
Postmortem Brains from Subjects with Diabetes Mellitus Display Reduced GLUT4 Expression and Soma Area in Hippocampal Neurons: Potential Involvement of Inflammation


 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Immunohistochemistry (IHC) for Quantification of Proteins in Human Hippocampal Area
2.3. Cell Culture and Treatments
2.4. Quantitative Polymerase Chain Reaction (qPCR) for Gene Expression Analysis
2.5. Protein Extraction and Western Blotting for Protein Expression Analysis
2.6. Chromatin Immunoprecipitation Assay (ChIP Assay) for p65 Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinical and Demographic Characteristics of the Subjects Studied
3.2. GLUT4 Protein Detection in Hippocampus of Human Brain
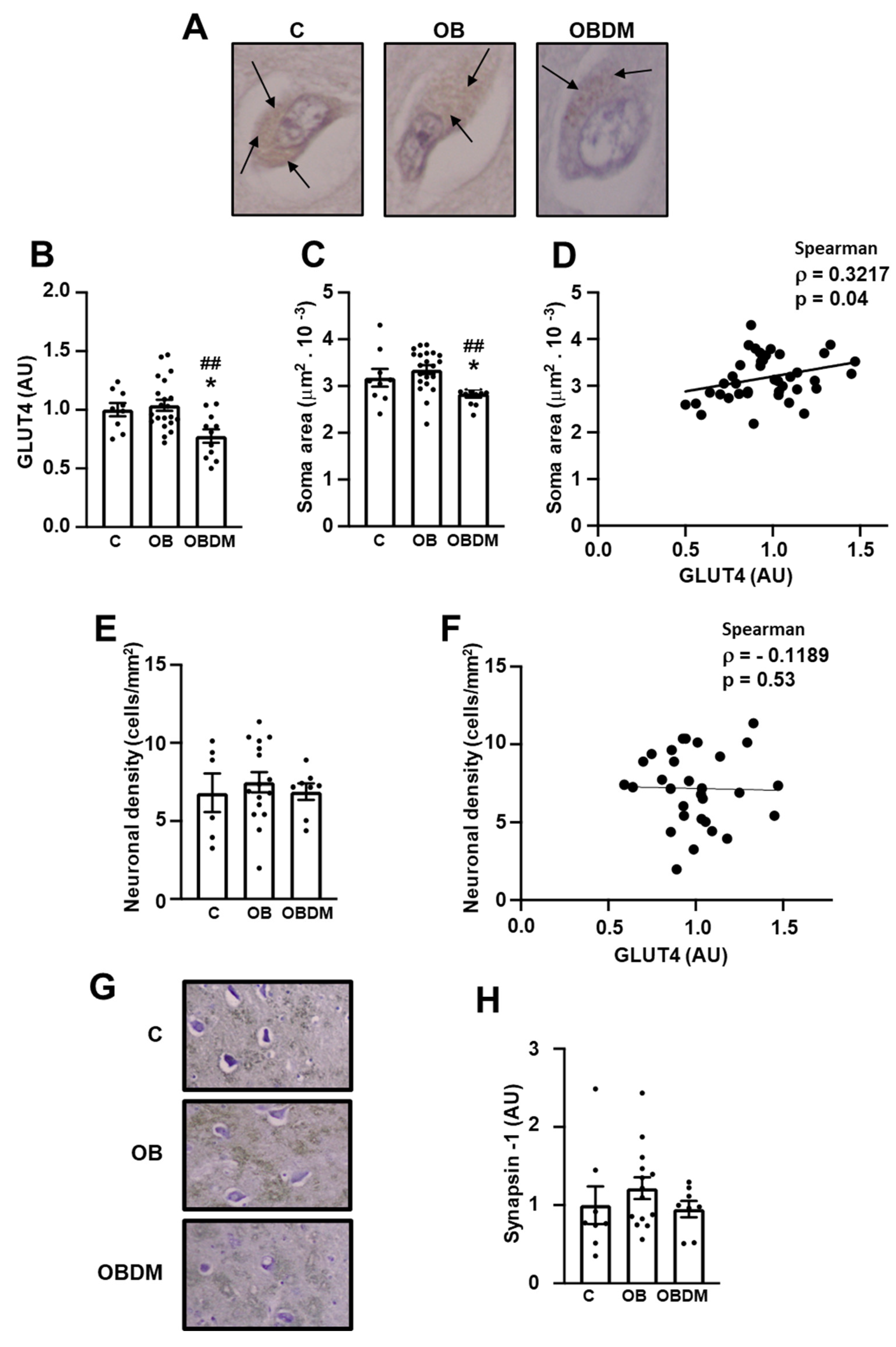
3.3. Diabetes Decreases Neuronal GLUT4 Protein Content and Soma Area in Hippocampal CA4/Hilus Area
3.4. Obesity Increases NFKB Subunit p65 but Does Not Change Carboxymethyllysine (CML) Content in Neurons from Hippocampal CA4/Hilus Area
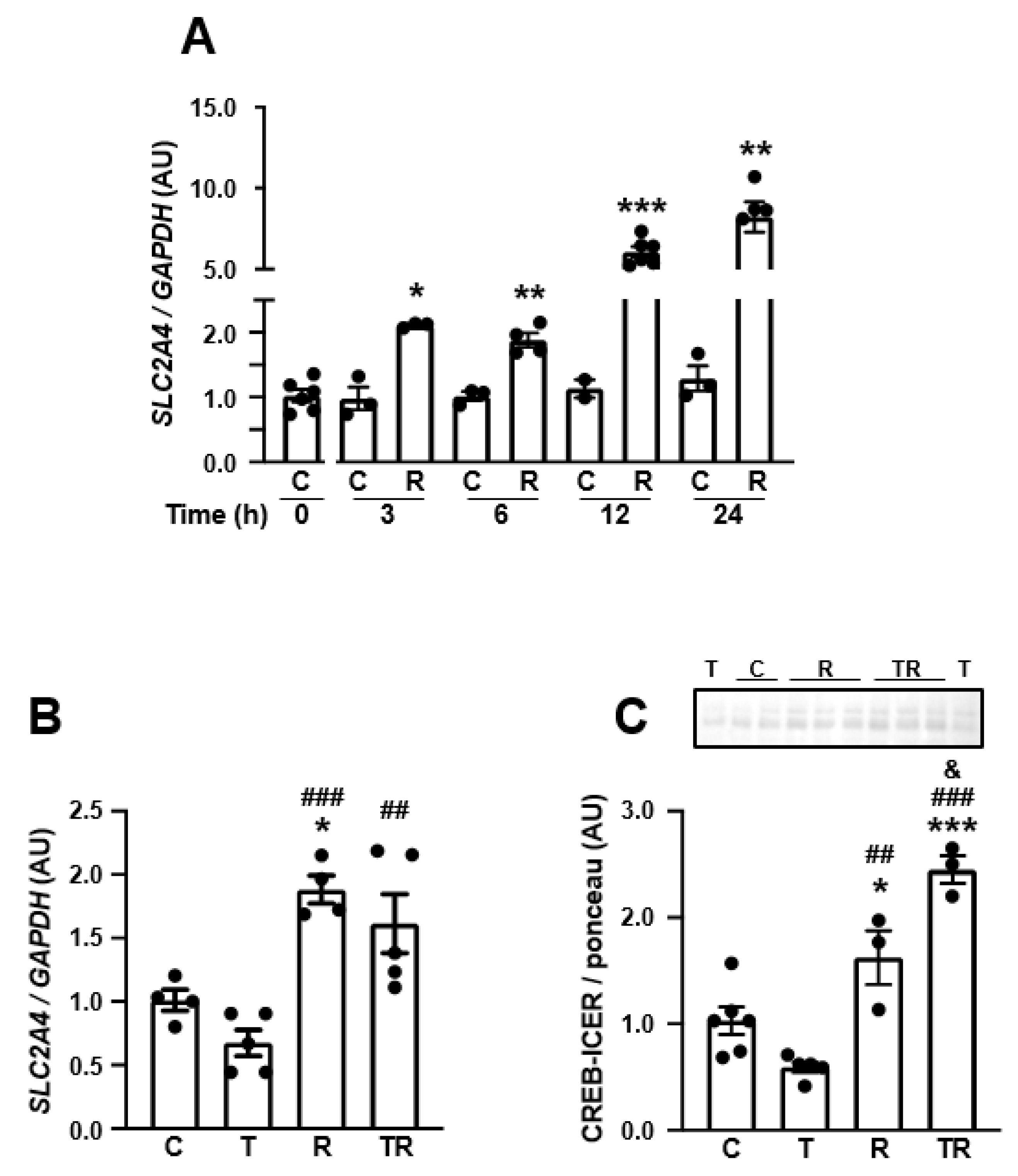
3.5. TNF Represses SLC2A4/GLUT4 Expression by a NFKB-Mediated Mechanism in Human Neuronal SH-SY5Y Cells
3.6. TNF Represses the Expression of Neuronal Markers in Human Neuronal SH-SY5Y Cells
3.7. Inhibition of Histone Deacetylase 3 (HDAC3) Increases SLC2A4 Expression, Abrogates TNF-Induced Re-Pression of SLC2A4 Repression and Enhances Nuclear Content of CREB in Human Neuronal SH-SY5Y Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADGs | AD-related genes |
| AGEs | advanced glycation end products |
| BBB | blood–brain barrier |
| BAS | Biobank for Aging Study |
| BMI | body mass index |
| CA1 | cornu ammonis 1 area |
| CA4 | cornu ammonis 4 area |
| CHIP | chromatin Immunoprecipitation |
| CML | carboxymethyllysine |
| CNS | central nervous system |
| CREB/ICER | CRE-binding proteins |
| DAB | 3-3-diaminobenzidine tetrahydrochloride |
| DM | diabetes mellitus |
| GA | glycated albumin |
| GLUT1 | glucose transporter type 1 protein |
| GLUT3 | glucose transporter type 3 protein |
| GLUT4 | glucose transporter type 4 protein |
| GVD | granulovacuolar degeneration |
| H3Kac | lysine acetylation of histone 3 |
| H4Kac | lysine acetylation of histone 4 |
| H3K9ac | lysine 9 acetylation of histone 3 |
| HDAC3 | histone deacetylase 3 |
| IHC | immunohistochemistry |
| IR | insulin resistance |
| MTT | tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| OB | obese subject |
| OBDM | obese diabetic subject |
| p65 | nuclear factor NF-kappa-B subunit p65 |
| PB | phosphate buffer |
| qPCR | quantitative polymerase chain reaction |
| RAGE | advanced glycation end product receptor |
| RGFP-996 | HDAC3 inhibitor |
| SYP | synaptophysin protein |
| SYP | synaptophysin gene (human) |
| SYN1 | synapsin-1 (protein) |
| SYN1 | synapsin-1 gene (human) |
| Slc2a4 | solute carrier family 2 member 4 gene (mouse) |
| SLC2A4 | solute carrier family 2 member 4 gene (human |
| SPAS | Sao Paulo Autopsy Service |
| TNF | tumor necrosis factor |
| T1D | type 1 diabetes |
| T2D | type 2 diabetes |
| TH | tyrosine hydroxylase protein |
| TH | tyrosine hydroxylase gene (human) |
References
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’adamio, L.; Grassi, C.; Devanand, D.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M.B. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef]
- Simó, R.; Ciudin, A.; Simó-Servat, O.; Hernández, C. Cognitive impairment and dementia: A new emerging complication of type 2 diabetes—The diabetologist’s perspective. Acta Diabetol. 2017, 54, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: The Hisayama Study. Neurology 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, N.; Hata, J.; Ohara, T.; Mukai, N.; Nagata, M.; Shibata, M.; Gotoh, S.; Furuta, Y.; Yamashita, F.; Yoshihara, K.; et al. Association Between Diabetes and Hippocampal Atrophy in Elderly Japanese: The Hisayama Study. Diabetes Care 2016, 39, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-Analysis of Alzheimer’s Disease Risk with Obesity, Diabetes, and Related Disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; Carvalho, M.D.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s disease and type 2 diabetes mellitus: A systematic review of proteomic studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef] [PubMed]
- Heijer, T.D.; Vermeer, S.E.; Van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Koekkoek, P.S.; Kappelle, L.J.; van den Berg, E.; Rutten, G.E.H.M.; Biessels, G.J. Cognitive function in patients with diabetes mellitus: Guidance for daily care. Lancet Neurol. 2015, 14, 329–340. [Google Scholar] [CrossRef]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef]
- Luchsinger, J.A.M.; Cheng, D.M.; Tang, M.X.; Schupf, N.; Mayeux, R. Central Obesity in the Elderly is Related to Late-onset Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 101–105. [Google Scholar] [CrossRef]
- Mueller, K.; Sacher, J.; Arelin, K.; Holiga, Š.; Kratzsch, J.; Villringer, A.; Schroeter, M.L. Overweight and obesity are associated with neuronal injury in the human cerebellum and hippocampus in young adults: A combined MRI, serum marker and gene expression study. Transl. Psychiatry 2012, 2, e200. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults With Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Gammazza, A.; Cappello, F.; Mulè, F.; Carlo, M. Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Nisar, O.; Pervez, H.; Mandalia, B.; Waqas, M.; Sra, H.K. Type 3 Diabetes Mellitus: A Link Between Alzheimer’s Disease and Type 2 Diabetes Mellitus. Cureus 2020, 12, e11703. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Sadgrove, M.P.; Beaver, C.J.; Turner, D.A. Effects of relative hypoglycemia on LTP and NADH imaging in rat hippocampal slices. Brain Res. 2007, 1165, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Galeffi, F.; Shetty, P.K.; Sadgrove, M.P.; Turner, D.A. Age-related metabolic fatigue during low glucose conditions in rat hippocampus. Neurobiol. Aging 2015, 36, 982–992. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- McEwen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e3. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef]
- Jang, S.; Nelson, J.C.; Bend, E.G.; Rodríguez-Laureano, L.; Tueros, F.G.; Cartagenova, L.; Underwood, K.; Jorgensen, E.M.; Colón-Ramos, D.A. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 2016, 90, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Park, S.H.; De Vivo, D.C.; Monani, U.R. Therapeutic strategies for glucose transporter 1 deficiency syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 1923–1932. [Google Scholar] [CrossRef]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.W.; Geary, B.R.; Engelstad, K.M.; Natarajan, A.; Yang, H.; De Vivo, D.C. Glucose transporter type I deficiency syndrome: Epilepsy phenotypes and outcomes. Epilepsia 2012, 53, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, A.M.; Szylberg, Ł.; Kasperska, A.; Marszałek, A. The diagnosis and management of congenital and adult-onset hyperinsulinism (nesidioblastosis)—Literature review. Pol. J. Pathol. 2017, 2, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Giannis, D.; Moris, D.; Karachaliou, G.S.; Tsilimigras, D.; Karaolanis, G.; Papalampros, A.; Felekouras, E. Insulinomas: From diagnosis to treatment. A review of the literature. J. BUON 2020, 25, 1302–1314. [Google Scholar]
- Sheen, Y.-J.; Sheu, W.H. Association between hypoglycemia and dementia in patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2016, 116, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.; Moheet, A.; Seaquist, E.R. Central Mechanisms of Glucose Sensing and Counterregulation in Defense of Hypoglycemia. Endocr. Rev. 2018, 40, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef]
- De Felice, F.G.; Benedict, C. A Key Role of Insulin Receptors in Memory. Diabetes 2015, 64, 3653–3655. [Google Scholar] [CrossRef]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and Systemic Endotoxin Challenges Exacerbate the Local Inflammatory Response and Increase Neuronal Death during Chronic Neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, D.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNF-alpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Fraga, V.G.; Magalhães, C.A.; Loures, C.D.M.G.; de Souza, L.C.; Guimarães, H.C.; Zauli, D.A.G.; Carvalho, M.D.G.; Ferreira, C.N.; Caramelli, P.; de Sousa, L.P.; et al. Inflammatory and Pro-resolving Mediators in Frontotemporal Dementia and Alzheimer’s Disease. Neuroscience 2019, 421, 123–135. [Google Scholar] [CrossRef]
- Furuya, D.; Neri, E.; Poletto, A.; Anhê, G.; Freitas, H.; Campello, R.; Rebouças, N.; Machado, U. Identification of nuclear factor-κB sites in the Slc2a4 gene promoter. Mol. Cell. Endocrinol. 2013, 370, 87–95. [Google Scholar] [CrossRef]
- Moraes, P.A.; Yonamine, C.Y.; Junior, D.C.P.; Esteves, J.V.D.; Machado, U.F.; Mori, R.C. Insulin acutely triggers transcription of Slc2a4 gene: Participation of the AT-rich, E-box and NFKB-binding sites. Life Sci. 2014, 114, 36–44. [Google Scholar] [CrossRef]
- Ebersbach-Silva, P.; Poletto, A.C.; David-Silva, A.; Seraphim, P.; Anhe, G.; Passarelli, M.; Furuya, D.T.; Machado, U.F. Palmitate-induced Slc2a4/GLUT4 downregulation in L6 muscle cells: Evidence of inflammatory and endoplasmic reticulum stress involvement. Lipids Heal. Dis. 2018, 17, 64. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Yonamine, C.Y.; Alves-Wagner, A.B.; Esteves, J.V.; Okamoto, M.M.; Correa-Giannella, M.L.; Giannella-Neto, D.; Machado, U.F. Diabetes induces tri-methylation at lysine 9 of histone 3 at Slc2a4 gene in skeletal muscle: A new target to improve glycemic control. Mol. Cell. Endocrinol. 2019, 481, 26–34. [Google Scholar] [CrossRef]
- Roohk, H.V.; Zaidi, A.R.; Patel, D. Glycated albumin (GA) and inflammation: Role of GA as a potential marker of inflammation. Inflamm. Res. 2018, 67, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M.; Aria, F.; Premoli, M.; Maccarinelli, G.; Mastinu, A.; Bonini, S.; Memo, M.; Uberti, D.; Abate, G. Methylglyoxal affects cognitive behaviour and modulates RAGE and Presenilin-1 expression in hippocampus of aged mice. Food Chem. Toxicol. 2021, 158, 112608. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xin, Y.; Chu, T.; Liu, C.; Xu, A. Dexmedetomidine attenuates perioperative neurocognitive disorders by suppressing hippocampal neuroinflammation and HMGB1/RAGE/NF-κB signaling pathway. Biomed. Pharmacother. 2022, 150, 113006. [Google Scholar] [CrossRef] [PubMed]
- Corrêa-Giannella, M.L.; Machado, U.F. SLC2A4 gene: A promising target for pharmacogenomics of insulin resistance. Pharmacogenomics 2013, 14, 847–850. [Google Scholar] [CrossRef]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.T.; Lucena Ferretti, R.E.; Farfel, J.M.; Leite, R.; Pasqualucci, C.A.; Rosemberg, S.; Nitrini, R.; Saldiva, P.H.N.; Filho, W.J.; Brazilian Aging Brain Study Group. Brain bank of the Brazilian aging brain study group—A milestone reached and more than 1,600 collected brains. Cell Tissue Bank. 2007, 8, 151–162. [Google Scholar] [CrossRef]
- Passarelli, M.; Machado, U.F.F. AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus. Cells 2021, 11, 104. [Google Scholar] [CrossRef]
- Stephens, J.; Pekala, P. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J. Biol. Chem. 1992, 267, 13580–13584. [Google Scholar] [CrossRef]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor Necrosis Factor-α Suppresses Adipocyte-Specific Genes and Activates Expression of Preadipocyte Genes in 3T3-L1 Adipocytes. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef]
- Benomar, Y.; Naour, N.; Aubourg, A.; Bailleux, V.; Gertler, A.; Djiane, J.; Guerre-Millo, M.; Taouis, M. Insulin and Leptin Induce Glut4 Plasma Membrane Translocation and Glucose Uptake in a Human Neuronal Cell Line by a Phosphatidylinositol 3-Kinase- Dependent Mechanism. Endocrinology 2006, 147, 2550–2556. [Google Scholar] [CrossRef]
- Cohen, L.D.; Zuchman, R.; Sorokina, O.; Müller, A.; Dieterich, D.C.; Armstrong, D.; Ziv, T.; Ziv, N.E. Metabolic Turnover of Synaptic Proteins: Kinetics, Interdependencies and Implications for Synaptic Maintenance. PLoS ONE 2013, 8, e63191. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of Advanced Glycation End-Products for Cultured Cortical Neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, H.; Osler, M.E.; Krook, A.; Zierath, J. Epigenetic flexibility in metabolic regulation: Disease cause and prevention? Trends Cell Biol. 2013, 23, 203–209. [Google Scholar] [CrossRef]
- Esteves, J.V.; Yonamine, C.Y.; Machado, U.F. SLC2A4 expression and its epigenetic regulation as biomarkers for insulin resistance treatment in diabetes mellitus. Biomarkers Med. 2020, 14, 413–416. [Google Scholar] [CrossRef]
- Shyamasundar, S.; Jadhav, S.P.; Bay, B.H.; Tay, S.S.W.; Kumar, S.D.; Rangasamy, D.; Dheen, S.T. Analysis of Epigenetic Factors in Mouse Embryonic Neural Stem Cells Exposed to Hyperglycemia. PLoS ONE 2013, 8, e65945. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Zhang, Q.; Wan, C.; Li, D.; Zhang, T.; Li, H. CBP/p300 inhibitor C646 prevents high glucose exposure induced neuroepithelial cell proliferation. Birth Defects Res. 2018, 110, 1118–1128. [Google Scholar] [CrossRef]
- Zheng, S.; Rollet, M.; Pan, Y.-X. Protein restriction during gestation alters histone modifications at the glucose transporter 4 (GLUT4) promoter region and induces GLUT4 expression in skeletal muscle of female rat offspring. J. Nutr. Biochem. 2012, 23, 1064–1071. [Google Scholar] [CrossRef]
- Mantamadiotis, T.; Lemberger, T.; Bleckmann, S.C.; Kern, H.; Kretz, O.; Martin-Villalba, A.; Tronche, F.; Kellendonk, C.; Gau, D.; Kapfhammer, J.P.; et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002, 31, 47–54. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Tabunoki, H.; Arima, K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis. Markers 2009, 27, 239–252. [Google Scholar] [CrossRef]
- Alves-Wagner, A.B.; Yonamine, C.Y.; De Fatima, L.A.; Festuccia, W.; Machado, U.F. Sympathetic Regulation of Slc2a4 Gene Expression: Participation of a Putative cAMP Responsive Element (CRE) Site in the Slc2a4 Promoter. Cell. Physiol. Biochem. 2019, 52, 580–594. [Google Scholar] [CrossRef]
- Hugais, M.M.; Cobos, S.N.; Bennett, S.A.; Paredes, J.; Foran, G.; Torrente, M.P. Changes in Histone H3 Acetylation on Lysine 9 Ac-company Aβ 1-40 Overexpression in an Alzheimer’s Disease Yeast Model. MicroPubl. Biol. 2021, 2021, 1–40. [Google Scholar] [CrossRef]
- Li, T.; Yin, L.; Kang, X.; Xue, W.; Wang, N.; Zhang, J.; Yuan, P.; Lin, L.; Li, Y. TFEB acetylation promotes lysosome biogenesis and ameliorates Alzheimer’s disease–relevant phenotypes in mice. J. Biol. Chem. 2022, 298, 102649. [Google Scholar] [CrossRef]
- Amin, S.A.; Adhikari, N.; Kotagiri, S.; Jha, T.; Ghosh, B. Histone deacetylase 3 inhibitors in learning and memory processes with special emphasis on benzamides. Eur. J. Med. Chem. 2019, 166, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone Deacetylase (HDAC) Inhibition as a Novel Treatment for Diabetes Mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Jensen, A.; Jones, K.; Raghavan, S.; Phillips, L.S.; Hung, A.; Sun, Y.V.; Li, G.; Reaven, P.; Zhou, H.; et al. A platform for phenotyping disease progression and associated longitudinal risk factors in large-scale EHRs, with application to incident diabetes complications in the UK Biobank. JAMIA Open 2023, 6, ooad006. [Google Scholar] [CrossRef]
- Nianogo, R.A.; Onyebuchi, A.; Arah, O.A. Forecasting Obesity and Type 2 Diabetes Incidence and Burden: The ViLA-Obesity Simulation Model. Front. Public Health 2022, 10, 646. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Tsai, C.C.W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 34, 5851–5859. [Google Scholar] [CrossRef]
- Herman, R.; Kravos, N.A.; Jensterle, M.; Janež, A.; Dolžan, V. Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. Int. J. Mol. Sci. 2022, 23, 1264. [Google Scholar] [CrossRef]
- Obafemi, T.O.; Olasehinde, O.R.; Olaoye, O.; Jaiyesimi, K.F.; Adewumi, F.D.; Adewale, O.; Afolabi, B.A. Metformin/Donepezil combination modulates brain antioxidant status and hippocampal endoplasmic reticulum stress in type 2 diabetic rats. J. Diabetes Metab. Disord. 2020, 19, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Zhang, X.-Y.; Sun, Y.-Q.; Lv, R.-H.; Chen, M.; Li, M. Metformin use is associated with a reduced risk of cognitive impairment in adults with diabetes mellitus: A systematic review and meta-analysis. Front. Neurosci. 2022, 16, 984559. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Control n = 9 | Obese n = 21 | Obese with DM n = 11 |
|---|---|---|---|
| Age (years), mean ± SD | 60.22 ± 9.52 | 61.95 ± 12.55 | 69.73 ± 10.54 |
| BMI (kg/m2), mean ± SD | 27.21 ± 1.49 | 31.99 ± 1.38 * | 32.29 ± 1.67 * |
| Male, n (%) | 3 (33.3) | 11 (52.4) | 4 (36.4) |
| Ethnicity, n (%) | |||
| White | 4 (44.4) | 17 (80.9) | 7 (63.6) |
| Brown | 3 (33.3) | 1 (4.8) | 2 (18.2) |
| Black | 2 (22.2) | 3 (14.3) | 2 (18.2) |
| Socioeconomic class, n (%) | |||
| High | 0 (0) | 7 (33.3) | 2 (18.2) |
| Middle | 3 (33.3) | 6 (28.6) | 5 (45,5) |
| Low | 6 (66.7) | 8 (38.1) | 4 (36.3) |
| Education (years), mean ± SD | 5.33 ± 2.95 | 6.9 ± 3.91 | 5.45 ± 4.9 |
| Dyslipidemia, n (%) | 0 (0) | 3 (14.3) | 1 (9) |
| Smoking, n (%) | 4 (44.4) | 5 (23.8) | 5 (45.5) |
| Alcohol use disorder, n (%) | 3 (33.3) | 1 (4.8) | 0 (0) |
| Coronary arterial disease, n (%) | 1 (11.1) | 4 (19.0) | 2 (18.2) |
| Hypertension, n (%) | 8 (89) | 16 (76.0) | 9 (81.8) |
| Heart failure, n (%) | 0 (0) | 5 (23.8) | 3 (27.3) |
| Cerebrovascular disease, n (%) | 1 (11.1) | 1 (4.8) | 4 (36.4) |
| Alzheimer’s disease, n (%) | 0 (0) | 0 (0) | 1 (9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yonamine, C.Y.; Passarelli, M.; Suemoto, C.K.; Pasqualucci, C.A.; Jacob-Filho, W.; Alves, V.A.F.; Marie, S.K.N.; Correa-Giannella, M.L.; Britto, L.R.; Machado, U.F. Postmortem Brains from Subjects with Diabetes Mellitus Display Reduced GLUT4 Expression and Soma Area in Hippocampal Neurons: Potential Involvement of Inflammation. Cells 2023, 12, 1250. https://doi.org/10.3390/cells12091250
Yonamine CY, Passarelli M, Suemoto CK, Pasqualucci CA, Jacob-Filho W, Alves VAF, Marie SKN, Correa-Giannella ML, Britto LR, Machado UF. Postmortem Brains from Subjects with Diabetes Mellitus Display Reduced GLUT4 Expression and Soma Area in Hippocampal Neurons: Potential Involvement of Inflammation. Cells. 2023; 12(9):1250. https://doi.org/10.3390/cells12091250
Chicago/Turabian StyleYonamine, Caio Yogi, Marisa Passarelli, Claudia Kimie Suemoto, Carlos Augusto Pasqualucci, Wilson Jacob-Filho, Venâncio Avancini Ferreira Alves, Suely Kazue Nagahashi Marie, Maria Lucia Correa-Giannella, Luiz Roberto Britto, and Ubiratan Fabres Machado. 2023. "Postmortem Brains from Subjects with Diabetes Mellitus Display Reduced GLUT4 Expression and Soma Area in Hippocampal Neurons: Potential Involvement of Inflammation" Cells 12, no. 9: 1250. https://doi.org/10.3390/cells12091250
APA StyleYonamine, C. Y., Passarelli, M., Suemoto, C. K., Pasqualucci, C. A., Jacob-Filho, W., Alves, V. A. F., Marie, S. K. N., Correa-Giannella, M. L., Britto, L. R., & Machado, U. F. (2023). Postmortem Brains from Subjects with Diabetes Mellitus Display Reduced GLUT4 Expression and Soma Area in Hippocampal Neurons: Potential Involvement of Inflammation. Cells, 12(9), 1250. https://doi.org/10.3390/cells12091250