
Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunofluorescent Staining
2.3. Image Analysis
2.4. Western Blot Analysis
2.5. Weight Gain and Longevity
2.6. Statistical Analysis
3. Results
3.1. Rapamycin Upregulates Autophagy
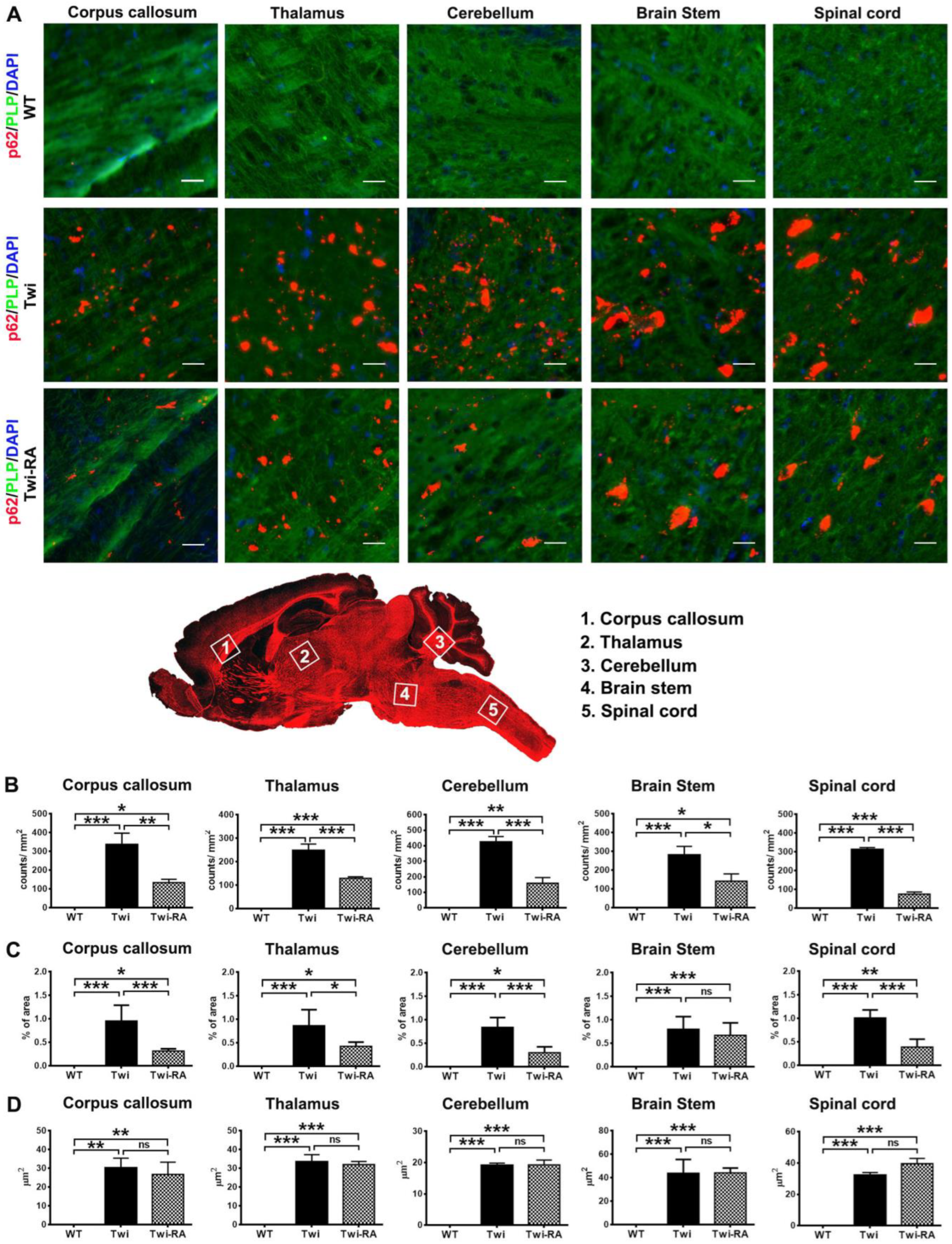
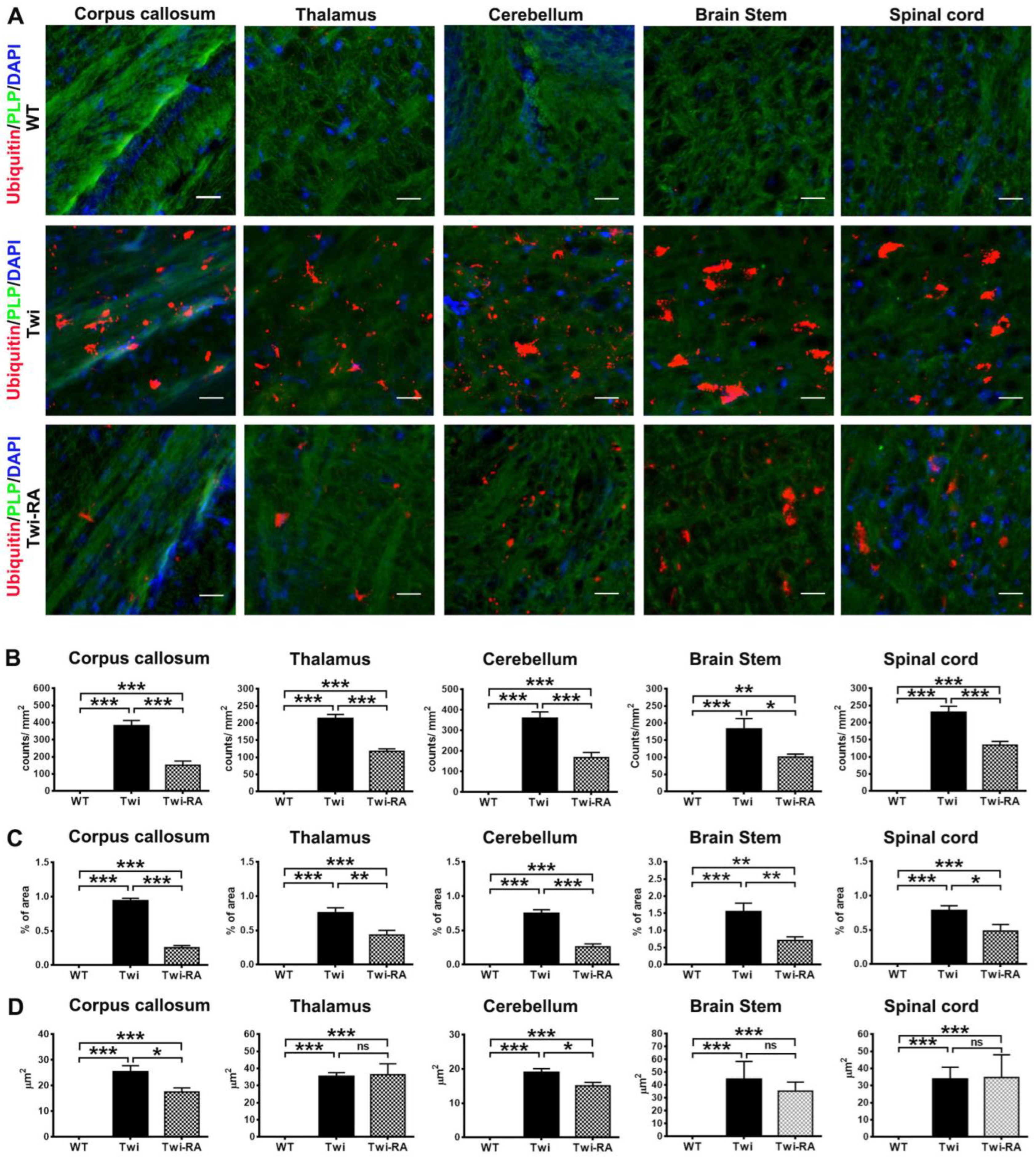
3.2. Reduction of Ubiquitin- and p62-Aggregates
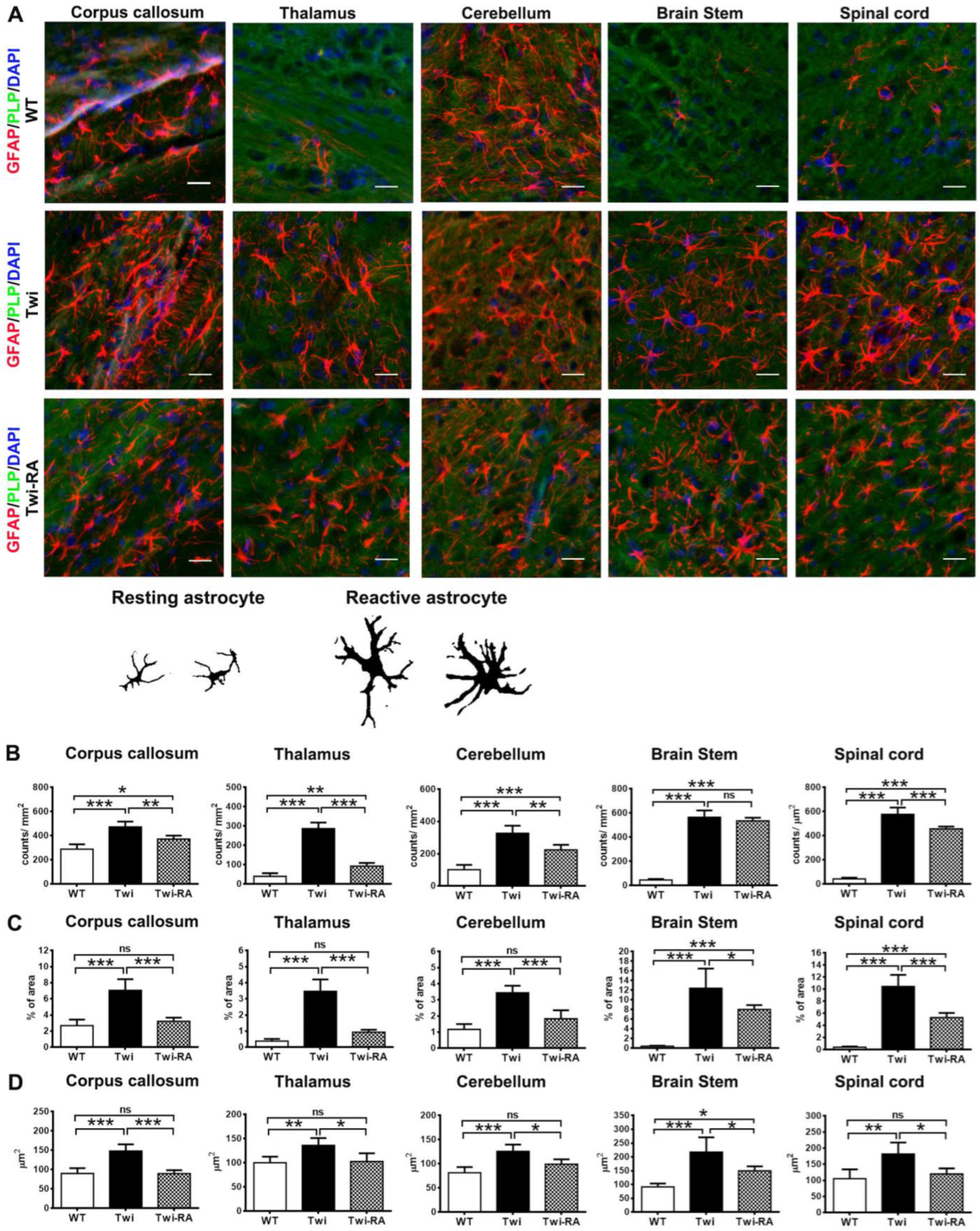
3.3. Alleviation of Reactive Astrocytes
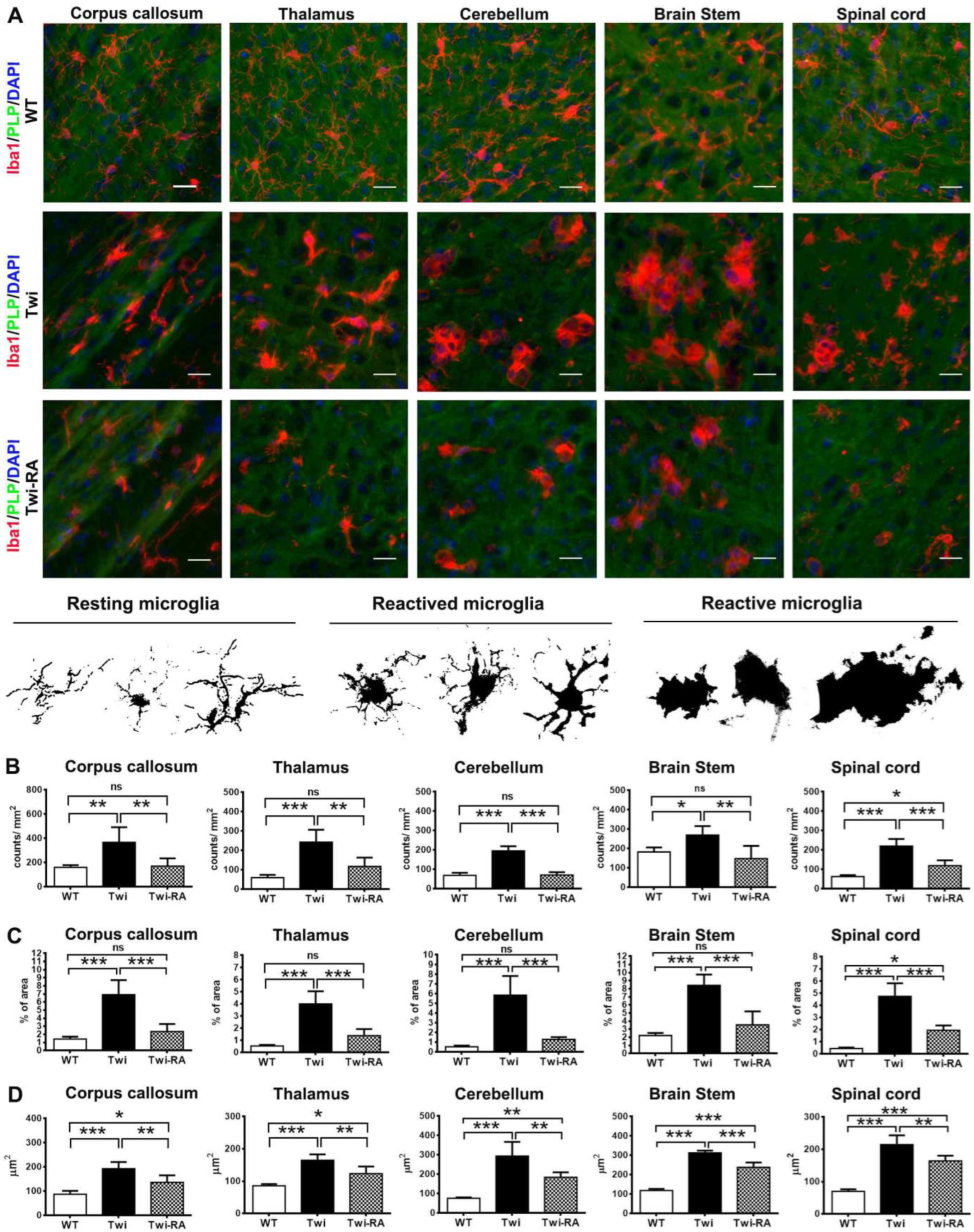
3.4. Alleviation of Microglia Activation
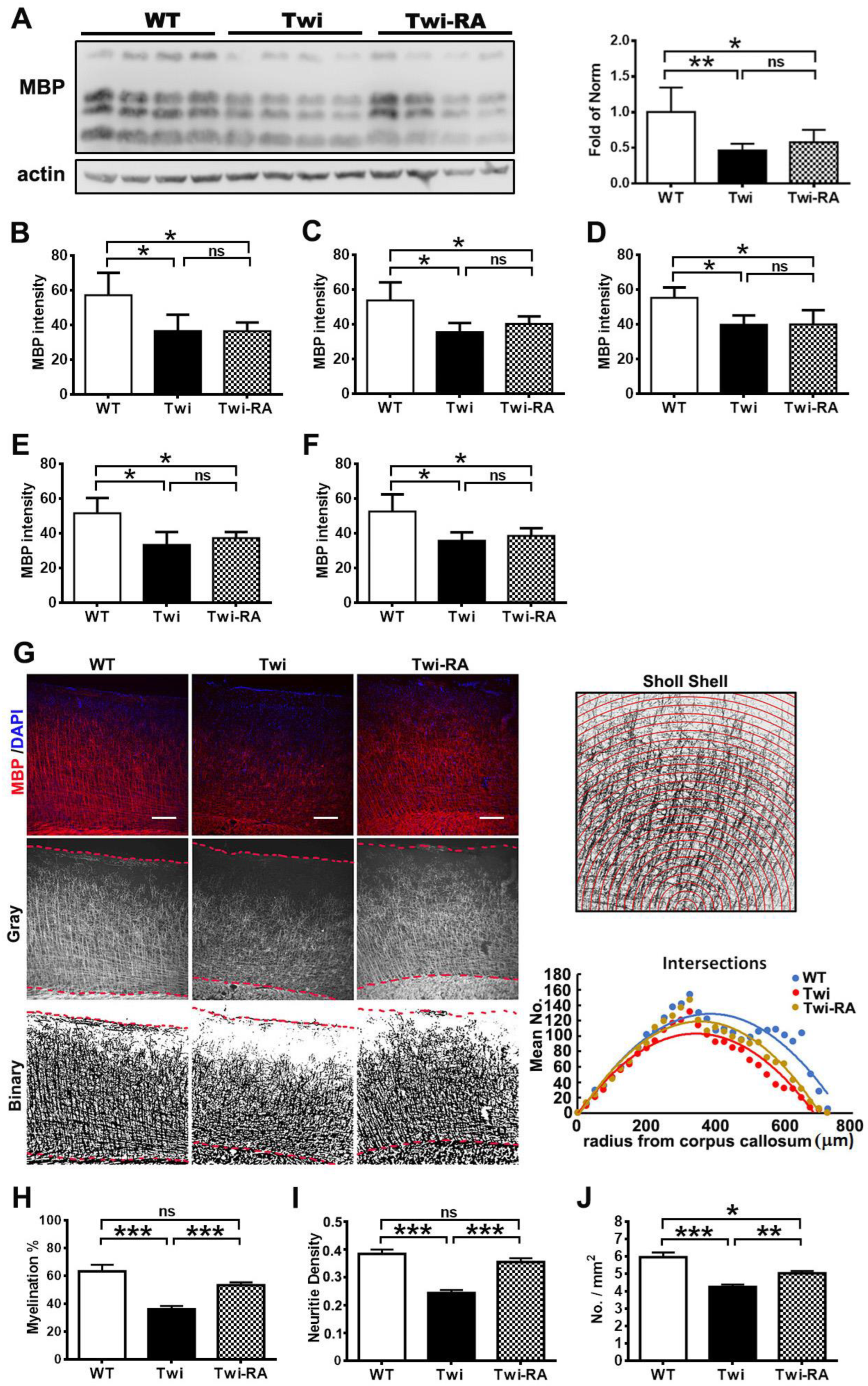
3.5. Rapamycin Preserves Cortical Myeloarchitecture
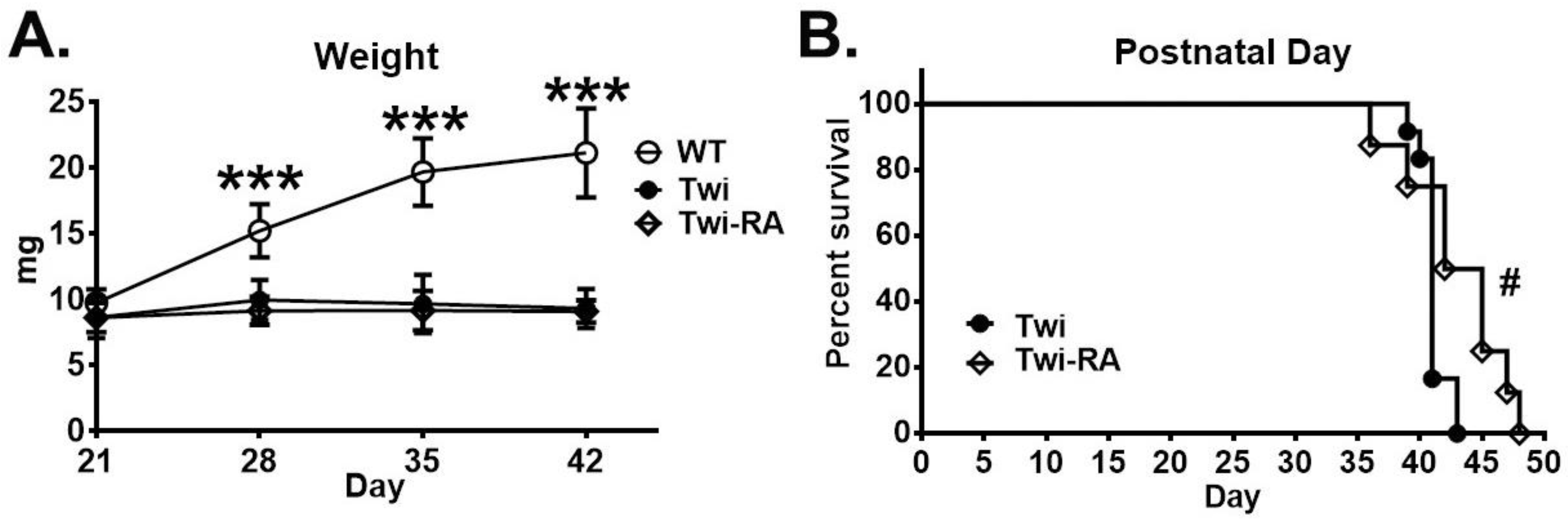
3.6. Body Weight Was Not Altered by Rapamycin
3.7. Prolonged Lifespan by Rapamycin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wenger, D.A.; Rafi, M.A.; Luzi, P. Krabbe disease: One Hundred years from the bedside to the bench to the bedside. J. Neurosci. Res. 2016, 94, 982–989. [Google Scholar] [CrossRef]
- Duffner, P.K.; Jalal, K.; Carter, R.L. The Hunter’s Hope Krabbe family database. Pediatr. Neurol. 2009, 40, 13–18. [Google Scholar] [CrossRef]
- Suzuki, K.; Suzuki, Y. Globoid cell leucodystrophy (Krabbe’s disease): Deficiency of galactocerebroside beta-galactosidase. Proc. Natl. Acad. Sci. USA 1970, 66, 302–329. [Google Scholar] [CrossRef]
- Wenger, D.A.; Rafi, M.A.; Luzi, P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): Diagnostic and clinical implications. Hum. Mutat. 1997, 10, 268–279. [Google Scholar] [CrossRef]
- Vanier, M.-T.; Svennerholm, L. Chemical Pathology of Krabbe Disease: The Occurrence of Psychosine and Other Neutral Sphingoglycolipids. Adv. Exp. Med. Biol. 1976, 68, 115–126. [Google Scholar] [CrossRef]
- Svennerholm, L.; Vanier, M.T.; Månsson, J.E. Krabbe disease: A galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 1980, 21, 53–64. [Google Scholar] [CrossRef]
- Miyatake, T.; Suzuki, K. Globoid cell leukodystrophy: Additional deficiency of psychosine galactosidase. Biochem. Biophys. Res. Commun. 1972, 48, 538–543. [Google Scholar] [CrossRef]
- Suzuki, K.; Suzuki, K. The twitcher mouse: A model for Krabbe disease and for experimental therapies. Brain Pathol. 1995, 5, 249–258. [Google Scholar] [CrossRef]
- Miyatake, T.; Suzuki, K. Galactosylsphingosine galactosyl hydrolase. Partial purification and properties of the enzyme in rat brain. J. Biol. Chem. 1972, 247, 5398–5403. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103. [Google Scholar] [CrossRef]
- Inamura, N.; Kito, M.; Go, S.; Kishi, S.; Hosokawa, M.; Asai, K.; Takakura, N.; Takebayashi, H.; Matsuda, J.; Enokido, Y. Developmental defects and aberrant accumulation of endogenous psychosine in oligodendrocytes in a murine model of Krabbe disease. Neurobiol. Dis. 2018, 120, 51–62. [Google Scholar] [CrossRef]
- Cantuti Castelvetri, L.; Givogri, M.I.; Hebert, A.; Smith, B.; Song, Y.; Kaminska, A.; Lopez-Rosas, A.; Morfini, G.; Pigino, G.; Sands, M.; et al. The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3beta and deregulation of molecular motors. J. Neurosci. 2013, 33, 10048–10056. [Google Scholar] [CrossRef]
- Wenger, D.A. Murine, canine and non-human primate models of Krabbe disease. Mol. Med. Today 2000, 6, 449–451. [Google Scholar] [CrossRef]
- Lin, D.-S.; Ho, C.-S.; Huang, Y.-W.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Wang, T.-J.; Yang, S.-J.; Chiang, M.-F. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Del Grosso, A.; Angella, L.; Tonazzini, I.; Moscardini, A.; Giordano, N.; Caleo, M.; Rocchiccioli, S.; Cecchini, M. Dysregulated autophagy as a new aspect of the molecular pathogenesis of Krabbe disease. Neurobiol. Dis. 2019, 129, 195–207. [Google Scholar] [CrossRef]
- Ribbens, J.J.; Moser, A.B.; Hubbard, W.C.; Bongarzone, E.R.; Maegawa, G.H. Characterization and application of a disease-cell model for a neurodegenerative lysosomal disease. Mol. Genet. Metab. 2014, 111, 172–183. [Google Scholar] [CrossRef]
- Del Grosso, A.; Antonini, S.; Angella, L.; Tonazzini, I.; Signore, G.; Cecchini, M. Lithium improves cell viability in psychosine-treated MO3.13 human oligodendrocyte cell line via autophagy activation. J. Neurosci. Res. 2016, 94, 1246–1260. [Google Scholar] [CrossRef]
- Pellegrini, D.; del Grosso, A.; Angella, L.; Giordano, N.; Dilillo, M.; Tonazzini, I.; Caleo, M.; Cecchini, M.; McDonnell, L.A. Quantitative Microproteomics Based Characterization of the Central and Peripheral Nervous System of a Mouse Model of Krabbe Disease. Mol. Cell. Proteom. 2019, 18, 1227–1241. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 860–864. [Google Scholar] [CrossRef]
- Sakai, N.; Inui, K.; Tatsumi, N.; Fukushima, H.; Nishigaki, T.; Taniike, M.; Nishimoto, J.; Tsukamoto, H.; Yanagihara, I.; Ozono, K.; et al. Molecular Cloning and Expression of cDNA for Murine Galactocerebrosidase and Mutation Analysis of the Twitcher Mouse, a Model of Krabbe’s Disease. J. Neurochem. 1996, 66, 1118–1124. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Bongarzone, E.R.; Crocker, S.J. A microglial hypothesis of globoid cell leukodystrophy pathology. J. Neurosci. Res. 2016, 94, 1049–1061. [Google Scholar] [CrossRef]
- Snook, E.R.; Fisher-Perkins, J.M.; Sansing, H.A.; Lee, K.M.; Alvarez, X.; MacLean, A.G.; Peterson, K.E.; Lackner, A.A.; Bunnell, B.A. Innate Immune Activation in the Pathogenesis of a Murine Model of Globoid Cell Leukodystrophy. Am. J. Pathol. 2014, 184, 382–396. [Google Scholar] [CrossRef]
- van Tilborg, E.; van Kammen, C.M.; de Theije, C.G.M.; van Meer, M.P.A.; Dijkhuizen, R.M.; Nijboer, C.H. A quantitative method for microstructural analysis of myelinated axons in the injured rodent brain. Sci. Rep. 2017, 7, 16492. [Google Scholar] [CrossRef]
- Weinstock, N.I.; Kreher, C.; Favret, J.; Nguyen, D.; Bongarzone, E.R.; Wrabetz, L.; Feltri, M.L.; Shin, D. Brainstem development requires galactosylceramidase and is critical for pathogenesis in a model of Krabbe disease. Nat. Commun. 2020, 11, 5356. [Google Scholar] [CrossRef]
- Kobayashi, S.; Chiu, F.C.; Katayama, M.; Sacchi, R.S.; Suzuki, K.; Suzuki, K. Expression of glial fibrillary acidic protein in the CNS and PNS of murine globoid cell leukodystrophy, the twitcher. Am. J. Pathol. 1986, 125, 227–243. [Google Scholar] [PubMed]
- Potter, G.B.; Petryniak, M.A. Neuroimmune mechanisms in Krabbe’s disease. J. Neurosci. Res. 2016, 94, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Nutma, E.; Marzin, M.C.; Cillessen, S.A.; Amor, S. Autophagy in white matter disorders of the CNS : Mechanisms and therapeutic opportunities. J. Pathol. 2021, 253, 133–147. [Google Scholar] [CrossRef]
- Folts, C.J.; Scott-Hewitt, N.; Pröschel, C.; Mayer-Pröschel, M.; Noble, M. Lysosomal Re-acidification Prevents Lysosphingolipid-Induced Lysosomal Impairment and Cellular Toxicity. PLoS Biol. 2016, 14, e1002583. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. An evolutionarily conserved pathway controls proteasome homeostasis. Nature 2016, 536, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Quan, Z.; Rash, B.; Oliver, S.G. Synergistic effects of TOR and proteasome pathways on the yeast transcriptome and cell growth. Open Biol. 2013, 3, 120137. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.L.; Hall, M.N. Elucidating TOR Signaling and Rapamycin Action: Lessons from Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2002, 66, 579–591. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing Autophagy by Rapamycin Before, but Not After, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Y.; Yu, X.; Lu, J.; Jia, W.; Song, J.; Liu, L.; Wang, Y.; Huang, Y.; Xie, J.; et al. Corynoxine Protects Dopaminergic Neurons Through Inducing Autophagy and Diminishing Neuroinflammation in Rotenone-Induced Animal Models of Parkinson’s Disease. Front. Pharmacol. 2021, 12, 642900. [Google Scholar] [CrossRef]
- Sn, S.; Pandurangi, J.; Murumalla, R.; Dj, V.; Garimella, L.; Acharya, A.; Rai, S.; Paul, A.; Yarreiphang, H.; Pillai, M.S.; et al. Small molecule modulator of aggrephagy regulates neuroinflammation to curb pathogenesis of neurodegeneration. Ebiomedicine 2019, 50, 260–273. [Google Scholar] [CrossRef]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Kanner, S.; Zacs, M.; Frisca, F.; Pinto, A.R.; Currie, P.D.; Pinkas-Kramarski, R. Rapamycin increases neuronal survival, reduces inflammation and astrocyte proliferation after spinal cord injury. Mol. Cell. Neurosci. 2015, 68, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, X.; Wu, X.; Lei, M.; Wei, Z.; Zhang, X.; Wen, L.; Xu, P.; Li, S.; Qu, S. Rapamycin upregulates glutamate transporter and IL-6 expression in astrocytes in a mouse model of Parkinson’s disease. Cell Death Dis. 2017, 8, e2611. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Wenning, G.K.; Stefanova, N.; Jellinger, K.A.; Poewe, W.; Schlossmacher, M.G. Multiple system atrophy: A primary oligodendrogliopathy. Ann. Neurol. 2008, 64, 239–246. [Google Scholar] [CrossRef]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn, W.A.; Notterpek, L. Rapamycin Activates Autophagy and Improves Myelination in Explant Cultures from Neuropathic Mice. J. Neurosci. 2010, 30, 11388–11397. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A.; Joy, S.; Li, J.; Notterpek, L. Emerging Role for Autophagy in the Removal of Aggresomes in Schwann Cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef]
- Fortun, J.; Verrier, J.D.; Go, J.C.; Madorsky, I.; Dunn, W.A.; Notterpek, L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol. Dis. 2007, 25, 252–265. [Google Scholar] [CrossRef]
- Carson, R.P.; Van Nielen, D.L.; Winzenburger, P.A.; Ess, K.C. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 2012, 45, 369–380. [Google Scholar] [CrossRef]
- Shi, Q.; Saifetiarova, J.; Taylor, A.M.; Bhat, M.A. mTORC1 Activation by Loss of Tsc1 in Myelinating Glia Causes Downregulation of Quaking and Neurofascin 155 Leading to Paranodal Domain Disorganization. Front. Cell. Neurosci. 2018, 12, 201. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhang, B.; Liu, W.; Sun, M.-J.; Zhang, Y.-L.; Liu, H.; Wang, M.-X. Rapamycin Alleviates the Symptoms of Multiple Sclerosis in Experimental Autoimmune Encephalomyelitis (EAE) Through Mediating the TAM-TLRs-SOCS Pathway. Front. Neurol. 2020, 11, 590884. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, R.; Huang, Z.; Lin, J.; Ji, W.; Huang, Z.; Liu, Q.; Wu, X.; Wu, X.; Jiang, H.; et al. Rapamycin Preserves Neural Tissue, Promotes Schwann Cell Myelination and Reduces Glial Scar Formation After Hemi-Contusion Spinal Cord Injury in Mice. Front. Mol. Neurosci. 2020, 13, 574041. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M.; Pedchenko, T.V.; Bronshteyn, I.G.; Pinson, D.M. L-cycloserine slows the clinical and pathological course in mice with globoid cell leukodystrophy (twitcher mice). J. Neurosci. Res. 2000, 60, 231–236. [Google Scholar] [CrossRef]
- Lin, D.-S.; Hsiao, C.-D.; Liau, I.; Lin, S.-P.; Chiang, M.-F.; Chuang, C.-K.; Wang, T.-J.; Wu, T.-Y.; Jian, Y.-R.; Huang, S.-F.; et al. CNS-targeted AAV5 gene transfer results in global dispersal of vector and prevention of morphological and function deterioration in CNS of globoid cell leukodystrophy mouse model. Mol. Genet. Metab. 2011, 103, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Hawkins-Salsbury, J.A.; Shea, L.; Jiang, X.; Hunter, D.A.; Guzman, A.M.; Reddy, A.S.; Qin, E.Y.; Li, Y.; Gray, S.J.; Ory, D.S.; et al. Mechanism-Based Combination Treatment Dramatically Increases Therapeutic Efficacy in Murine Globoid Cell Leukodystrophy. J. Neurosci. 2015, 35, 6495–6505. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.-S.; Huang, Y.-W.; Lee, T.-H.; Chang, L.; Huang, Z.-D.; Wu, T.-Y.; Wang, T.-J.; Ho, C.-S. Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy. Cells 2023, 12, 993. https://doi.org/10.3390/cells12070993
Lin D-S, Huang Y-W, Lee T-H, Chang L, Huang Z-D, Wu T-Y, Wang T-J, Ho C-S. Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy. Cells. 2023; 12(7):993. https://doi.org/10.3390/cells12070993
Chicago/Turabian StyleLin, Dar-Shong, Yu-Wen Huang, Tsung-Han Lee, Lung Chang, Zon-Darr Huang, Tsu-Yen Wu, Tuan-Jen Wang, and Che-Sheng Ho. 2023. "Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy" Cells 12, no. 7: 993. https://doi.org/10.3390/cells12070993
APA StyleLin, D.-S., Huang, Y.-W., Lee, T.-H., Chang, L., Huang, Z.-D., Wu, T.-Y., Wang, T.-J., & Ho, C.-S. (2023). Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy. Cells, 12(7), 993. https://doi.org/10.3390/cells12070993