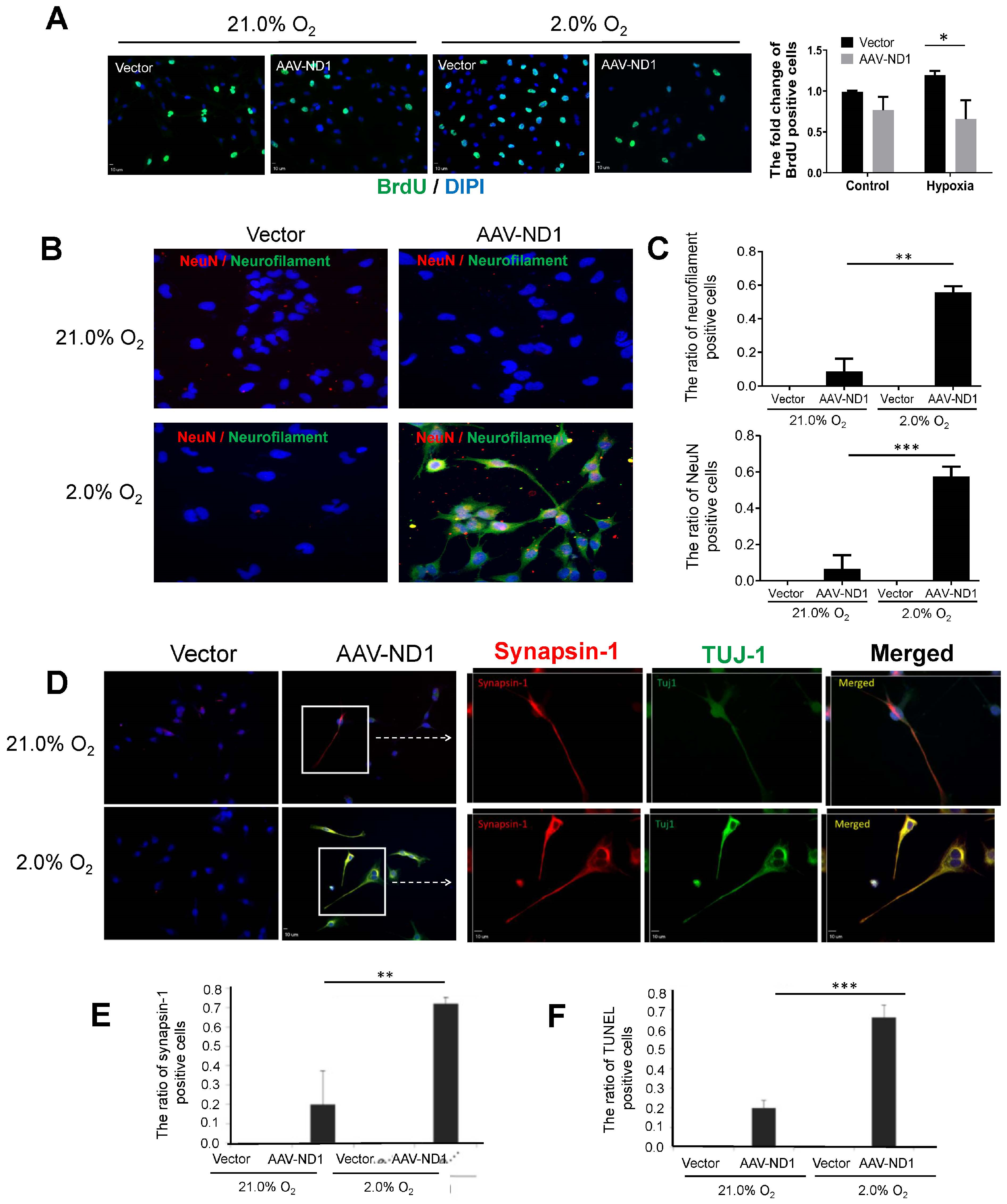
3.3. Promoting Effect of Hypoxia on ND1-Induced Neuronal Conversion of GBM Cells
3.4. Attenuated Cell Migration of ND1-Reprogrammed Cells
3.5. Regulatory Roles of the Wnt-3 Pathway in ND1-Induced Reprogramming and Neuronal Conversion of GBM Cells
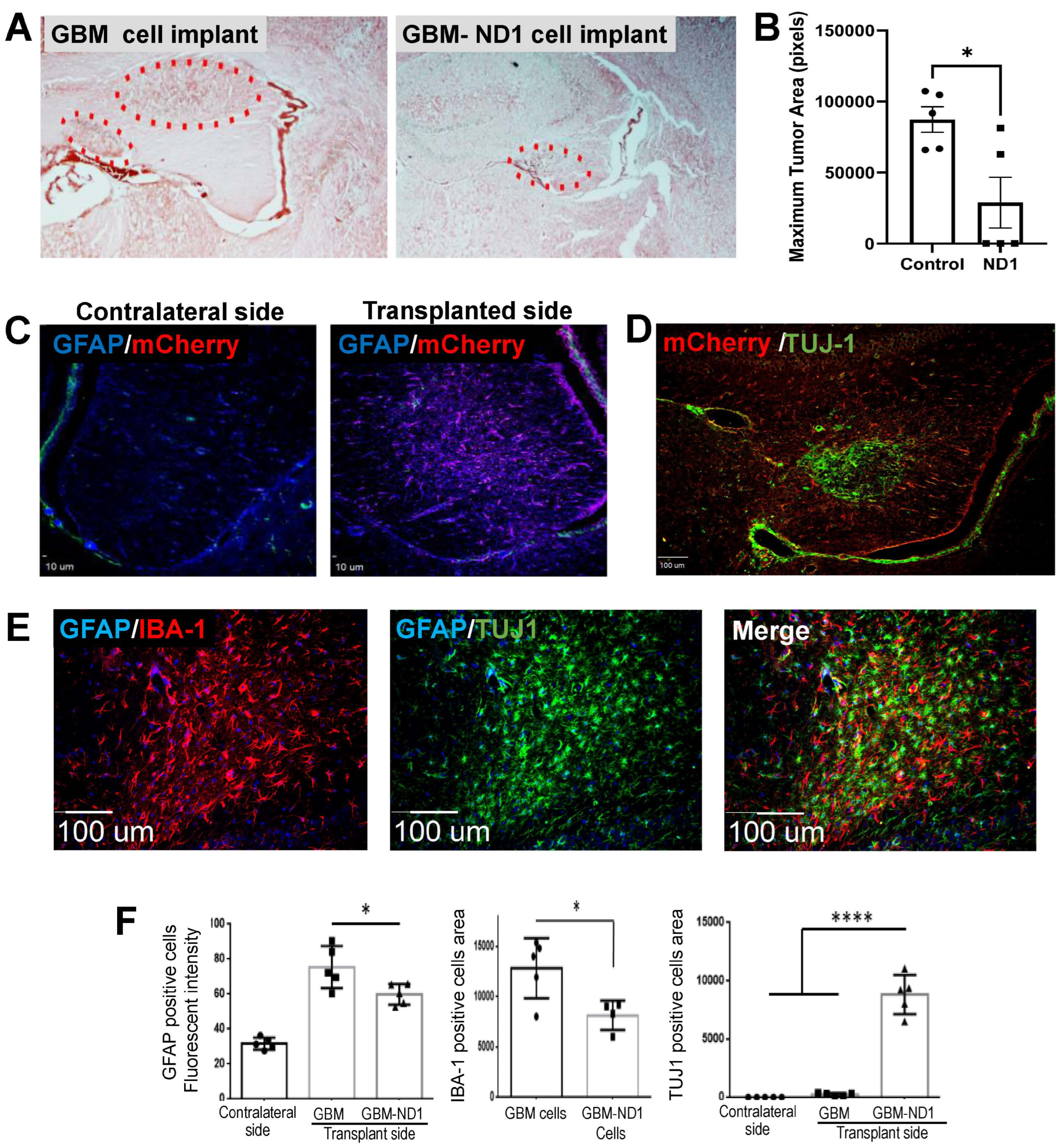
3.6. ND1 Reprogramming Therapy in a GBM Mouse Model
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fazeny-Dorner, B.; Wenzel, C.; Veitl, M.; Piribauer, M.; Rossler, K.; Dieckmann, K.; Ungersbock, K.; Marosi, C. Survival and prognostic factors of patients with unresectable glioblastoma multiforme. Anticancer Drugs 2003, 14, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2023, 25, iv1–iv99. [Google Scholar] [CrossRef]
- Liberski, P.P.; Kordek, R. Ultrastructural pathology of glial brain tumors revisited: A review. Ultrastruct. Pathol. 1997, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Carrillo-Perez, F.; Pizurica, M.; Heiland, D.H.; Gevaert, O. Spatial cellular architecture predicts prognosis in glioblastoma. Nat. Commun. 2023, 14, 4122. [Google Scholar] [CrossRef]
- Awad, A.W.; Karsy, M.; Sanai, N.; Spetzler, R.; Zhang, Y.; Xu, Y.; Mahan, M.A. Impact of removed tumor volume and location on patient outcome in glioblastoma. J. Neuro-Oncol. 2017, 135, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Domino, J.S.; Ormond, D.R.; Germano, I.M.; Sami, M.; Ryken, T.C.; Olson, J.J. Cytoreductive surgery in the management of newly diagnosed glioblastoma in adults: A systematic review and evidence-based clinical practice guideline update. J. Neuro-Oncol. 2020, 150, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Yadav, S.; Chekuri, G.; Karulkar, A.; Jaiswal, A.K.; Goda, J.S.; Purwar, R. Combinatorial approaches to effective therapy in glioblastoma (GBM): Current status and what the future holds. Int. Rev. Immunol. 2022, 41, 582–605. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J.S.; Melendez, B. Glioblastoma Biology, Genetics and Possible Therapies. Cells 2023, 12, 2063. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Xie, X.; Bao, S.; Zhao, H.; Li, L.; Fu, X. Efficacy and Safety of Bevacizumab for Treating Glioblastoma: A Systematic Review and Meta-Analysis of Phase II and III Randomized Controlled Trials. Cancer Investig. 2023, 41, 305–317. [Google Scholar] [CrossRef]
- Kamiya-Matsuoka, C.; Gilbert, M.R. Treating recurrent glioblastoma: An update. CNS Oncol. 2015, 4, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Wefel, J.S.; Armstrong, T.S.; Pugh, S.L.; Gilbert, M.R.; Wendland, M.M.; Brachman, D.G.; Roof, K.S.; Brown, P.D.; Crocker, I.R.; Robins, H.I.; et al. Neurocognitive, symptom, and health-related quality of life outcomes of a randomized trial of bevacizumab for newly diagnosed glioblastoma (NRG/RTOG 0825). Neuro Oncol. 2021, 23, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Franzmeier, S.; Liesche-Starnecker, F.; Schlegel, J. Enhanced Sensitivity to ALDH1A3-Dependent Ferroptosis in TMZ-Resistant Glioblastoma Cells. Cells 2023, 12, 2522. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.S.; Wefel, J.S.; Wang, M.; Gilbert, M.R.; Won, M.; Bottomley, A.; Mendoza, T.R.; Coens, C.; Werner-Wasik, M.; Brachman, D.G.; et al. Net clinical benefit analysis of radiation therapy oncology group 0525: A phase III trial comparing conventional adjuvant temozolomide with dose-intensive temozolomide in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2013, 31, 4076–4084. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Chan, P.; Rich, J.N.; Kay, S.A. Watching the clock in glioblastoma. Neuro Oncol. 2023, 25, 1932–1946. [Google Scholar] [CrossRef]
- Korja, M.; Raj, R.; Seppa, K.; Luostarinen, T.; Malila, N.; Seppala, M.; Maenpaa, H.; Pitkaniemi, J. Glioblastoma survival is improving despite increasing incidence rates: A nationwide study between 2000 and 2013 in Finland. Neuro Oncol. 2019, 21, 370–379. [Google Scholar] [CrossRef]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Wang, W.; Zhang, Y.; He, D.; Xiao, B.; Zhang, H.; Song, A.; Xing, Y.; Li, B. Reprogramming of Rat Fibroblasts into Induced Neurons by Small-Molecule Compounds In Vitro and In Vivo. ACS Chem. Neurosci. 2022, 13, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pu, J.; Zhang, B. Progress and Challenges of Cell Replacement Therapy for Neurodegenerative Diseases Based on Direct Neural Reprogramming. Hum. Gene Ther. 2016, 27, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Guichet, P.O.; Bieche, I.; Teigell, M.; Serguera, C.; Rothhut, B.; Rigau, V.; Scamps, F.; Ripoll, C.; Vacher, S.; Taviaux, S.; et al. Cell death and neuronal differentiation of glioblastoma stem-like cells induced by neurogenic transcription factors. Glia 2013, 61, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Efe, J.A.; Zhu, S.; Talantova, M.; Yuan, X.; Wang, S.; Lipton, S.A.; Zhang, K.; Ding, S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 7838–7843. [Google Scholar] [CrossRef]
- Meyer, S.; Worsdorfer, P.; Gunther, K.; Thier, M.; Edenhofer, F. Derivation of Adult Human Fibroblasts and their Direct Conversion into Expandable Neural Progenitor Cells. J. Vis. Exp. 2015, 101, 52831. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.R.; Szabo, E.; Benoit, Y.D.; Case, D.T.; Mechael, R.; Alamilla, J.; Lee, J.H.; Fiebig-Comyn, A.; Gillespie, D.C.; Bhatia, M. Activation of neural cell fate programs toward direct conversion of adult human fibroblasts into tri-potent neural progenitors using OCT-4. Stem Cells Dev. 2014, 23, 1937–1946. [Google Scholar] [CrossRef]
- Lee, J.H.; Mitchell, R.R.; McNicol, J.D.; Shapovalova, Z.; Laronde, S.; Tanasijevic, B.; Milsom, C.; Casado, F.; Fiebig-Comyn, A.; Collins, T.J.; et al. Single Transcription Factor Conversion of Human Blood Fate to NPCs with CNS and PNS Developmental Capacity. Cell Rep. 2015, 11, 1367–1376. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Zhang, D.; Jiao, J. Acquisition of functional neurons by direct conversion: Switching the developmental clock directly. J. Genet. Genom. 2019, 46, 459–465. [Google Scholar] [CrossRef]
- Lee, C.; Robinson, M.; Willerth, S.M. Direct Reprogramming of Glioblastoma Cells into Neurons Using Small Molecules. ACS Chem. Neurosci. 2018, 9, 3175–3185. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Poulin, G.; Turgeon, B.; Drouin, J. NeuroD1/beta2 contributes to cell-specific transcription of the proopiomelanocortin gene. Mol. Cell Biol. 1997, 17, 6673–6682. [Google Scholar] [CrossRef]
- Tutukova, S.; Tarabykin, V.; Hernandez-Miranda, L.R. The Role of Neurod Genes in Brain Development, Function, and Disease. Front. Mol. Neurosci. 2021, 14, 662774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.Q.; Yu, S.P.; Wei, Z.Z.; Zhong, W.; Cao, W.; Gu, X.; Wu, A.; McCrary, M.R.; Berglund, K.; Wei, L. Conversion of Reactive Astrocytes to Induced Neurons Enhances Neuronal Repair and Functional Recovery After Ischemic Stroke. Front. Aging Neurosci. 2021, 13, 612856. [Google Scholar] [CrossRef]
- Xu, X.; Chen, W.; Zhu, W.; Chen, J.; Ma, B.; Ding, J.; Wang, Z.; Li, Y.; Wang, Y.; Zhang, X. Adeno-associated virus (AAV)-based gene therapy for glioblastoma. Cancer Cell Int. 2021, 21, 76. [Google Scholar] [CrossRef]
- Bohman, L.E.; Swanson, K.R.; Moore, J.L.; Rockne, R.; Mandigo, C.; Hankinson, T.; Assanah, M.; Canoll, P.; Bruce, J.N. Magnetic resonance imaging characteristics of glioblastoma multiforme: Implications for understanding glioma ontogeny. Neurosurgery 2010, 67, 1319–1327; discussion 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Arnes, M.; Casas Tinto, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef]
- Han, M.; Wang, S.; Fritah, S.; Wang, X.; Zhou, W.; Yang, N.; Ni, S.; Huang, B.; Chen, A.; Li, G.; et al. Interfering with long non-coding RNA MIR22HG processing inhibits glioblastoma progression through suppression of Wnt/beta-catenin signalling. Brain 2020, 143, 512–530. [Google Scholar] [CrossRef]
- Rahmani, F.; Hashemian, P.; Tabrizi, A.T.; Ghorbani, Z.; Ziaeemehr, A.; Alijannejad, S.; Ferns, G.A.; Avan, A.; Shahidsales, S. Regulatory role of miRNAs on Wnt/beta-catenin signaling in tumorigenesis of glioblastoma. Indian. J. Cancer 2023, 60, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Taheriazam, A.; Bayanzadeh, S.D.; Heydari Farahani, M.; Mojtabavi, S.; Zandieh, M.A.; Gholami, S.; Heydargoy, M.H.; Jamali Hondori, M.; Kangarloo, Z.; Behroozaghdam, M.; et al. Non-coding RNA-based therapeutics in cancer therapy: An emphasis on Wnt/beta-catenin control. Eur. J. Pharmacol. 2023, 951, 175781. [Google Scholar] [CrossRef]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013, 4, e500. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Chinn, H.K.; Gardell, J.L.; Matsumoto, L.R.; Labadie, K.P.; Mihailovic, T.N.; Lieberman, N.A.P.; Davis, A.; Pillarisetty, V.G.; Crane, C.A. Hypoxia-inducible lentiviral gene expression in engineered human macrophages. J. Immunother. Cancer 2022, 10, e003770. [Google Scholar] [CrossRef]
- Yool, A.J.; Ramesh, S. Molecular Targets for Combined Therapeutic Strategies to Limit Glioblastoma Cell Migration and Invasion. Front. Pharmacol. 2020, 11, 358. [Google Scholar] [CrossRef]
- Xiong, Q.; Liu, B.; Ding, M.; Zhou, J.; Yang, C.; Chen, Y. Hypoxia and cancer related pathology. Cancer Lett. 2020, 486, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Shrivastava, V.; Joshi, D.; Singal, C.M.S.; Tyagi, S.; Bhat, M.A.; Jaiswal, P.; Sharma, J.B.; Palanichamy, J.K.; Sinha, S.; et al. Hypoxia Induces Early Neurogenesis in Human Fetal Neural Stem Cells by Activating the WNT Pathway. Mol. Neurobiol. 2023, 60, 2910–2921. [Google Scholar] [CrossRef]
- Mayes, D.A.; Hu, Y.; Teng, Y.; Siegel, E.; Wu, X.; Panda, K.; Tan, F.; Yung, W.K.; Zhou, Y.H. PAX6 suppresses the invasiveness of glioblastoma cells and the expression of the matrix metalloproteinase-2 gene. Cancer Res. 2006, 66, 9809–9817. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Liu, J.; Pang, H.; Tian, Y.; Yuan, K.; Li, Y.; Wang, J.; Bian, S.; Zheng, Y.; Dong, D.; et al. MicroRNA-365 suppressed cell proliferation and migration via targeting PAX6 in glioblastoma. Am. J. Transl. Res. 2019, 11, 361–369. [Google Scholar]
- Ng, N.; Newbery, M.; Maksour, S.; Dottori, M.; Sluyter, R.; Ooi, L. Transgene and Chemical Transdifferentiation of Somatic Cells for Rapid and Efficient Neurological Disease Cell Models. Front. Cell Neurosci. 2022, 16, 858432. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Stefani, F.R.; Li, J.; Zetterdahl, O.G.; Canals, I.; Ahlenius, H.; Bengzon, J. Transcription Factor-Forced Astrocytic Differentiation Impairs Human Glioblastoma Growth In Vitro and In Vivo. Mol. Cancer Ther. 2023, 22, 274–286. [Google Scholar] [CrossRef]
- Liu, X.; Guo, C.; Leng, T.; Fan, Z.; Mai, J.; Chen, J.; Xu, J.; Li, Q.; Jiang, B.; Sai, K.; et al. Differential regulation of H3K9/H3K14 acetylation by small molecules drives neuron-fate-induction of glioma cell. Cell Death Dis. 2023, 14, 142. [Google Scholar] [CrossRef]
- Yang, Y.; Tu, Y.; Lu, J.; Chen, Q.; Zhu, Z.; Peng, W.; Zhu, W.; Wen, S.; Zhang, J.; Yin, W.; et al. PT109, a novel multi-kinase inhibitor suppresses glioblastoma multiforme through cell reprogramming: Involvement of PTBP1/PKM1/2 pathway. Eur. J. Pharmacol. 2022, 920, 174837. [Google Scholar] [CrossRef] [PubMed]
- Reguera-Nunez, E.; Roca, C.; Hardy, E.; de la Fuente, M.; Csaba, N.; Garcia-Fuentes, M. Implantable controlled release devices for BMP-7 delivery and suppression of glioblastoma initiating cells. Biomaterials 2014, 35, 2859–2867. [Google Scholar] [CrossRef]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, M.; Bergstrom, T.; Jarvius, M.; Sundstrom, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal transition and increased therapy resistance of glioblastoma cells is related to astrocyte reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Bago, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.R.; Magness, S.T.; Hingtgen, S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef]
- Slyk, Z.; Wrzesien, R.; Barszcz, S.; Gawrychowski, K.; Malecki, M. Adeno-associated virus vector hydrogel formulations for brain cancer gene therapy applications. Biomed. Pharmacother. 2023, 170, 116061. [Google Scholar] [CrossRef]
- Chau, M.; Deveau, T.C.; Song, M.; Wei, Z.Z.; Gu, X.; Yu, S.P.; Wei, L. Transplantation of iPS cell-derived neural progenitors overexpressing SDF-1alpha increases regeneration and functional recovery after ischemic stroke. Oncotarget 2017, 8, 97537–97553. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, O.; Drury-Stewart, D.; Song, M.; Faulkner, B.; Chen, D.; Yu, S.P.; Wei, L. Vector-free and transgene-free human iPS cells differentiate into functional neurons and enhance functional recovery after ischemic stroke in mice. PLoS ONE 2013, 8, e64160. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.M.; Gaitsch, H.; Alomari, S.; Lubelski, D.; Tyler, B.M. Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers 2022, 14, 3743. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Syahir, A. Could We Address the Interplay Between CD133, Wnt/beta-Catenin, and TERT Signaling Pathways as a Potential Target for Glioblastoma Therapy? Front. Oncol. 2021, 11, 642719. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/beta-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhu, K.; Yang, Z.; Zhou, Y.; Xia, Z.; Ren, J.; Zhao, Y.; Wu, G.; Liu, C. Hypoxia-Induced Autophagy Is Involved in Radioresistance via HIF1A-Associated Beclin-1 in Glioblastoma Multiforme. Heliyon 2023, 9, e12820. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef]
- Yi, L.; Lu, C.; Hu, W.; Sun, Y.; Levine, A.J. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012, 72, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.-H.; Liu, X.; Rennert, O.M. Apoptosis: Reprogramming and the Fate of Mature Cells. ISRN Cell Biol. 2012, 2012, 685852. [Google Scholar] [CrossRef]
- Feng, B.; Ng, J.H.; Heng, J.C.; Ng, H.H. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell Stem Cell 2009, 4, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Luu, H.H.; Zhang, R.; Haydon, R.C.; Rayburn, E.; Kang, Q.; Si, W.; Park, J.K.; Wang, H.; Peng, Y.; Jiang, W.; et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr. Cancer Drug Targets 2004, 4, 653–671. [Google Scholar] [CrossRef]
- Yao, H.; Ashihara, E.; Maekawa, T. Targeting the Wnt/beta-catenin signaling pathway in human cancers. Expert. Opin. Ther. Targets 2011, 15, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Wu, Y.; Awadasseid, A.; Tanaka, Y.; Zhang, W. New Advances in Canonical Wnt/beta-Catenin Signaling in Cancer. Cancer Manag. Res. 2020, 12, 6987–6998. [Google Scholar] [CrossRef]
- Teng, J.; da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, E.A.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9, 85. [Google Scholar] [CrossRef]
- Fabbri, R.; Cacopardo, L.; Ahluwalia, A.; Magliaro, C. Advanced 3D Models of Human Brain Tissue Using Neural Cell Lines: State-of-the-Art and Future Prospects. Cells 2023, 12, 1181. [Google Scholar] [CrossRef]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef]
- da Hora, C.C.; Schweiger, M.W.; Wurdinger, T.; Tannous, B.A. Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells 2019, 8, 1177. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, M.Q.; Yu, S.P.; Estaba, T.; Choi, E.; Berglund, K.; Gu, X.; Wei, L. Reprogramming Glioblastoma Cells into Non-Cancerous Neuronal Cells as a Novel Anti-Cancer Strategy. Cells 2024, 13, 897. https://doi.org/10.3390/cells13110897
Jiang MQ, Yu SP, Estaba T, Choi E, Berglund K, Gu X, Wei L. Reprogramming Glioblastoma Cells into Non-Cancerous Neuronal Cells as a Novel Anti-Cancer Strategy. Cells. 2024; 13(11):897. https://doi.org/10.3390/cells13110897
Chicago/Turabian StyleJiang, Michael Q., Shan Ping Yu, Takira Estaba, Emily Choi, Ken Berglund, Xiaohuan Gu, and Ling Wei. 2024. "Reprogramming Glioblastoma Cells into Non-Cancerous Neuronal Cells as a Novel Anti-Cancer Strategy" Cells 13, no. 11: 897. https://doi.org/10.3390/cells13110897
APA StyleJiang, M. Q., Yu, S. P., Estaba, T., Choi, E., Berglund, K., Gu, X., & Wei, L. (2024). Reprogramming Glioblastoma Cells into Non-Cancerous Neuronal Cells as a Novel Anti-Cancer Strategy. Cells, 13(11), 897. https://doi.org/10.3390/cells13110897