



Selective PPAR-Delta/PPAR-Gamma Activation Improves Cognition in a Model of Alzheimer’s Disease
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
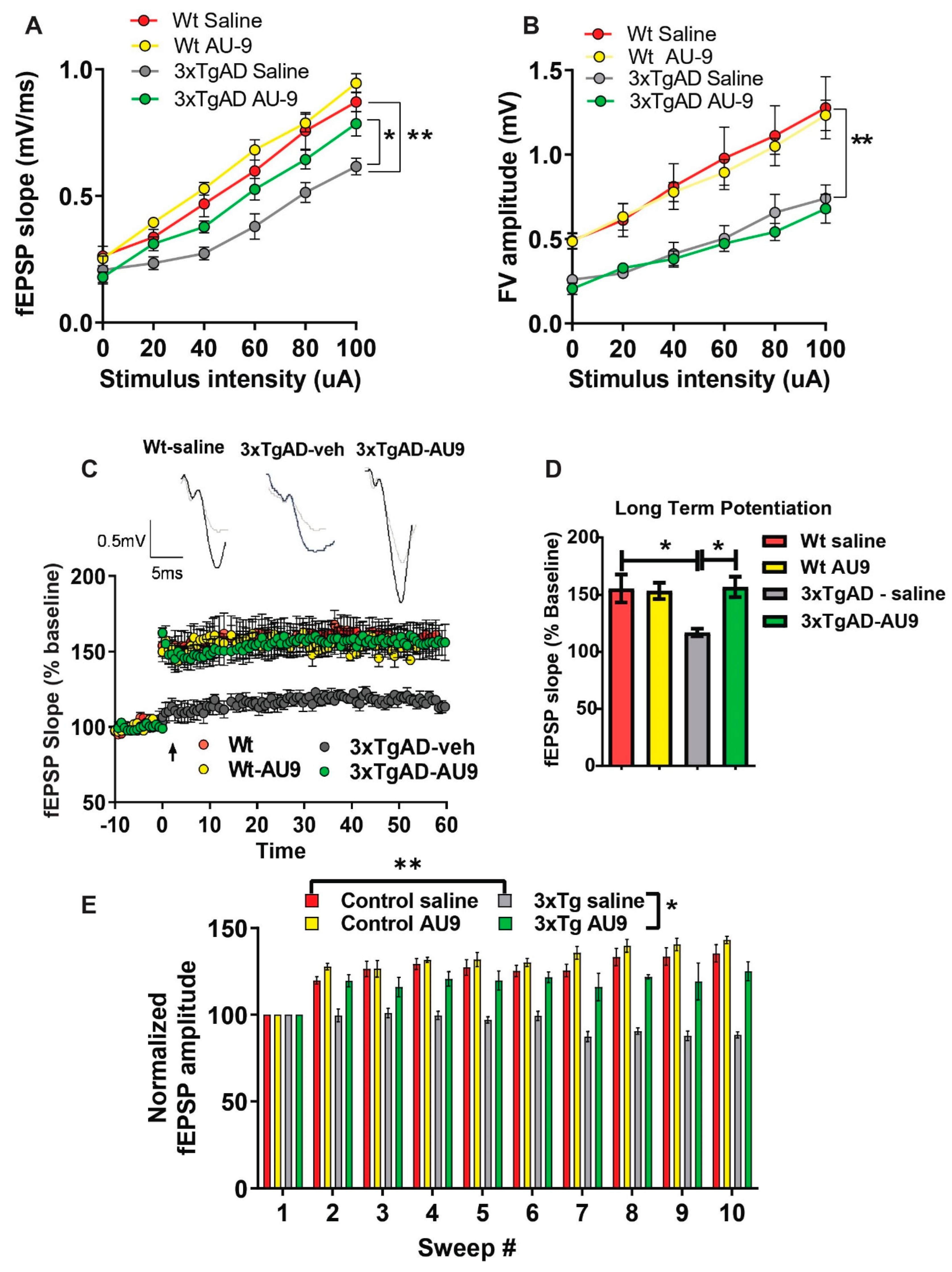
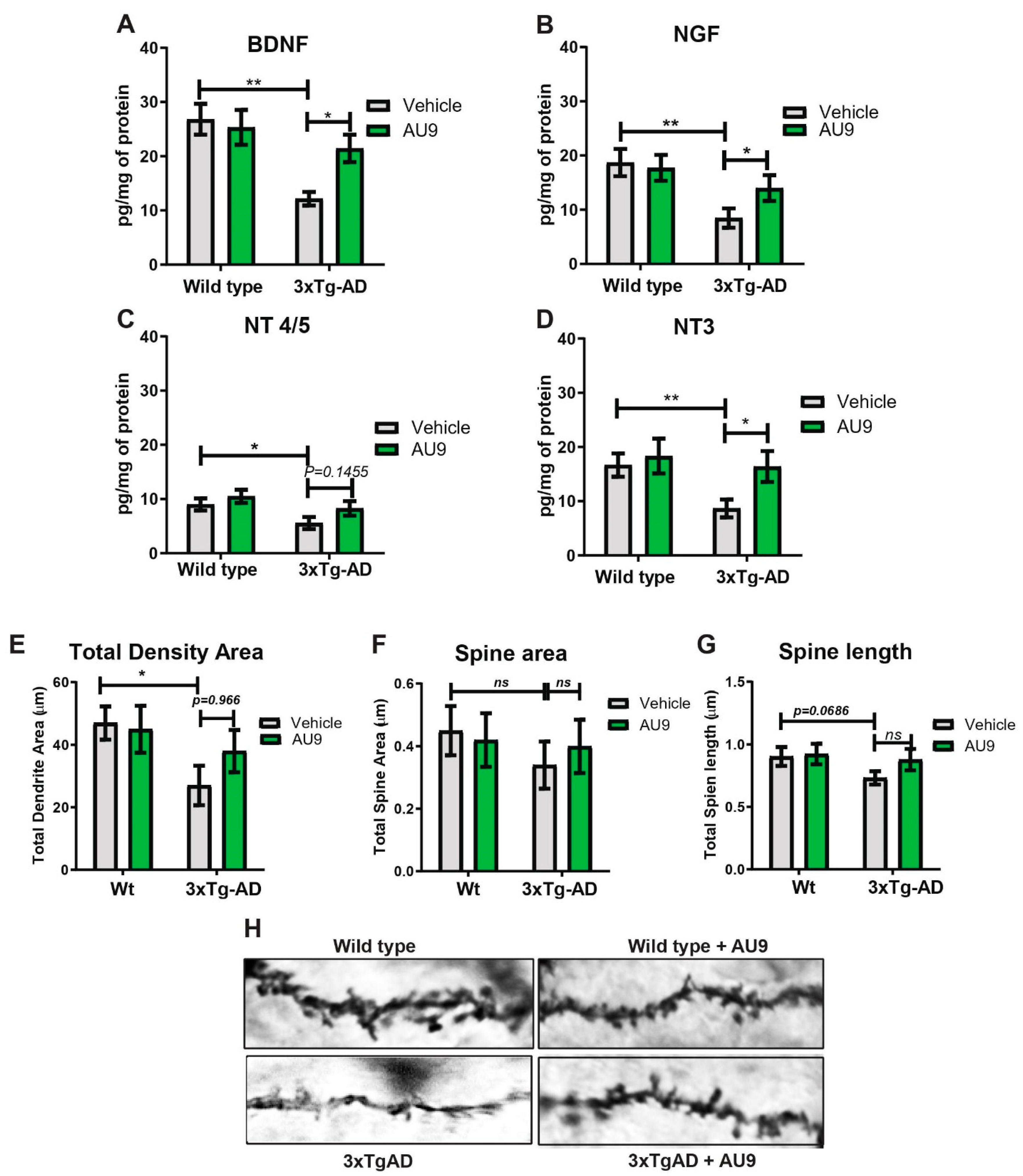
3. Results
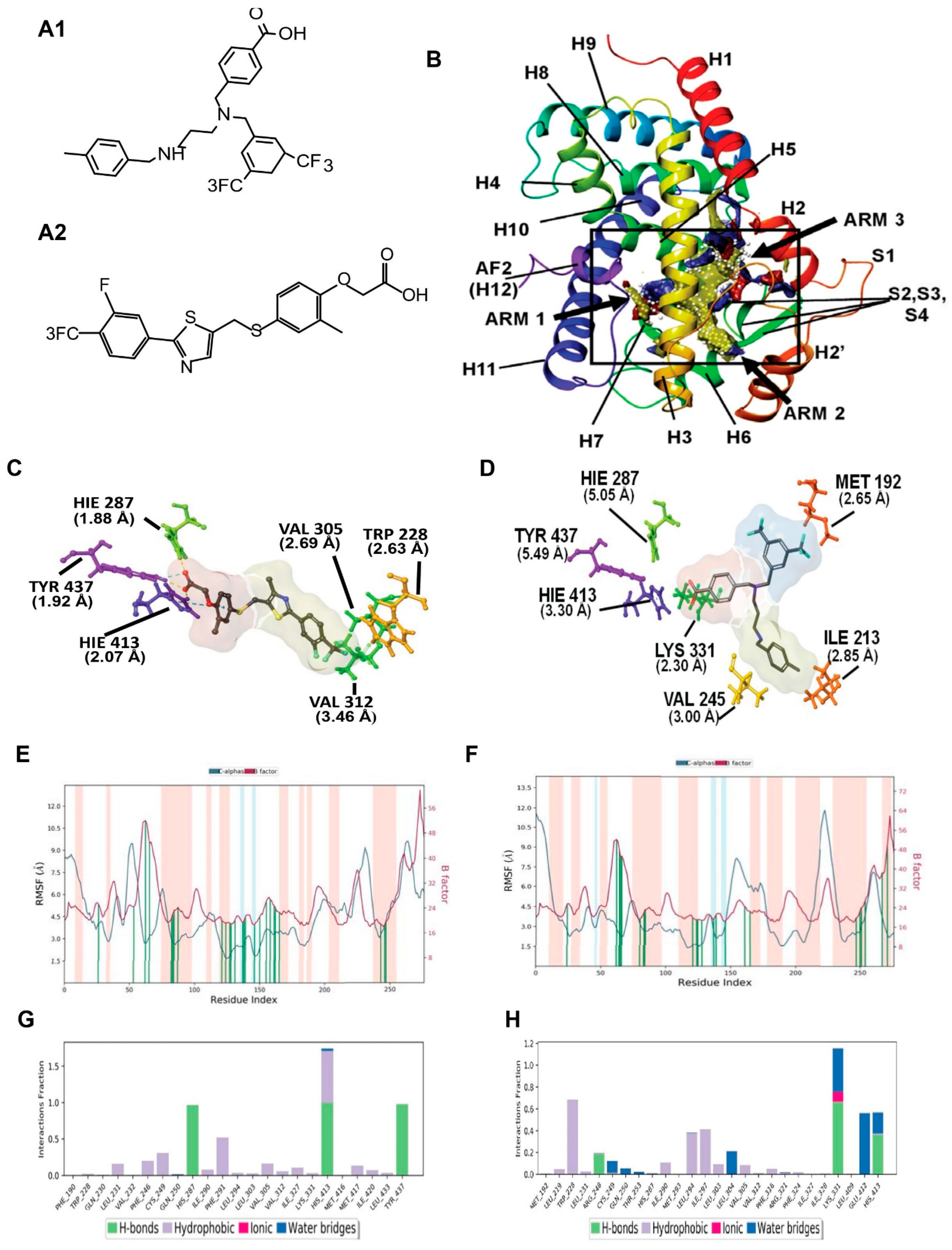
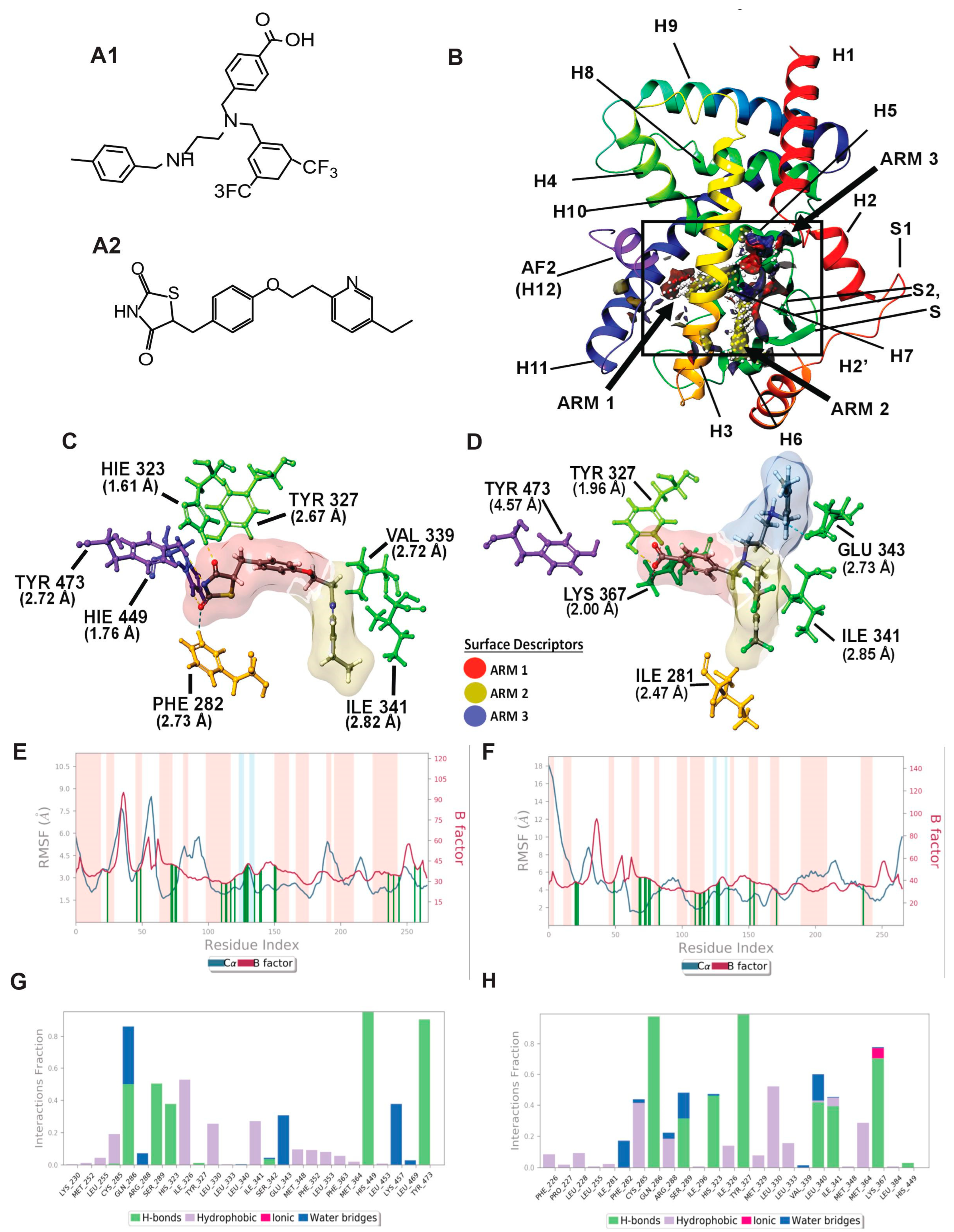
3.1. PPAR Delta Induced Fit Docking and Molecular Dynamics


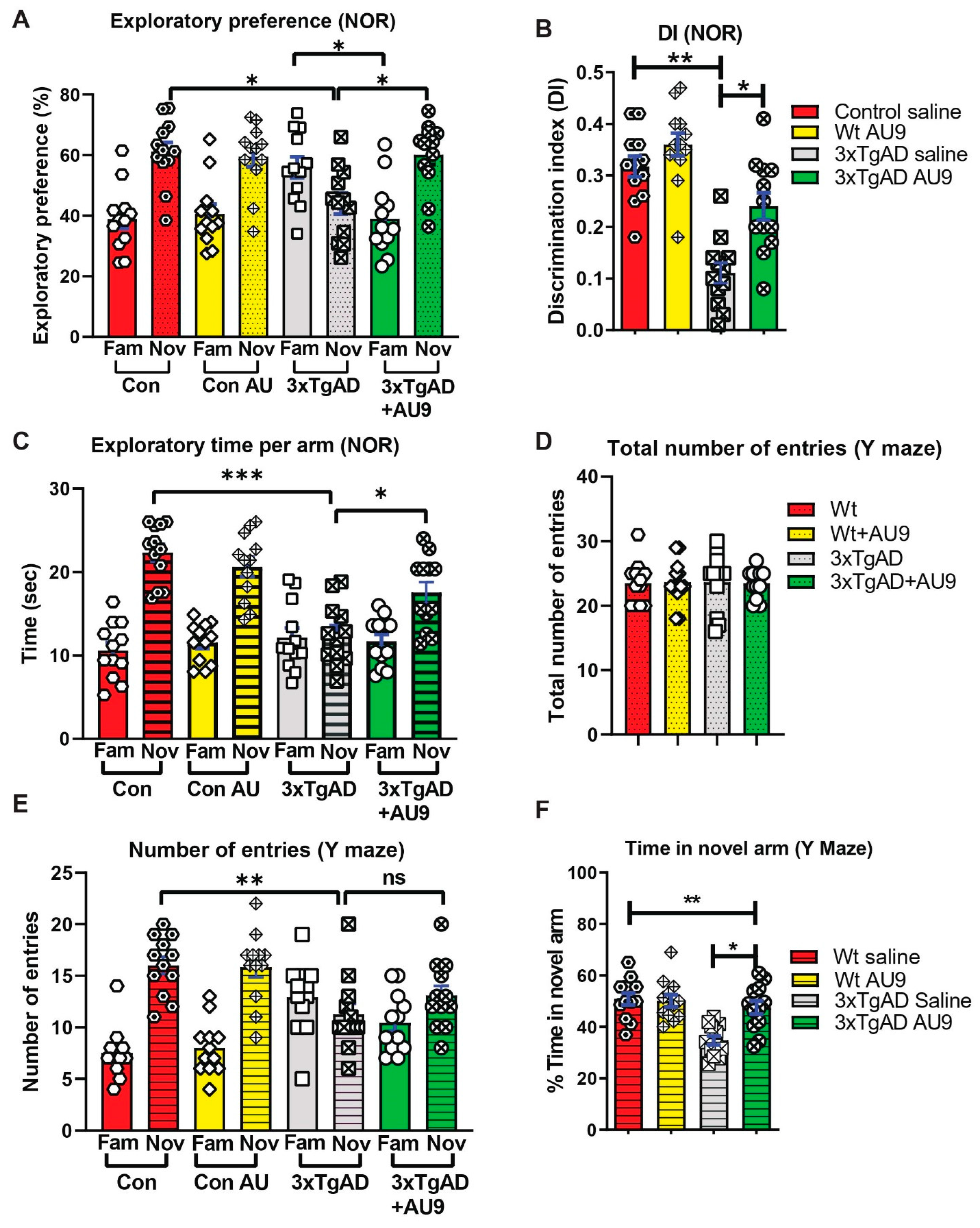
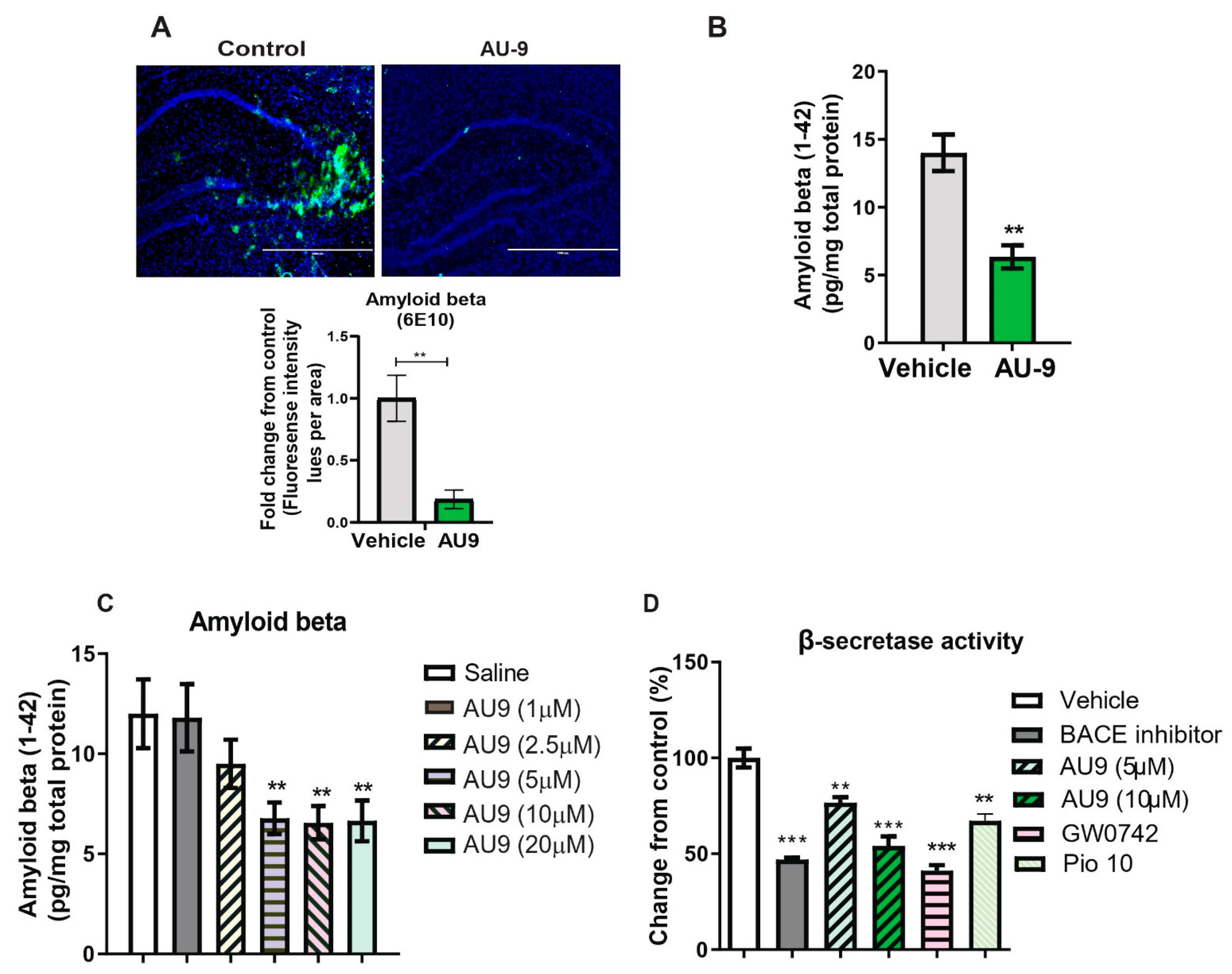
3.2. AU9 Reduces Aβ Levels in 3xTgAD Mice
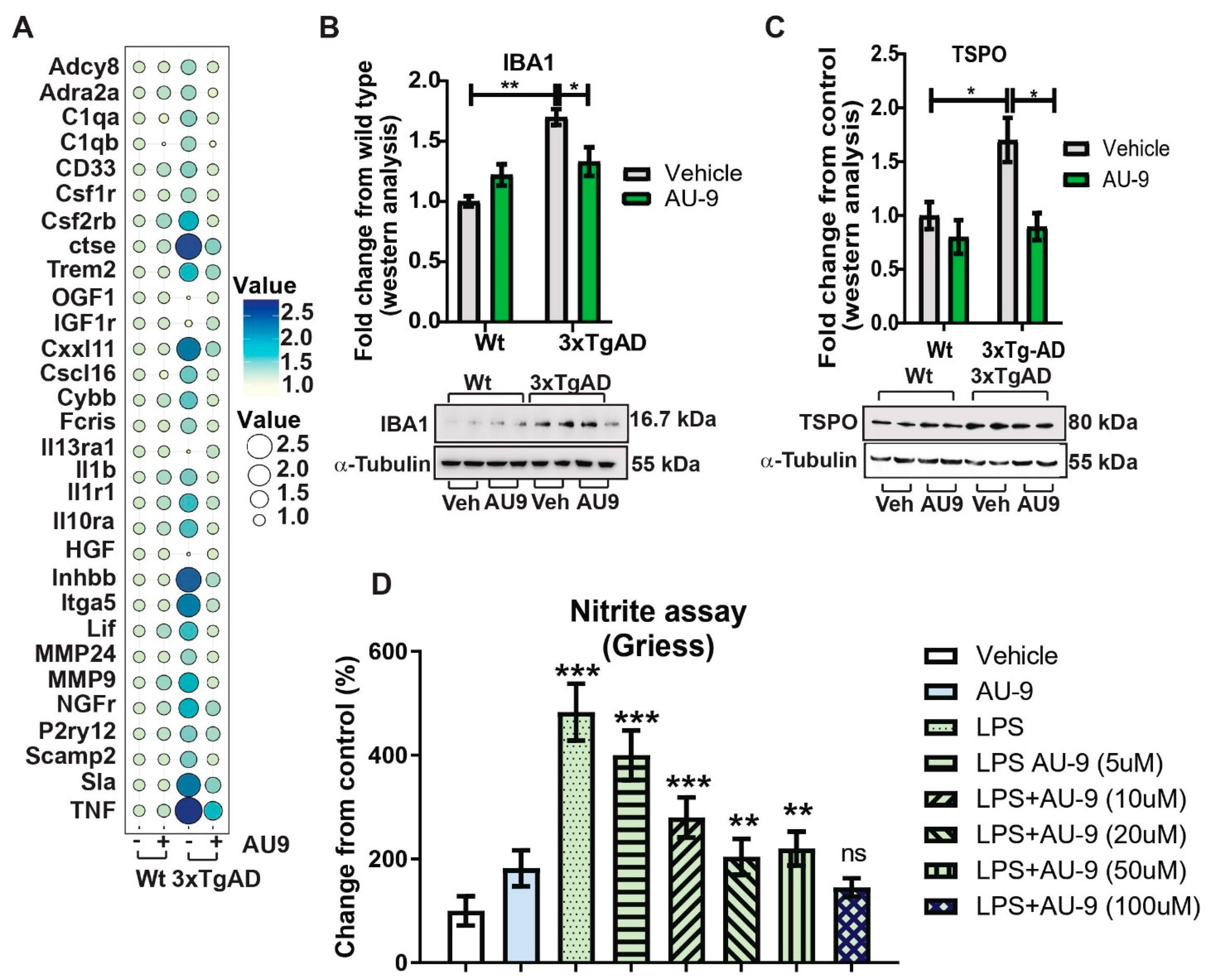
3.3. AU9 Reduces Neuroinflammation
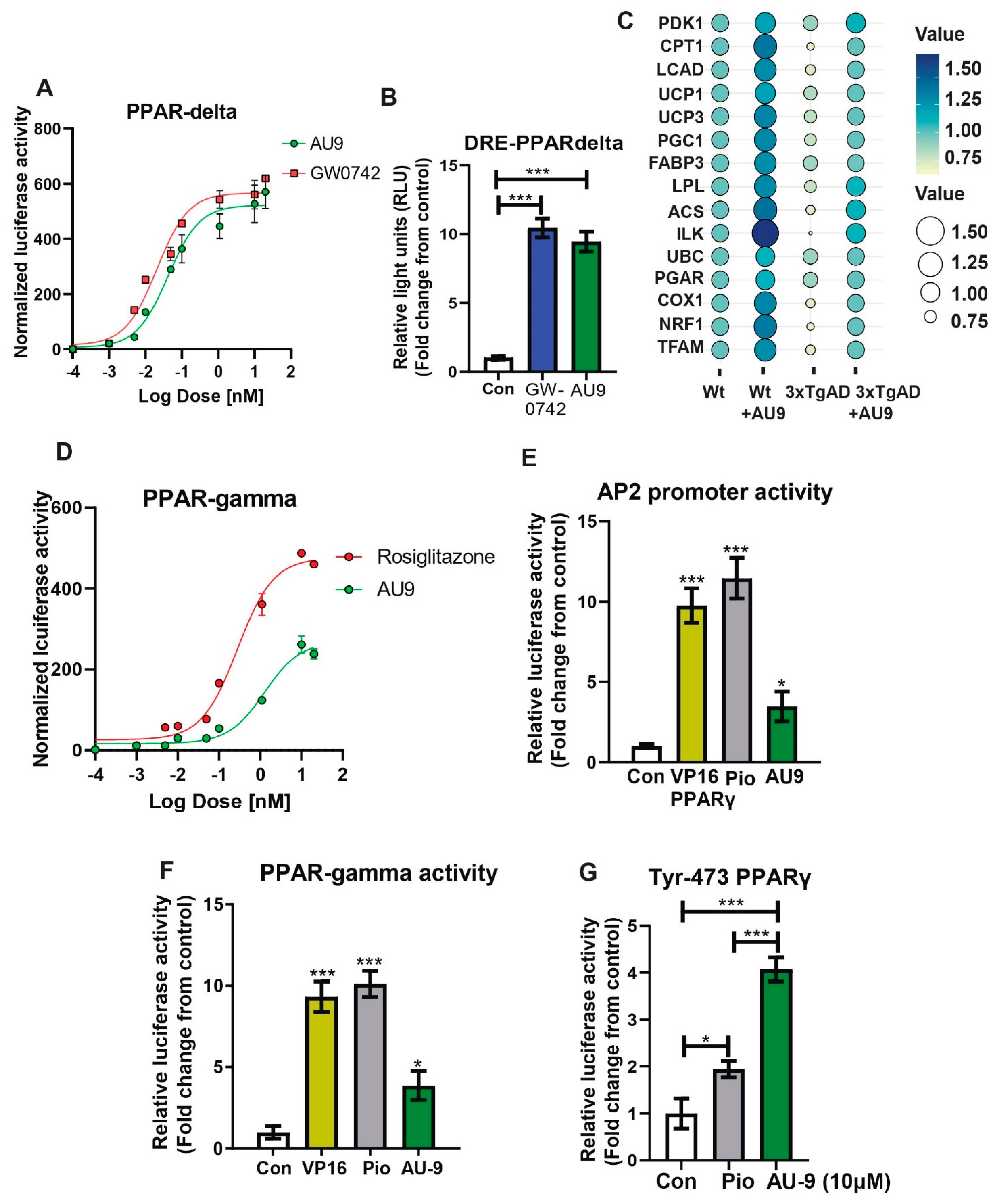
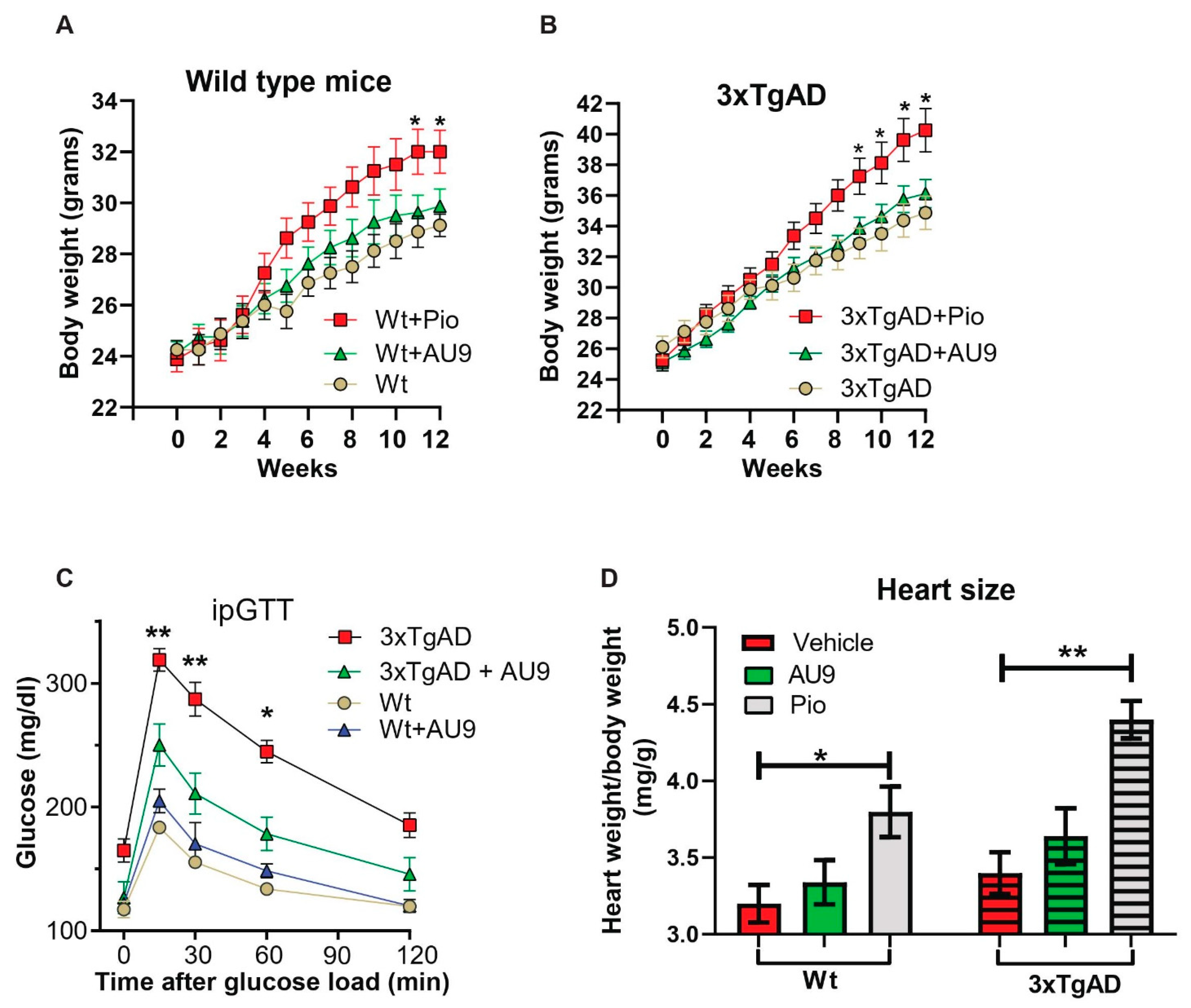
3.4. Peripheral Effects of AU9
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zieleniak, A.; Wojcik, M.; Wozniak, L.A. Structure and physiological functions of the human peroxisome proliferator-activated receptor gamma. Arch. Immunol. Ther. Exp. 2008, 56, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.E.; Berger, J.P. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. S3), S17–S21. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Pedersen, W.A.; McMillan, P.J.; Kulstad, J.J.; Leverenz, J.B.; Craft, S.; Haynatzki, G.R. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 2006, 199, 265–273. [Google Scholar] [CrossRef]
- Escribano, L.; Simon, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; Lopez de Maturana, R.; Garcia-Osta, A.; Ricobaraza, A.; Perez-Mediavilla, A.; Del Rio, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef]
- Costello, D.A.; O’Leary, D.M.; Herron, C.E. Agonists of peroxisome proliferator-activated receptor-gamma attenuate the Abeta-mediated impairment of LTP in the hippocampus in vitro. Neuropharmacology 2005, 49, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Pathan, A.R.; Gaikwad, A.B.; Viswanad, B.; Ramarao, P. Rosiglitazone attenuates the cognitive deficits induced by high fat diet feeding in rats. Eur. J. Pharm. Pharmacol. 2008, 589, 176–179. [Google Scholar] [CrossRef]
- Jiang, L.Y.; Tang, S.S.; Wang, X.Y.; Liu, L.P.; Long, Y.; Hu, M.; Liao, M.X.; Ding, Q.L.; Hu, W.; Li, J.C.; et al. PPARgamma agonist pioglitazone reverses memory impairment and biochemical changes in a mouse model of type 2 diabetes mellitus. CNS Neurosci. Ther. 2012, 18, 659–666. [Google Scholar] [CrossRef]
- Masciopinto, F.; Di Pietro, N.; Corona, C.; Bomba, M.; Pipino, C.; Curcio, M.; Di Castelnuovo, A.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.; et al. Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 2012, 3, e448. [Google Scholar] [CrossRef]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef]
- Chamberlain, S.; Gabriel, H.; Strittmatter, W.; Didsbury, J. An Exploratory Phase IIa Study of the PPAR delta/gamma Agonist T3D-959 Assessing Metabolic and Cognitive Function in Subjects with Mild to Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1085–1103. [Google Scholar] [CrossRef]
- Gathiaka, S.; Nanayakkara, G.; Boncher, T.; Acevedo, O.; Wyble, J.; Patel, S.; Patel, A.; Shane, M.E.; Bonkowski, B.; Wieczorek, J.; et al. Design, development and evaluation of novel dual PPARdelta/PPARgamma agonists. Bioorg. Med. Chem. Lett. 2013, 23, 873–879. [Google Scholar] [CrossRef]
- Hieke, M.; Ness, J.; Steri, R.; Dittrich, M.; Greiner, C.; Werz, O.; Baumann, K.; Schubert-Zsilavecz, M.; Weggen, S.; Zettl, H. Design, synthesis, and biological evaluation of a novel class of gamma-secretase modulators with PPARgamma activity. J. Med. Chem. 2010, 53, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Bloemer, J.; Pinky, P.D.; Smith, W.D.; Bhattacharya, D.; Chauhan, A.; Govindarajulu, M.; Hong, H.; Dhanasekaran, M.; Judd, R.; Amin, R.H.; et al. Adiponectin Knockout Mice Display Cognitive and Synaptic Deficits. Front. Endocrinol. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Leger, M.; Quiedeville, A.; Bouet, V.; Haelewyn, B.; Boulouard, M.; Schumann-Bard, P.; Freret, T. Object recognition test in mice. Nat. Protoc. 2013, 8, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- Stover, K.R.; Campbell, M.A.; Van Winssen, C.M.; Brown, R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 2015, 289, 29–38. [Google Scholar] [CrossRef]
- Parameshwaran, K.; Buabeid, M.A.; Karuppagounder, S.S.; Uthayathas, S.; Thiruchelvam, K.; Shonesy, B.; Dityatev, A.; Escobar, M.C.; Dhanasekaran, M.; Suppiramaniam, V. Developmental nicotine exposure induced alterations in behavior and glutamate receptor function in hippocampus. Cell. Mol. Life Sci. 2012, 69, 829–841. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. In Pre-Clinical Models. Methods in Molecular Biology; Guest, P., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1916, pp. 105–111. [Google Scholar]
- Sarnyai, Z.; Sibille, E.L.; Pavlides, C.; Fenster, R.J.; McEwen, B.S.; Toth, M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 14731–14736. [Google Scholar] [CrossRef]
- Conrad, C.D.; Galea, L.A.; Kuroda, Y.; McEwen, B.S. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav. Neurosci. 1996, 110, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.S.; Abraham, W.C. Cooperative interactions among afferents govern the induction of homosynaptic long-term depression in the hippocampus. Proc. Natl. Acad. Sci. USA 1995, 92, 11637–11641. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, M.; Bukalo, O.; Chazal, G.; Wang, L.; Goridis, C.; Schachner, M.; Gerardy-Schahn, R.; Cremer, H.; Dityatev, A. Mice deficient in the polysialyltransferase ST8SiaIV/PST-1 allow discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity. J. Neurosci. 2000, 20, 5234–5244. [Google Scholar] [CrossRef]
- Graves, R.A.; Tontonoz, P.; Spiegelman, B.M. Analysis of a tissue-specific enhancer: ARF6 regulates adipogenic gene expression. Mol. Cell. Biol. 1992, 12, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar] [CrossRef]
- Risher, W.C.; Ustunkaya, T.; Singh Alvarado, J.; Eroglu, C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE 2014, 9, e107591. [Google Scholar] [CrossRef]
- Kolbeck, R.; Bartke, I.; Eberle, W.; Barde, Y.A. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J. Neurochem. 1999, 72, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Kilroy, G.E.; Zhang, X.; Floyd, Z.E. PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity 2009, 17, 665–673. [Google Scholar] [CrossRef]
- Shang, J.; Kojetin, D.J. Structural mechanism underlying ligand binding and activation of PPARgamma. Structure 2021, 29, 940–950.e4. [Google Scholar] [CrossRef]
- Markt, P.; Schuster, D.; Kirchmair, J.; Laggner, C.; Langer, T. Pharmacophore modeling and parallel screening for PPAR ligands. J. Comput. Aided Mol. Des. 2007, 21, 575–590. [Google Scholar] [CrossRef]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; Xu, H.E.; Montana, V.G.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)--synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sakamoto, J.; Kimura, H.; Moriyama, S.; Odaka, H.; Momose, Y.; Sugiyama, Y.; Sawada, H. Activation of Human Peroxisome Proliferator-Activated Receptor (PPAR) Subtypes by Pioglitazone. Biochem. Biophys. Res. Commun. 2000, 278, 704–711. [Google Scholar] [CrossRef]
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Le Winter, M.; Porte, D.; Semenkovich, C.F.; Smith, S.; et al. Thiazolidinedione use, fluid retention, and congestive heart failure: A consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004, 27, 256–263. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef]
- Denner, L.A.; Rodriguez-Rivera, J.; Haidacher, S.J.; Jahrling, J.B.; Carmical, J.R.; Hernandez, C.M.; Zhao, Y.; Sadygov, R.G.; Starkey, J.M.; Spratt, H.; et al. Cognitive enhancement with rosiglitazone links the hippocampal PPARgamma and ERK MAPK signaling pathways. J. Neurosci. 2012, 32, 16725–16735. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, E.A.; Bhattacharya, S.; Buabeid, M.; Majrashi, M.; Bloemer, J.; Tao, Y.X.; Dhanasekaran, M.; Escobar, M.; Amin, R.; Suppiramaniam, V. PPAR-delta Activation Ameliorates Diabetes-Induced Cognitive Dysfunction by Modulating Integrin-linked Kinase and AMPA Receptor Function. J. Am. Coll. Nutr. 2019, 38, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.F.; Ho, H.; Greger, I.H. Synaptic transmission and plasticity require AMPA receptor anchoring via its N-terminal domain. eLife 2017, 6, e23024. [Google Scholar] [CrossRef] [PubMed]
- Kariharan, T.; Nanayakkara, G.; Parameshwaran, K.; Bagasrawala, I.; Ahuja, M.; Abdel-Rahman, E.; Amin, A.T.; Dhanasekaran, M.; Suppiramaniam, V.; Amin, R.H. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol. Aging 2015, 36, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Gomez-Palacio-Schjetnan, A.; Escobar, M.L. Neurotrophins and synaptic plasticity. Curr. Top. Behav. Neurosci. 2013, 15, 117–136. [Google Scholar] [CrossRef] [PubMed]
- McAllister, A.K.; Katz, L.C.; Lo, D.C. Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci. 1999, 22, 295–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cui, Y.; Yang, J.; Zhang, J.; Yuan, D.; Wei, Y.; Li, Y.; Duo, Y.; Li, S.; Zhu, W.; et al. Combining serum and urine biomarkers in the early diagnosis of mild cognitive impairment that evolves into Alzheimer’s disease in patients with the apolipoprotein E ϵ4 genotype. Biomarkers 2015, 20, 84–88. [Google Scholar] [CrossRef]
- Siuda, J.; Patalong-Ogiewa, M.; Zmuda, W.; Targosz-Gajniak, M.; Niewiadomska, E.; Matuszek, I.; Jedrzejowska-Szypulka, H.; Lewin-Kowalik, J.; Rudzinska-Bar, M. Cognitive impairment and BDNF serum levels. Neurol. Neurochir. Polska 2017, 51, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Dursun, E.; Hanagasi, H.; Bilgic, B.; Lohman, E.; Araz, O.S.; Atasoy, I.L.; Alaylioglu, M.; Onal, B.; Gurvit, H.; et al. BDNF, TNFalpha, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J. Alzheimer’s Dis. 2013, 37, 185–195. [Google Scholar] [CrossRef]
- Peng, S.; Garzon, D.J.; Marchese, M.; Klein, W.; Ginsberg, S.D.; Francis, B.M.; Mount, H.T.; Mufson, E.J.; Salehi, A.; Fahnestock, M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2009, 29, 9321–9329. [Google Scholar] [CrossRef]
- Javonillo, D.I.; Tran, K.M.; Phan, J.; Hingco, E.; Kramar, E.A.; da Cunha, C.; Forner, S.; Kawauchi, S.; Milinkeviciute, G.; Gomez-Arboledas, A.; et al. Systematic Phenotyping and Characterization of the 3xTg-AD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 1829. [Google Scholar] [CrossRef] [PubMed]
- Vandal, M.; White, P.J.; Chevrier, G.; Tremblay, C.; St-Amour, I.; Planel, E.; Marette, A.; Calon, F. Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. FASEB J. 2015, 29, 4273–4284. [Google Scholar] [CrossRef]
- Saunders, A.M.; Burns, D.K.; Gottschalk, W.K. Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666958. [Google Scholar] [CrossRef]
- Searcy, J.L.; Phelps, J.T.; Pancani, T.; Kadish, I.; Popovic, J.; Anderson, K.L.; Beckett, T.L.; Murphy, M.P.; Chen, K.C.; Blalock, E.M.; et al. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 30, 943–961. [Google Scholar] [CrossRef]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Burns, D.K.; Alexander, R.C.; Welsh-Bohmer, K.A.; Culp, M.; Chiang, C.; O’Neil, J.; Evans, R.M.; Harrigan, P.; Plassman, B.L.; Burke, J.R.; et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Einstein, M.; Akiyama, T.E.; Castriota, G.A.; Wang, C.F.; McKeever, B.; Mosley, R.T.; Becker, J.W.; Moller, D.E.; Meinke, P.T.; Wood, H.B.; et al. The differential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is a physical basis for their unique biological activities. Mol. Pharmacol. 2008, 73, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Burris, T.P. Small molecule modulation of nuclear receptor conformational dynamics: Implications for function and drug discovery. Mol. Pharmacol. 2013, 83, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.M.; Fairall, L.; Schwabe, J.W. Negative regulation by nuclear receptors: A plethora of mechanisms. Trends Endocrinol. Metab. 2011, 22, 87–93. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Zhou, H.X. From induced fit to conformational selection: A continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J. 2010, 98, L15–L17. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Reimers, J.M.; Loweth, J.A.; Wolf, M.E. BDNF contributes to both rapid and homeostatic alterations in AMPA receptor surface expression in nucleus accumbens medium spiny neurons. Eur. J. Neurosci. 2014, 39, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Sri, S.; Pegasiou, C.M.; Cave, C.A.; Hough, K.; Wood, N.; Gomez-Nicola, D.; Deinhardt, K.; Bannerman, D.; Perry, V.H.; Vargas-Caballero, M. Emergence of synaptic and cognitive impairment in a mature-onset APP mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Makela, J.; Tselykh, T.V.; Kukkonen, J.P.; Eriksson, O.; Korhonen, L.T.; Lindholm, D. Peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist is neuroprotective and stimulates PGC-1alpha expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 2016, 102, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Thouennon, E.; Cheng, Y.; Falahatian, V.; Cawley, N.X.; Loh, Y.P. Rosiglitazone-activated PPARgamma induces neurotrophic factor-alpha1 transcription contributing to neuroprotection. J. Neurochem. 2015, 134, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; D’Angelo, B.; Cristiano, L.; Di Giacomo, E.; Fanelli, F.; Moreno, S.; Cecconi, F.; Fidoamore, A.; Antonosante, A.; Falcone, R.; et al. Involvement of peroxisome proliferator-activated receptor beta/delta (PPAR beta/delta) in BDNF signaling during aging and in Alzheimer disease: Possible role of 4-hydroxynonenal (4-HNE). Cell. Cycle 2014, 13, 1335–1344. [Google Scholar] [CrossRef]
- Sherwood, N.T.; Lo, D.C. Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J. Neurosci. 1999, 19, 7025–7036. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef]
- Assaf, N.; El-Shamarka, M.E.; Salem, N.A.; Khadrawy, Y.A.; El Sayed, N.S. Neuroprotective effect of PPAR alpha and gamma agonists in a mouse model of amyloidogenesis through modulation of the Wnt/beta catenin pathway via targeting alpha- and beta-secretases. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109793. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinke, I.; Govindarajulu, M.; Pinky, P.D.; Bloemer, J.; Yoo, S.; Ward, T.; Schaedig, T.; Young, T.; Wibowo, F.S.; Suppiramaniam, V.; et al. Selective PPAR-Delta/PPAR-Gamma Activation Improves Cognition in a Model of Alzheimer’s Disease. Cells 2023, 12, 1116. https://doi.org/10.3390/cells12081116
Steinke I, Govindarajulu M, Pinky PD, Bloemer J, Yoo S, Ward T, Schaedig T, Young T, Wibowo FS, Suppiramaniam V, et al. Selective PPAR-Delta/PPAR-Gamma Activation Improves Cognition in a Model of Alzheimer’s Disease. Cells. 2023; 12(8):1116. https://doi.org/10.3390/cells12081116
Chicago/Turabian StyleSteinke, Ian, Manoj Govindarajulu, Priyanka Das Pinky, Jenna Bloemer, Sieun Yoo, Tracey Ward, Taylor Schaedig, Taylor Young, Fajar Setyo Wibowo, Vishnu Suppiramaniam, and et al. 2023. "Selective PPAR-Delta/PPAR-Gamma Activation Improves Cognition in a Model of Alzheimer’s Disease" Cells 12, no. 8: 1116. https://doi.org/10.3390/cells12081116
APA StyleSteinke, I., Govindarajulu, M., Pinky, P. D., Bloemer, J., Yoo, S., Ward, T., Schaedig, T., Young, T., Wibowo, F. S., Suppiramaniam, V., & Amin, R. H. (2023). Selective PPAR-Delta/PPAR-Gamma Activation Improves Cognition in a Model of Alzheimer’s Disease. Cells, 12(8), 1116. https://doi.org/10.3390/cells12081116