
Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress


Abstract
1. Introduction
2. Oxidative Stress
3. The MEK5/ERK5 Signalling Pathway
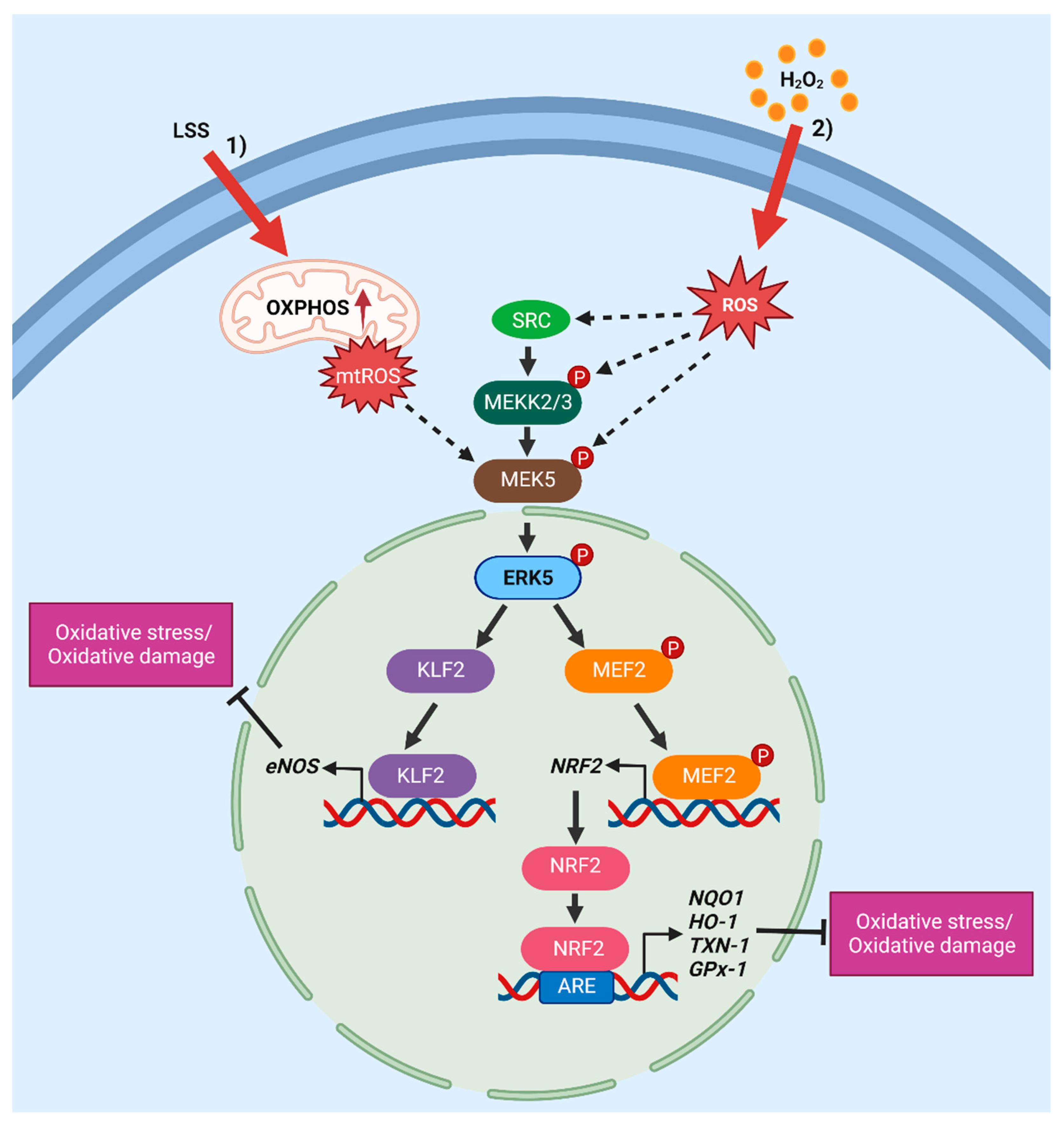
4. Activation of the MEK5/ERK5 Pathway by Oxidative Stress

5. Role of the MEK5/ERK5 Pathway in ROS-Dependent Effects on Human Diseases
5.1. Pathophysiology of Blood Vessels
5.1.1. Endothelial Cells
5.1.2. Atherosclerosis
5.1.3. Hypertension
5.2. Pathophysiology of the Heart
5.2.1. Ischemic Heart Disease
5.2.2. Myocardial Disease
5.3. Pathophysiology of the Lung
5.4. Pathophysiology of the Lymphohematopoietic System
5.5. Pathophysiology of the Kidney
5.6. Pathophysiology of the Liver
5.7. Pathophysiology of the Central Nervous System
5.7.1. Degenerative Diseases
5.7.2. Cerebrovascular Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.D.; Ulevitch, R.J.; Han, J. Primary structure of BMK1: A new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef]
- Paudel, R.; Fusi, L.; Schmidt, M. The MEK5/ERK5 Pathway in Health and Disease. Int. J. Mol. Sci. 2021, 22, 7594. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. Int. J. Mol. Sci. 2023, 24, 1841. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Kruk, J.; Aboul-Enein, H.Y. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Melov, S.; Doctrow, S.R.; Schneider, J.A.; Haberson, J.; Patel, M.; Coskun, P.E.; Huffman, K.; Wallace, D.C.; Malfroy, B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J. Neurosci. 2001, 21, 8348–8353. [Google Scholar] [CrossRef]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef]
- Chu, F.F.; Esworthy, R.S.; Chu, P.G.; Longmate, J.A.; Huycke, M.M.; Wilczynski, S.; Doroshow, J.H. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004, 64, 962–968. [Google Scholar] [CrossRef]
- Kang, S.W.; Rhee, S.G.; Chang, T.S.; Jeong, W.; Choi, M.H. 2-Cys peroxiredoxin function in intracellular signal transduction: Therapeutic implications. Trends Mol. Med. 2005, 11, 571–578. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef]
- Cavanaugh, J.E.; Ham, J.; Hetman, M.; Poser, S.; Yan, C.; Xia, Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J. Neurosci. 2001, 21, 434–443. [Google Scholar] [CrossRef]
- Finegan, K.G.; Wang, X.; Lee, E.J.; Robinson, A.C.; Tournier, C. Regulation of neuronal survival by the extracellular signal-regulated protein kinase 5. Cell Death Differ. 2009, 16, 674–683. [Google Scholar] [CrossRef]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Abe, J.; Kusuhara, M.; Ulevitch, R.J.; Berk, B.C.; Lee, J.D. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 1996, 271, 16586–16590. [Google Scholar] [CrossRef]
- Suzaki, Y.; Yoshizumi, M.; Kagami, S.; Koyama, A.H.; Taketani, Y.; Houchi, H.; Tsuchiya, K.; Takeda, E.; Tamaki, T. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: Potential role in cell survival following oxidative insults. J. Biol. Chem. 2002, 277, 9614–9621. [Google Scholar] [CrossRef]
- Yan, C.; Takahashi, M.; Okuda, M.; Lee, J.D.; Berk, B.C. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J. Biol. Chem. 1999, 274, 143–150. [Google Scholar] [CrossRef]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef]
- Erazo, T.; Espinosa-Gil, S.; Diéguez-Martínez, N.; Gómez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. [Google Scholar] [CrossRef]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Dello Sbarba, P.; Rovida, E. Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 2020, 21, 938. [Google Scholar] [CrossRef]
- Yan, C.; Luo, H.; Lee, J.D.; Abe, J.; Berk, B.C. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 2001, 276, 10870–10878. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Morimoto, H.; Kondoh, K.; Nishimoto, S.; Terasawa, K.; Nishida, E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [Google Scholar] [CrossRef]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 is associated with ERK5 nuclear localization and ERK5-dependent transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef]
- Zhuang, K.; Zhang, J.; Xiong, M.; Wang, X.; Luo, X.; Han, L.; Meng, Y.; Zhang, Y.; Liao, W.; Liu, S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5-AP-1 axis. Cell Death Dis. 2016, 7, e2415. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, E.; Pandiella, A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010, 123, 3146–3156. [Google Scholar] [CrossRef]
- Inesta-Vaquera, F.A.; Campbell, D.G.; Tournier, C.; Gomez, N.; Lizcano, J.M.; Cuenda, A. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell. Signal. 2010, 22, 1829–1837. [Google Scholar] [CrossRef]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017, 392, 51–59. [Google Scholar] [CrossRef]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodriguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gomez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Tucker, J.A.; Tatum, N.J.; Wang, J.; Oxley, D.; Kidger, A.M.; Johnson, V.P.; Cassidy, M.A.; Gray, N.S.; Noble, M.E.M.; et al. Paradoxical activation of the protein kinase-transcription factor ERK5 by ERK5 kinase inhibitors. Nat. Commun. 2020, 11, 1383. [Google Scholar] [CrossRef]
- Lin, E.C.; Amantea, C.M.; Nomanbhoy, T.K.; Weissig, H.; Ishiyama, J.; Hu, Y.; Sidique, S.; Li, B.; Kozarich, J.W.; Rosenblum, J.S. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 11865–11870. [Google Scholar] [CrossRef]
- Nguyen, D.; Lemos, C.; Wortmann, L.; Eis, K.; Holton, S.J.; Boemer, U.; Moosmayer, D.; Eberspaecher, U.; Weiske, J.; Lechner, C.; et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido [3,2-d]pyrimidine Based in Vitro Probe BAY-885 for the Kinase ERK5. J. Med. Chem. 2019, 62, 928–940. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Clark, J.; Wang, L.Z.; Gilmour, L.; Squires, M.; Gilley, R.; Foxton, C.; Newell, D.R.; Wedge, S.R.; Cook, S.J. Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle 2016, 15, 506–518. [Google Scholar] [CrossRef]
- You, I.; Donovan, K.A.; Krupnick, N.M.; Boghossian, A.S.; Rees, M.G.; Ronan, M.M.; Roth, J.A.; Fischer, E.S.; Wang, E.S.; Gray, N.S. Acute pharmacological degradation of ERK5 does not inhibit cellular immune response or proliferation. Cell Chem. Biol. 2022, 29, 1630–1638.e7. [Google Scholar] [CrossRef]
- Abe, J.; Takahashi, M.; Ishida, M.; Lee, J.D.; Berk, B.C. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef]
- Zhao, J.; Kyotani, Y.; Itoh, S.; Nakayama, H.; Isosaki, M.; Yoshizumi, M. Big mitogen-activated protein kinase 1 protects cultured rat aortic smooth muscle cells from oxidative damage. J. Pharmacol. Sci. 2011, 116, 173–180. [Google Scholar] [CrossRef]
- Caselli, A.; Marzocchini, R.; Camici, G.; Manao, G.; Moneti, G.; Pieraccini, G.; Ramponi, G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem. 1998, 273, 32554–32560. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Buschbeck, M.; Hofbauer, S.; Di Croce, L.; Keri, G.; Ullrich, A. Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 2005, 6, 63–69. [Google Scholar] [CrossRef]
- Sun, W.; Wei, X.; Kesavan, K.; Garrington, T.P.; Fan, R.; Mei, J.; Anderson, S.M.; Gelfand, E.W.; Johnson, G.L. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell Biol. 2003, 23, 2298–2308. [Google Scholar] [CrossRef]
- Wu, N.; Sun, H.; Zhao, X.; Zhang, Y.; Tan, J.; Qi, Y.; Wang, Q.; Ng, M.; Liu, Z.; He, L.; et al. MAP3K2-regulated intestinal stromal cells define a distinct stem cell niche. Nature 2021, 592, 606–610. [Google Scholar] [CrossRef]
- Maurya, A.K.; Vinayak, M. PI-103 and Quercetin Attenuate PI3K-AKT Signaling Pathway in T-Cell Lymphoma Exposed to Hydrogen Peroxide. PLoS ONE 2016, 11, e0160686. [Google Scholar] [CrossRef]
- Sun, X.; Majumder, P.; Shioya, H.; Wu, F.; Kumar, S.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J. Biol. Chem. 2000, 275, 17237–17240. [Google Scholar] [CrossRef]
- Yoshida, K.; Yamaguchi, T.; Natsume, T.; Kufe, D.; Miki, Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell Biol. 2005, 7, 278–285. [Google Scholar] [CrossRef]
- Heppner, D.E.; van der Vliet, A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2016, 8, 24–27. [Google Scholar] [CrossRef]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef]
- Garcia, L.; Garcia, F.; Llorens, F.; Unzeta, M.; Itarte, E.; Gómez, N. PP1/PP2A phosphatases inhibitors okadaic acid and calyculin A block ERK5 activation by growth factors and oxidative stress. FEBS Lett. 2002, 523, 90–94. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef]
- Coon, B.G.; Timalsina, S.; Astone, M.; Zhuang, Z.W.; Fang, J.; Han, J.; Themen, J.; Chung, M.; Yang-Klingler, Y.J.; Jain, M.; et al. A mitochondrial contribution to anti-inflammatory shear stress signaling in vascular endothelial cells. J. Cell Biol. 2022, 221, e202109144. [Google Scholar] [CrossRef]
- Broustas, C.G.; Duval, A.J.; Chaudhary, K.R.; Friedman, R.A.; Virk, R.K.; Lieberman, H.B. Targeting MEK5 impairs nonhomologous end-joining repair and sensitizes prostate cancer to DNA damaging agents. Oncogene 2020, 39, 2467–2477. [Google Scholar] [CrossRef]
- Jiang, W.; Jin, G.; Cai, F.; Chen, X.; Cao, N.; Zhang, X.; Liu, J.; Chen, F.; Wang, F.; Dong, W.; et al. Extracellular signal-regulated kinase 5 increases radioresistance of lung cancer cells by enhancing the DNA damage response. Exp. Mol. Med. 2019, 51, 1–20. [Google Scholar] [CrossRef]
- Ulrich-Merzenich, G.; Zeitler, H.; Panek, D.; Bokemeyer, D.; Vetter, H. Vitamin C promotes human endothelial cell growth via the ERK-signaling pathway. Eur. J. Nutr. 2007, 46, 87–94. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.H. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [CrossRef]
- Verhamme, P.; Hoylaerts, M.F. The pivotal role of the endothelium in haemostasis and thrombosis. Acta Clin. Belg. 2006, 61, 213–219. [Google Scholar] [CrossRef]
- Higashi, Y.; Sasaki, S.; Kurisu, S.; Yoshimizu, A.; Sasaki, N.; Matsuura, H.; Kajiyama, G.; Oshima, T. Regular aerobic exercise augments endothelium-dependent vascular relaxation in normotensive as well as hypertensive subjects: Role of endothelium-derived nitric oxide. Circulation 1999, 100, 1194–1202. [Google Scholar] [CrossRef]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Kimura, M.; Noma, K.; Sasaki, S.; Hara, K.; Matsuura, H.; Goto, C.; Oshima, T.; et al. Low body mass index is a risk factor for impaired endothelium-dependent vasodilation in humans: Role of nitric oxide and oxidative stress. J. Am. Coll. Cardiol. 2003, 42, 256–263. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef]
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, E85–E90. [Google Scholar] [CrossRef]
- Sowers, J.R. Hypertension, angiotensin II, and oxidative stress. N. Engl. J. Med. 2002, 346, 1999–2001. [Google Scholar] [CrossRef]
- Leung, S.W.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization: Age, gender and blood pressure, do they matter? Acta Physiol. 2017, 219, 108–123. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef]
- Boulanger, C.M. Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e26–e31. [Google Scholar] [CrossRef]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, S.W.; Imanaka-Yoshida, K.; Yoshida, T.; Abel, E.D.; Eliceiri, B.; Yang, Y.; Ulevitch, R.J.; Lee, J.D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Investig. 2004, 113, 1138–1148. [Google Scholar] [CrossRef]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef]
- Yan, L.; Carr, J.; Ashby, P.R.; Murry-Tait, V.; Thompson, C.; Arthur, J.S. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 2003, 3, 11. [Google Scholar] [CrossRef]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Müller, J.; Cross, M.J. The role of ERK5 in endothelial cell function. Biochem. Soc. Trans. 2014, 42, 1584–1589. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., III; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef]
- Tatake, R.J.; O’Neill, M.M.; Kennedy, C.A.; Wayne, A.L.; Jakes, S.; Wu, D.; Kugler, S.Z., Jr.; Kashem, M.A.; Kaplita, P.; Snow, R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [Google Scholar] [CrossRef]
- Pardo-Andreu, G.L.; Paim, B.A.; Castilho, R.F.; Velho, J.A.; Delgado, R.; Vercesi, A.E.; Oliveira, H.C. Mangifera indica L. extract (Vimang) and its main polyphenol mangiferin prevent mitochondrial oxidative stress in atherosclerosis-prone hypercholesterolemic mouse. Pharmacol. Res. 2008, 57, 332–338. [Google Scholar] [CrossRef]
- Xia, G.; Li, X.; Zhu, X.; Yin, X.; Ding, H.; Qiao, Y. Mangiferin protects osteoblast against oxidative damage by modulation of ERK5/Nrf2 signaling. Biochem. Biophys. Res. Commun. 2017, 491, 807–813. [Google Scholar] [CrossRef]
- Lee, E.S.; Boldo, L.S.; Fernandez, B.O.; Feelisch, M.; Harmsen, M.C. Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF-beta1. Sci. Rep. 2017, 7, 42487. [Google Scholar] [CrossRef]
- Gan, W.; Dang, Y.; Han, X.; Ling, S.; Duan, J.; Liu, J.; Xu, J.W. ERK5/HDAC5-mediated, resveratrol-, and pterostilbene-induced expression of MnSOD in human endothelial cells. Mol. Nutr. Food Res. 2016, 60, 266–277. [Google Scholar] [CrossRef]
- Fava, C.; Montagnana, M. Atherosclerosis Is an Inflammatory Disease which Lacks a Common Anti-inflammatory Therapy: How Human Genetics Can Help to This Issue. A Narrative Review. Front. Pharmacol. 2018, 9, 55. [Google Scholar] [CrossRef]
- Vaccarezza, M.; Balla, C.; Rizzo, P. Atherosclerosis as an inflammatory disease: Doubts? No more. Int. J. Cardiol. Heart Vasc. 2018, 19, 1–2. [Google Scholar] [CrossRef]
- Kamal, K.; Du, W.; Mills, I.; Sumpio, B.E. Antiproliferative effect of elevated glucose in human microvascular endothelial cells. J. Cell. Biochem. 1998, 71, 491–501. [Google Scholar] [CrossRef]
- Liu, W.; Schoenkerman, A.; Lowe, W.L., Jr. Activation of members of the mitogen-activated protein kinase family by glucose in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E782–E790. [Google Scholar] [CrossRef]
- Woo, C.H.; Shishido, T.; McClain, C.; Lim, J.H.; Li, J.D.; Yang, J.; Yan, C.; Abe, J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 2008, 102, 538–545. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Jeune, B.; Pedersen, A.N.; Skinhøj, P.; Pedersen, B.K. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J. Gerontol. A Biol. Sci. Med. Sci. 1999, 54, M357–M364. [Google Scholar] [CrossRef]
- Ohnesorge, N.; Viemann, D.; Schmidt, N.; Czymai, T.; Spiering, D.; Schmolke, M.; Ludwig, S.; Roth, J.; Goebeler, M.; Schmidt, M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4). J. Biol. Chem. 2010, 285, 26199–26210. [Google Scholar] [CrossRef]
- Wu, K.; Tian, S.; Zhou, H.; Wu, Y. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochem. Pharmacol. 2013, 85, 1753–1760. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Yang, M.; Zhang, M.; Xiao, M.; Li, X. Butyrate mitigates TNF-α-induced attachment of monocytes to endothelial cells. J. Bioenerg. Biomembr. 2020, 52, 247–256. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, S.; Du, Z.; Ke, A.; Liang, Q.; Li, X. KLF2 Protects against Osteoarthritis by Repressing Oxidative Response through Activation of Nrf2/ARE Signaling In Vitro and In Vivo. Oxid. Med. Cell. Longev. 2019, 2019, 8564681. [Google Scholar] [CrossRef]
- Zhong, M.; Zhang, X.; Shi, X.; Zheng, C. Halofuginone inhibits LPS-induced attachment of monocytes to HUVECs. Int. Immunopharmacol. 2020, 87, 106753. [Google Scholar] [CrossRef]
- Sun, Z.; Han, Y.; Song, S.; Chen, T.; Han, Y.; Liu, Y. Activation of GPR81 by lactate inhibits oscillatory shear stress-induced endothelial inflammation by activating the expression of KLF2. IUBMB Life 2019, 71, 2010–2019. [Google Scholar] [CrossRef]
- Alipui, C.; Ramos, K.; Tenner, T.E., Jr. Alterations of rabbit aortic smooth muscle cell proliferation in diabetes mellitus. Cardiovasc. Res. 1993, 27, 1229–1232. [Google Scholar] [CrossRef]
- Natarajan, R.; Gonzales, N.; Xu, L.; Nadler, J.L. Vascular smooth muscle cells exhibit increased growth in response to elevated glucose. Biochem. Biophys. Res. Commun. 1992, 187, 552–560. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef]
- Yu, S.H.; Yu, J.M.; Yoo, H.J.; Lee, S.J.; Kang, D.H.; Cho, Y.J.; Kim, D.M. Anti-Proliferative Effects of Rutin on OLETF Rat Vascular Smooth Muscle Cells Stimulated by Glucose Variability. Yonsei Med. J. 2016, 57, 373–381. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Reckless, J.P. Fluvastatin. Expert Opin. Pharmacother. 2002, 3, 1631–1641. [Google Scholar] [CrossRef]
- Hwang, A.R.; Han, J.H.; Lim, J.H.; Kang, Y.J.; Woo, C.H. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PLoS ONE 2017, 12, e0178278. [Google Scholar] [CrossRef]
- Wang, C.C.; Sharma, G.; Draznin, B. Early growth response gene-1 expression in vascular smooth muscle cells effects of insulin and oxidant stress. Am. J. Hypertens. 2006, 19, 366–372. [Google Scholar] [CrossRef]
- Blaschke, F.; Bruemmer, D.; Law, R.E. Egr-1 is a major vascular pathogenic transcription factor in atherosclerosis and restenosis. Rev. Endocr. Metab. Disord. 2004, 5, 249–254. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar]
- Schiffrin, E.L. A critical review of the role of endothelial factors in the pathogenesis of hypertension. J. Cardiovasc. Pharmacol. 2001, 38 (Suppl. 2), S3–S6. [Google Scholar] [CrossRef]
- Kubo, T.; Ibusuki, T.; Chiba, S.; Kambe, T.; Fukumori, R. Mitogen-activated protein kinase activity regulation role of angiotensin and endothelin systems in vascular smooth muscle cells. Eur. J. Pharmacol. 2001, 411, 27–34. [Google Scholar] [CrossRef]
- Touyz, R.M.; He, G.; El Mabrouk, M.; Diep, Q.; Mardigyan, V.; Schiffrin, E.L. Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. J. Hypertens. 2001, 19, 553–559. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, Y.; Gorospe, M.; Udelsman, R.; Holbrook, N.J. Acute hypertension activates mitogen-activated protein kinases in arterial wall. J. Clin. Investig. 1996, 97, 508–514. [Google Scholar] [CrossRef]
- Touyz, R.M.; Cruzado, M.; Tabet, F.; Yao, G.; Salomon, S.; Schiffrin, E.L. Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: Role of receptor tyrosine kinase transactivation. Can. J. Physiol. Pharmacol. 2003, 81, 159–167. [Google Scholar] [CrossRef]
- Touyz, R.M.; Yao, G.; Viel, E.; Amiri, F.; Schiffrin, E.L. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J. Hypertens. 2004, 22, 1141–1149. [Google Scholar] [CrossRef]
- Morimoto, H.; Kanastu-Shinohara, M.; Ogonuki, N.; Kamimura, S.; Ogura, A.; Yabe-Nishimura, C.; Mori, Y.; Morimoto, T.; Watanabe, S.; Otsu, K.; et al. ROS amplification drives mouse spermatogonial stem cell self-renewal. Life Sci. Alliance 2019, 2, e201900374. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef]
- Devary, Y.; Gottlieb, R.A.; Smeal, T.; Karin, M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell 1992, 71, 1081–1091. [Google Scholar] [CrossRef]
- Baas, A.S.; Berk, B.C. Differential activation of mitogen-activated protein kinases by H2O2 and O2− in vascular smooth muscle cells. Circ. Res. 1995, 77, 29–36. [Google Scholar] [CrossRef]
- Takeishi, Y.; Abe, J.I.; Lee, J.D.; Kawakatsu, H.; Walsh, R.A.; Berk, B.C. Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ. Res. 1999, 85, 1164–1172. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Devereux, R.B.; Roman, M.J.; Paranicas, M.; O’Grady, M.J.; Lee, E.T.; Welty, T.K.; Fabsitz, R.R.; Robbins, D.; Rhoades, E.R.; Howard, B.V. Impact of diabetes on cardiac structure and function: The strong heart study. Circulation 2000, 101, 2271–2276. [Google Scholar] [CrossRef]
- Yan, C.; Ding, B.; Shishido, T.; Woo, C.H.; Itoh, S.; Jeon, K.I.; Liu, W.; Xu, H.; McClain, C.; Molina, C.A.; et al. Activation of extracellular signal-regulated kinase 5 reduces cardiac apoptosis and dysfunction via inhibition of a phosphodiesterase 3A/inducible cAMP early repressor feedback loop. Circ. Res. 2007, 100, 510–519. [Google Scholar] [CrossRef]
- Kimura, T.E.; Jin, J.; Zi, M.; Prehar, S.; Liu, W.; Oceandy, D.; Abe, J.; Neyses, L.; Weston, A.H.; Cartwright, E.J.; et al. Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ. Res. 2010, 106, 961–970. [Google Scholar] [CrossRef]
- Wang, Y.S.; Zhou, J.; Liang, C.; Hong, K.; Cheng, X.S.; Wu, Z.G. ERK5 knock down aggravates detrimental effects of hypothermal stimulation on cardiomyocytes via Bim upregulation. Environ. Toxicol. Pharmacol. 2013, 36, 724–731. [Google Scholar] [CrossRef]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Muñoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol. Rep. 2006, 16, 845–850. [Google Scholar] [CrossRef]
- Yu, B.; Zhao, Y.; Zhang, H.; Xie, D.; Nie, W.; Shi, K. Inhibition of microRNA-143-3p attenuates myocardial hypertrophy by inhibiting inflammatory response. Cell Biol. Int. 2018, 42, 1584–1593. [Google Scholar] [CrossRef]
- Cameron, S.J.; Ture, S.K.; Mickelsen, D.; Chakrabarti, E.; Modjeski, K.L.; McNitt, S.; Seaberry, M.; Field, D.J.; Le, N.T.; Abe, J.; et al. Platelet Extracellular Regulated Protein Kinase 5 Is a Redox Switch and Triggers Maladaptive Platelet Responses and Myocardial Infarct Expansion. Circulation 2015, 132, 47–58. [Google Scholar] [CrossRef]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef]
- Singh, M.V.; Kotla, S.; Le, N.T.; Ae Ko, K.; Heo, K.S.; Wang, Y.; Fujii, Y.; Thi Vu, H.; McBeath, E.; Thomas, T.N.; et al. Senescent Phenotype Induced by p90RSK-NRF2 Signaling Sensitizes Monocytes and Macrophages to Oxidative Stress in HIV-Positive Individuals. Circulation 2019, 139, 1199–1216. [Google Scholar] [CrossRef]
- Li, D.; Jin, L.; Alesi, G.N.; Kim, Y.M.; Fan, J.; Seo, J.H.; Wang, D.; Tucker, M.; Gu, T.L.; Lee, B.H.; et al. The prometastatic ribosomal S6 kinase 2-cAMP response element-binding protein (RSK2-CREB) signaling pathway up-regulates the actin-binding protein fascin-1 to promote tumor metastasis. J. Biol. Chem. 2013, 288, 32528–32538. [Google Scholar] [CrossRef]
- Kotla, S.; Zhang, A.; Imanishi, M.; Ko, K.A.; Lin, S.H.; Gi, Y.J.; Moczygemba, M.; Isgandarova, S.; Schadler, K.L.; Chung, C.; et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemoradiation. Redox Biol. 2021, 47, 102132. [Google Scholar] [CrossRef]
- Henricks, P.A.; Nijkamp, F.P. Reactive oxygen species as mediators in asthma. Pulm. Pharmacol. Ther. 2001, 14, 409–420. [Google Scholar] [CrossRef]
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- Nohl, H.; Kozlov, A.V.; Gille, L.; Staniek, K. Cell respiration and formation of reactive oxygen species: Facts and artefacts. Biochem. Soc. Trans. 2003, 31, 1308–1311. [Google Scholar] [CrossRef]
- Liu, P.L.; Chen, Y.L.; Chen, Y.H.; Lin, S.J.; Kou, Y.R. Wood smoke extract induces oxidative stress-mediated caspase-independent apoptosis in human lung endothelial cells: Role of AIF and EndoG. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2005, 289, L739–L749. [Google Scholar] [CrossRef]
- Vayssier-Taussat, M.; Camilli, T.; Aron, Y.; Meplan, C.; Hainaut, P.; Polla, B.S.; Weksler, B. Effects of tobacco smoke and benzo[a]pyrene on human endothelial cell and monocyte stress responses. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1293–H1300. [Google Scholar] [CrossRef]
- Mossman, B.T.; Kamp, D.W.; Weitzman, S.A. Mechanisms of carcinogenesis and clinical features of asbestos-associated cancers. Cancer Investig. 1996, 14, 466–480. [Google Scholar] [CrossRef]
- Scapoli, L.; Ramos-Nino, M.E.; Martinelli, M.; Mossman, B.T. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene 2004, 23, 805–813. [Google Scholar] [CrossRef]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef]
- Lopez-Royuela, N.; Rathore, M.G.; Allende-Vega, N.; Annicotte, J.S.; Fajas, L.; Ramachandran, B.; Gulick, T.; Villalba, M. Extracellular-signal-regulated kinase 5 modulates the antioxidant response by transcriptionally controlling Sirtuin 1 expression in leukemic cells. Int. J. Biochem. Cell Biol. 2014, 53, 253–261. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lin, H.R.; Lee, Y.H.; Huang, P.H.; Wei, H.C.; Stern, A.; Chiu, D.T. A novel fine tuning scheme of miR-200c in modulating lung cell redox homeostasis. Free Radic. Res. 2017, 51, 591–603. [Google Scholar] [CrossRef]
- Xue, Z.; Gao, X.; Yu, W.; Zhang, Q.; Song, W.; Li, S.; Zheng, X.; Kou, X. Biochanin A alleviates oxidative damage caused by the urban particulate matter. Food Funct. 2021, 12, 1958–1972. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef]
- Cao, Y.; Fang, Y.; Cai, J.; Li, X.; Xu, F.; Yuan, N.; Zhang, S.; Wang, J. ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology 2016, 21, 613–618. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguiló, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef]
- Susuki, T.; Yang, J. Hydrogen peroxide activation of ERK5 confers resistance to Jurkat cells against apoptosis induced by the extrinsic pathway. Biochem. Biophys. Res. Commun. 2014, 444, 248–253. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.H.; et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. eBioMedicine 2015, 3, 43–53. [Google Scholar] [CrossRef]
- Morena, M.; Cristol, J.P.; Senécal, L.; Leray-Moragues, H.; Krieter, D.; Canaud, B. Oxidative stress in hemodialysis patients: Is NADPH oxidase complex the culprit? Kidney Int. 2002, 61, S109–S114. [Google Scholar] [CrossRef]
- Ling, X.C.; Kuo, K.L. Oxidative stress in chronic kidney disease. Ren. Replace. Ther. 2018, 4, 53. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yao, L.; Nagai, Y.; Miyata, K.; Yoshizumi, M.; Kagami, S.; Kondo, S.; Kiyomoto, H.; Shokoji, T.; Kimura, S.; et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 2004, 43, 841–848. [Google Scholar] [CrossRef]
- Ishizawa, K.; Izawa-Ishizawa, Y.; Dorjsuren, N.; Miki, E.; Kihira, Y.; Ikeda, Y.; Hamano, S.; Kawazoe, K.; Minakuchi, K.; Tomita, S.; et al. Angiotensin II receptor blocker attenuates PDGF-induced mesangial cell migration in a receptor-independent manner. Nephrol. Dial. Transplant. 2010, 25, 364–372. [Google Scholar] [CrossRef]
- Urushihara, M.; Takamatsu, M.; Shimizu, M.; Kondo, S.; Kinoshita, Y.; Suga, K.; Kitamura, A.; Matsuura, S.; Yoshizumi, M.; Tamaki, T.; et al. ERK5 activation enhances mesangial cell viability and collagen matrix accumulation in rat progressive glomerulonephritis. Am. J. Physiol. Renal. Physiol. 2010, 298, F167–F176. [Google Scholar] [CrossRef]
- Ha, H.; Lee, H.B. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int. Suppl. 2000, 58, S19–S25. [Google Scholar] [CrossRef]
- Tomlinson, D.R. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia 1999, 42, 1271–1281. [Google Scholar] [CrossRef]
- Suzaki, Y.; Yoshizumi, M.; Kagami, S.; Nishiyama, A.; Ozawa, Y.; Kyaw, M.; Izawa, Y.; Kanematsu, Y.; Tsuchiya, K.; Tamaki, T. BMK1 is activated in glomeruli of diabetic rats and in mesangial cells by high glucose conditions. Kidney Int. 2004, 65, 1749–1760. [Google Scholar] [CrossRef]
- Kawakami, T.; Park, S.W.; Kaku, R.; Yang, J. Extracellular-regulated-kinase 5-mediated renal protection against ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 2012, 418, 603–608. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Lin, S.Y.; Dan, X.; Du, X.X.; Ran, C.L.; Lu, X.; Ren, S.J.; Tang, Z.T.; Yin, L.Z.; He, C.L.; Yuan, Z.X.; et al. Protective Effects of Salidroside against Carbon Tetrachloride (CCl4)-Induced Liver Injury by Initiating Mitochondria to Resist Oxidative Stress in Mice. Int. J. Mol. Sci. 2019, 20, 3187. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Shukla, V.; Mishra, S.K.; Pant, H.C. Oxidative stress in neurodegeneration. Adv. Pharmacol. Sci. 2011, 2011, 572634. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Liu, L.; Cavanaugh, J.E.; Wang, Y.; Sakagami, H.; Mao, Z.; Xia, Z. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc. Natl. Acad. Sci. USA 2003, 100, 8532–8537. [Google Scholar] [CrossRef]
- Shalizi, A.; Lehtinen, M.; Gaudilliere, B.; Donovan, N.; Han, J.; Konishi, Y.; Bonni, A. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J. Neurosci. 2003, 23, 7326–7336. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Nikam, S.; Nikam, P.; Ahaley, S.K.; Sontakke, A.V. Oxidative stress in Parkinson’s disease. Indian J. Clin. Biochem. 2009, 24, 98–101. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef]
- Su, C.; Sun, F.; Cunningham, R.L.; Rybalchenko, N.; Singh, M. ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age 2014, 36, 9685. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef]
- Liu, X.W.; Zi, Y.; Liu, Y.E.; Zhang, Y.B.; Xiang, L.B.; Hou, M.X. Melatonin exerts protective effect on N2a cells under hypoxia conditions through Zip1/ERK pathway. Neurosci. Lett. 2015, 595, 74–80. [Google Scholar] [CrossRef]
- Potdar, S.; Parmar, M.S.; Ray, S.D.; Cavanaugh, J.E. Protective effects of the resveratrol analog piceid in dopaminergic SH-SY5Y cells. Arch. Toxicol. 2018, 92, 669–677. [Google Scholar] [CrossRef]
- Cavanaugh, J.E.; Jaumotte, J.D.; Lakoski, J.M.; Zigmond, M.J. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J. Neurosci. Res. 2006, 84, 1367–1375. [Google Scholar] [CrossRef]
- Racette, B.A.; Aschner, M.; Guilarte, T.R.; Dydak, U.; Criswell, S.R.; Zheng, W. Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology 2012, 33, 881–886. [Google Scholar] [CrossRef]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef]
- Ding, H.; Wang, F.; Su, L.; Zhao, L.; Hu, B.; Zheng, W.; Yao, S.; Li, Y. Involvement of MEK5/ERK5 signaling pathway in manganese-induced cell injury in dopaminergic MN9D cells. J. Trace Elem. Med. Biol. 2020, 61, 126546. [Google Scholar] [CrossRef]
- Wang, R.M.; Zhang, Q.G.; Zhang, G.Y. Activation of ERK5 is mediated by N-methyl-D-aspartate receptor and L-type voltage-gated calcium channel via Src involving oxidative stress after cerebral ischemia in rat hippocampus. Neurosci. Lett. 2004, 357, 13–16. [Google Scholar] [CrossRef]
- Tubita, A.; Tusa, I.; Rovida, E. Playing the Whack-A-Mole Game: ERK5 Activation Emerges Among the Resistance Mechanisms to RAF-MEK1/2-ERK1/2-Targeted Therapy. Front. Cell Dev. Biol. 2021, 9, 647311. [Google Scholar] [CrossRef]

{kind=link}
| Tissue/System | Pathological Condition | Biological Outcome | Role | References |
|---|---|---|---|---|
| Cardiovascular system | Myocardial disease | ERK5 KD boosts hypothermia-induced injury/apoptosis of rat cardiomyocytes in vitro. | Protective | [134] |
| Cardiomyocyte-specific ERK5 KO in mice leads to dampened contractility. | Protective | [135] | ||
| Platelet-specific ERK5 KO mice show enhanced hyperlipidaemia-induced thrombosis. | Detrimental | [139] | ||
| Central nervous system | Degenerative disease | H2O2-induced SRC/ERK5 activation counteracts oxidative stress-induced damage through MEF2C in PC12 cells. | Protective | [36] |
| ERK5/KLF4 activation mediates PC- and NGF-induced neuroprotection against high H2O2 dose-induced cell death in PC12 cells and mouse primary hippocampal neurons in vitro. | Protective | [181] | ||
| Piceid protects SH-SY5Y cells from dopamine-induced cell death via ERK5 activation. | Protective | [185] | ||
| Pharmacological inhibition of MEK5 increases Mn-dependent cell death and ROS production in MN9D cells. | Protective | [189] | ||
| Urinary system | Glomerulonephritis | ERK5 activation enhances glomerular ECM accumulation in rats with GN. | Detrimental | [165] |
| ERK5 overexpression in mouse kidneys protects against renal I/R injury in vivo. | Protective | [169] | ||
| Blood vessel | Endothelial dysfunction | ERK5/NRF2 activation protects HUVECs from H2O2-induced cell death | Protective | [76] |
| ERK5 activation suppresses ROS/RNS generation caused by growth factor deprivation and shear stress in HUVECs. | Protective | [95] | ||
| Atherosclerosis | H2O2 and AGE inhibits ERK5/MEF2/KLF2 via ERK5 SUMOylation in HUVECs. | Protective | [101] | |
| LSS-induced mtROS promotes anti-inflammatory response via MEK5/ERK5/KLF2 in mouse ECs in vitro. | Protective | [72] | ||
| Statins inhibit TNF-α-induced ROS in human ECs via ERK5 activation in vitro. | Protective | [104] | ||
| Halofuginone promotes anti-inflammatory response via ERK5/NRF2 in HUVECs. | Protective | [108] | ||
| Lactate-induced GPR81 activation reduces OSS-induced oxidative stress via ERK5/KLF2 in HUVECs. | Protective | [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tusa, I.; Menconi, A.; Tubita, A.; Rovida, E. Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress. Cells 2023, 12, 1154. https://doi.org/10.3390/cells12081154
Tusa I, Menconi A, Tubita A, Rovida E. Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress. Cells. 2023; 12(8):1154. https://doi.org/10.3390/cells12081154
Chicago/Turabian StyleTusa, Ignazia, Alessio Menconi, Alessandro Tubita, and Elisabetta Rovida. 2023. "Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress" Cells 12, no. 8: 1154. https://doi.org/10.3390/cells12081154
APA StyleTusa, I., Menconi, A., Tubita, A., & Rovida, E. (2023). Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress. Cells, 12(8), 1154. https://doi.org/10.3390/cells12081154