
Residual Cystine Transport Activity for Specific Infantile and Juvenile CTNS Mutations in a PTEC-Based Addback Model

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
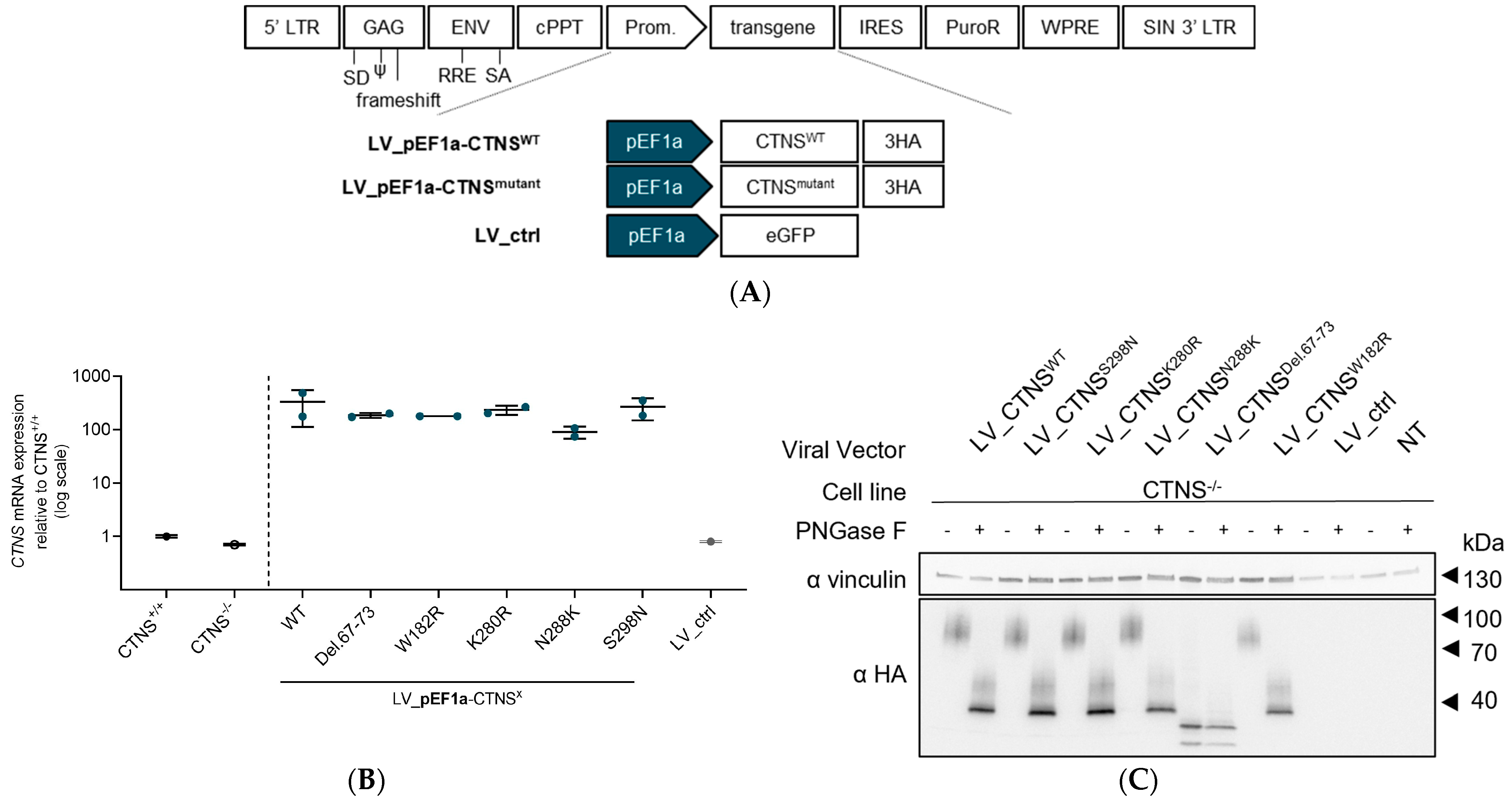
2.2. Generation of LV Transfer Plasmids for CTNSWT or CTNSmutants
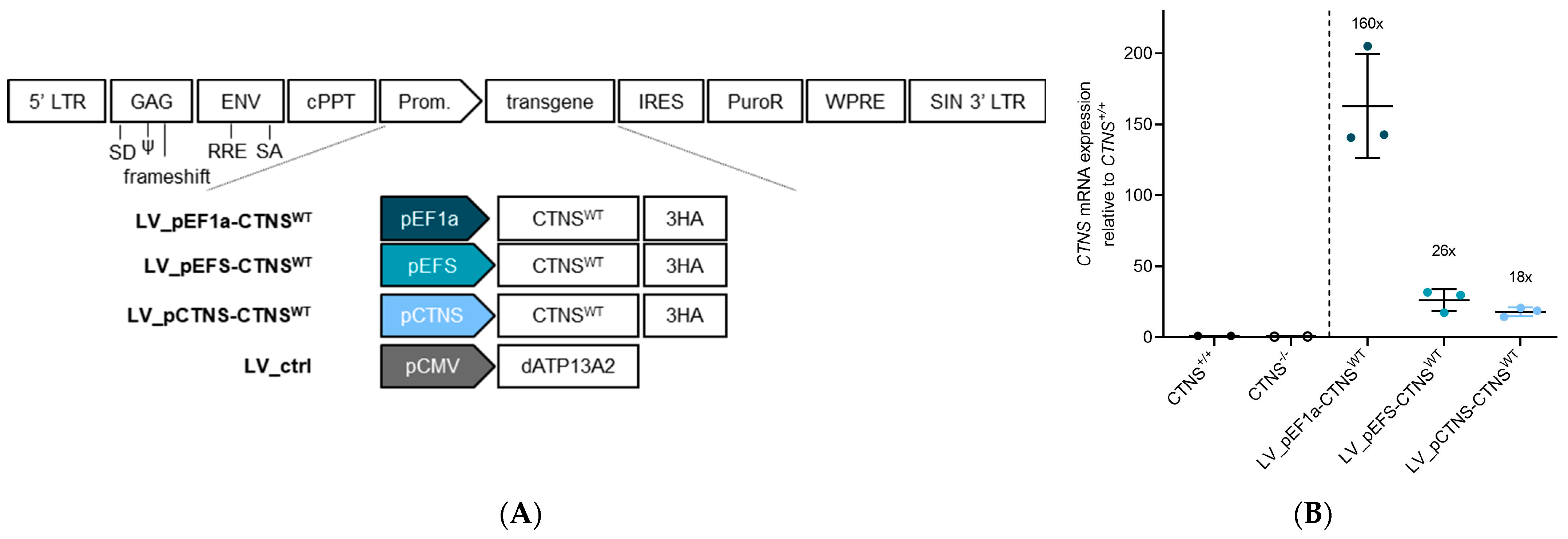
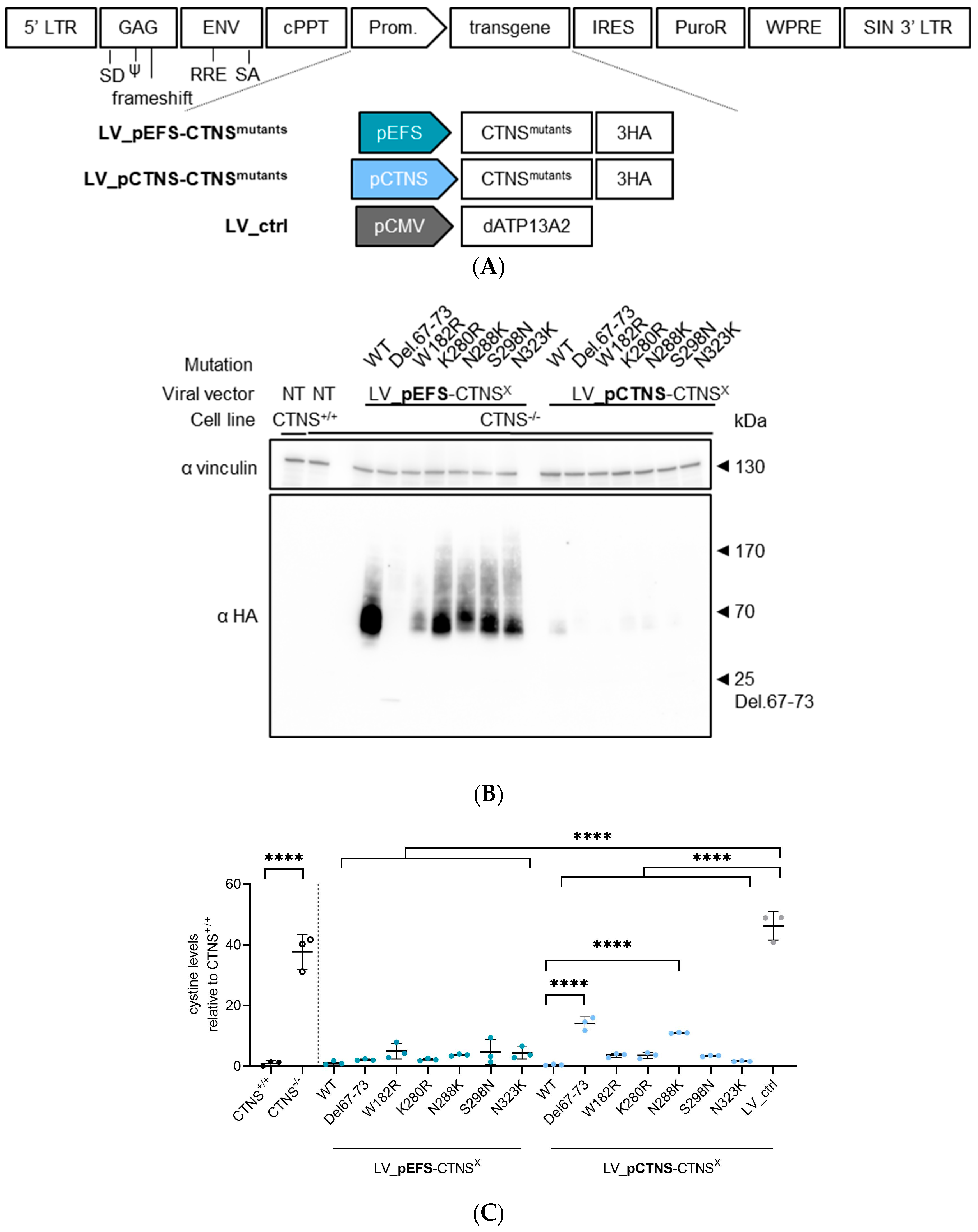
2.3. Production of LV Vectors and Generation of Stable Cell Lines Expressing CTNSWT or CTNSmutants Driven by Different Promoters
2.4. Determination of Integrated Copies
2.5. Quantification of CTNS-3HA mRNA Expression Levels
2.6. Metabolite and Cystine Measurements
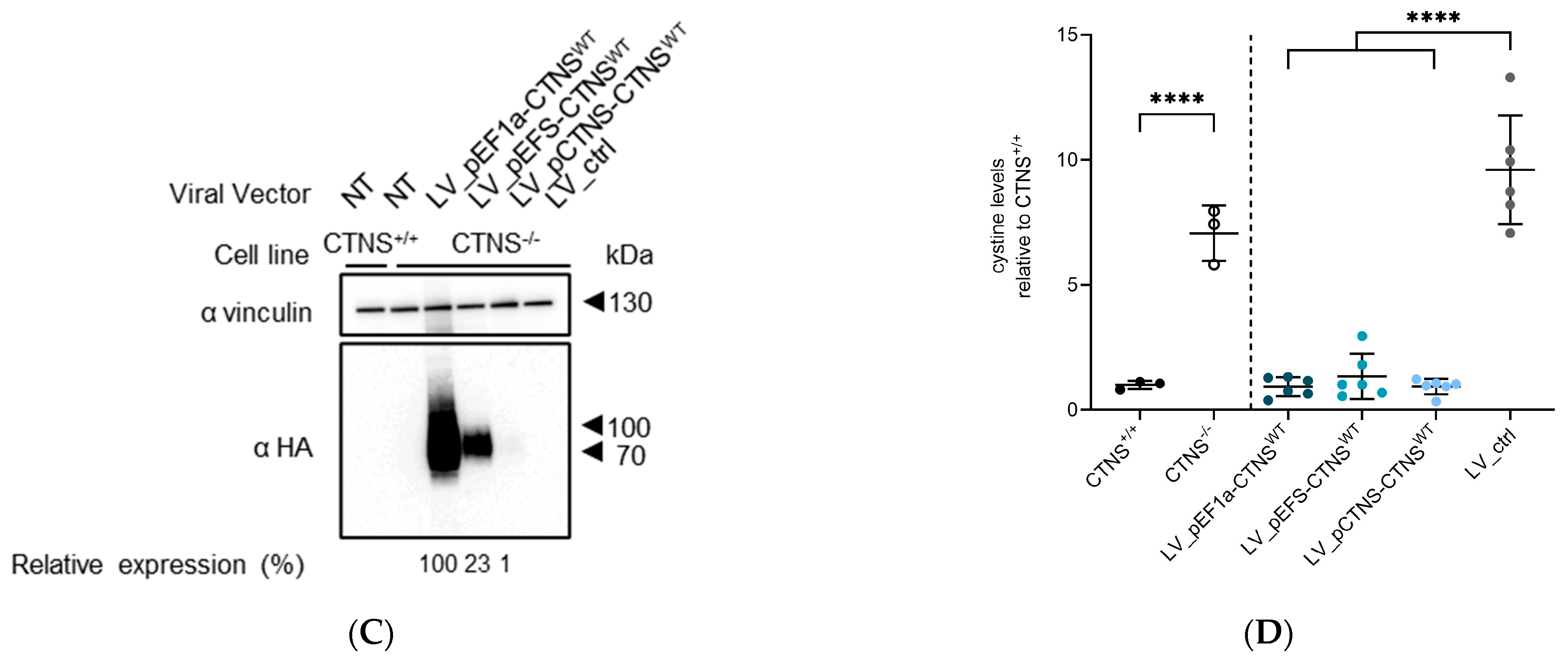
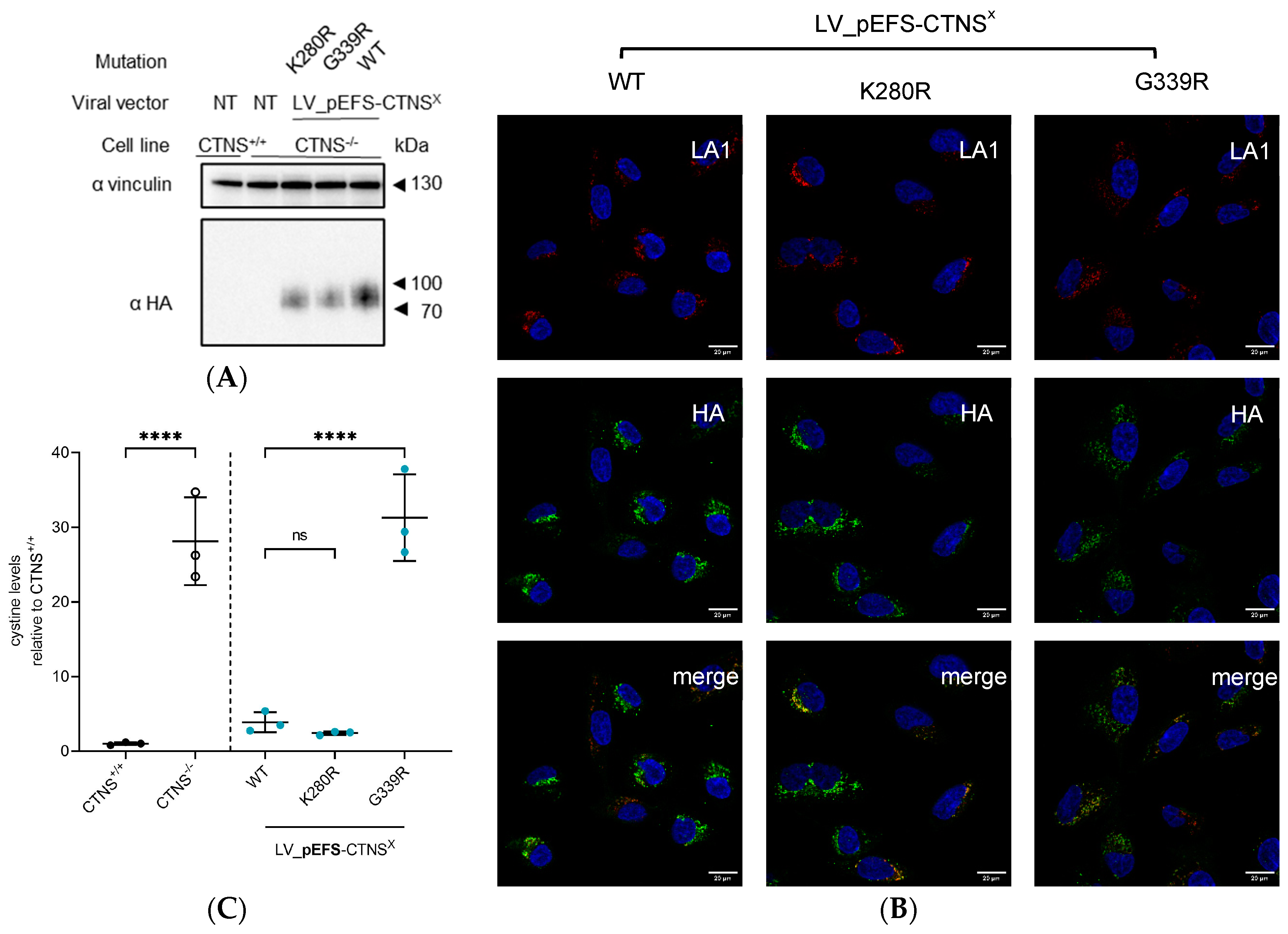
2.7. Protein Analysis by PAGE and Western Blot
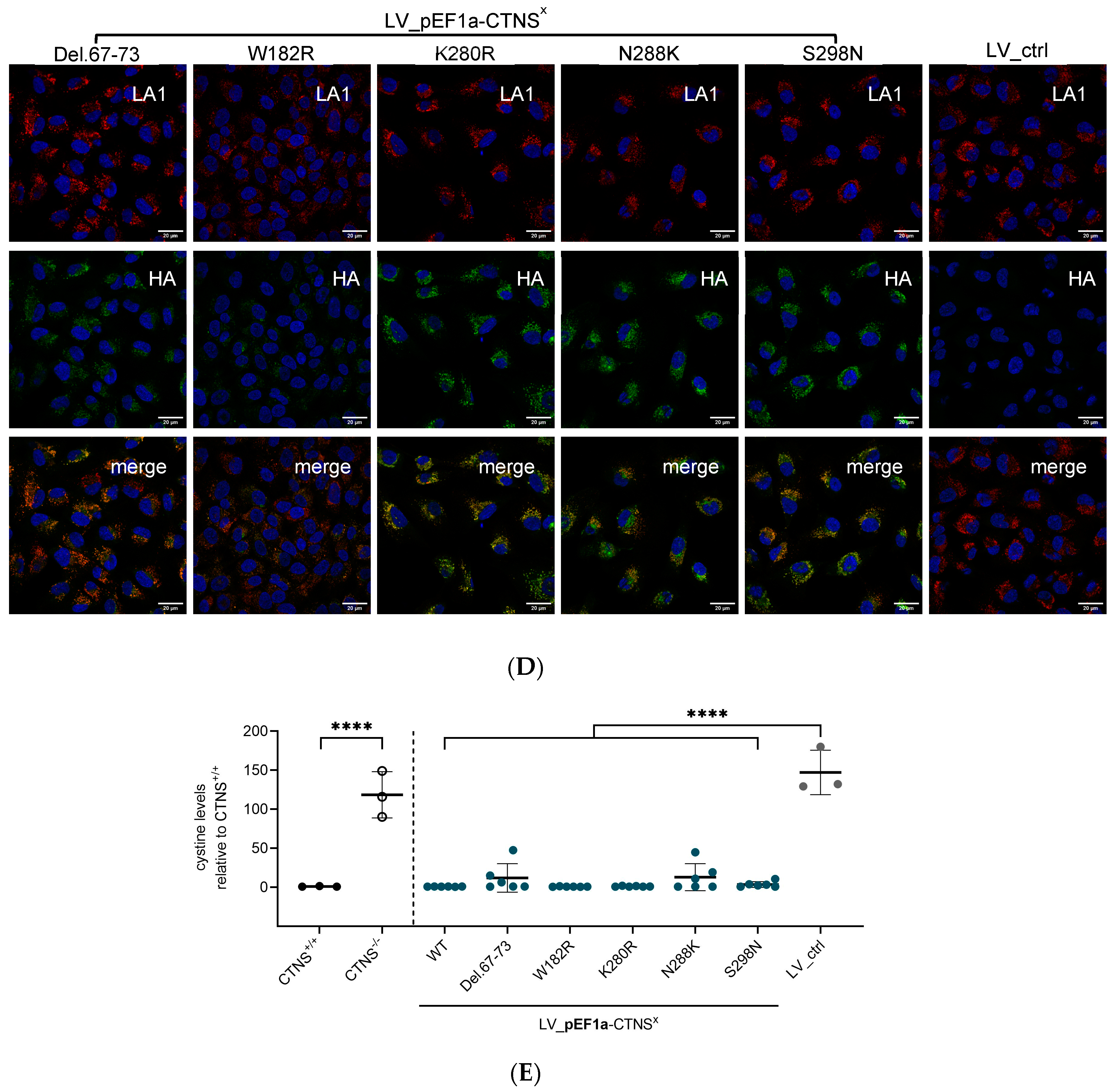
2.8. Immunocytochemistry Staining
2.9. AlphaMissense Pathogenicity and CADD Scores
2.10. Statistical Analysis
3. Results
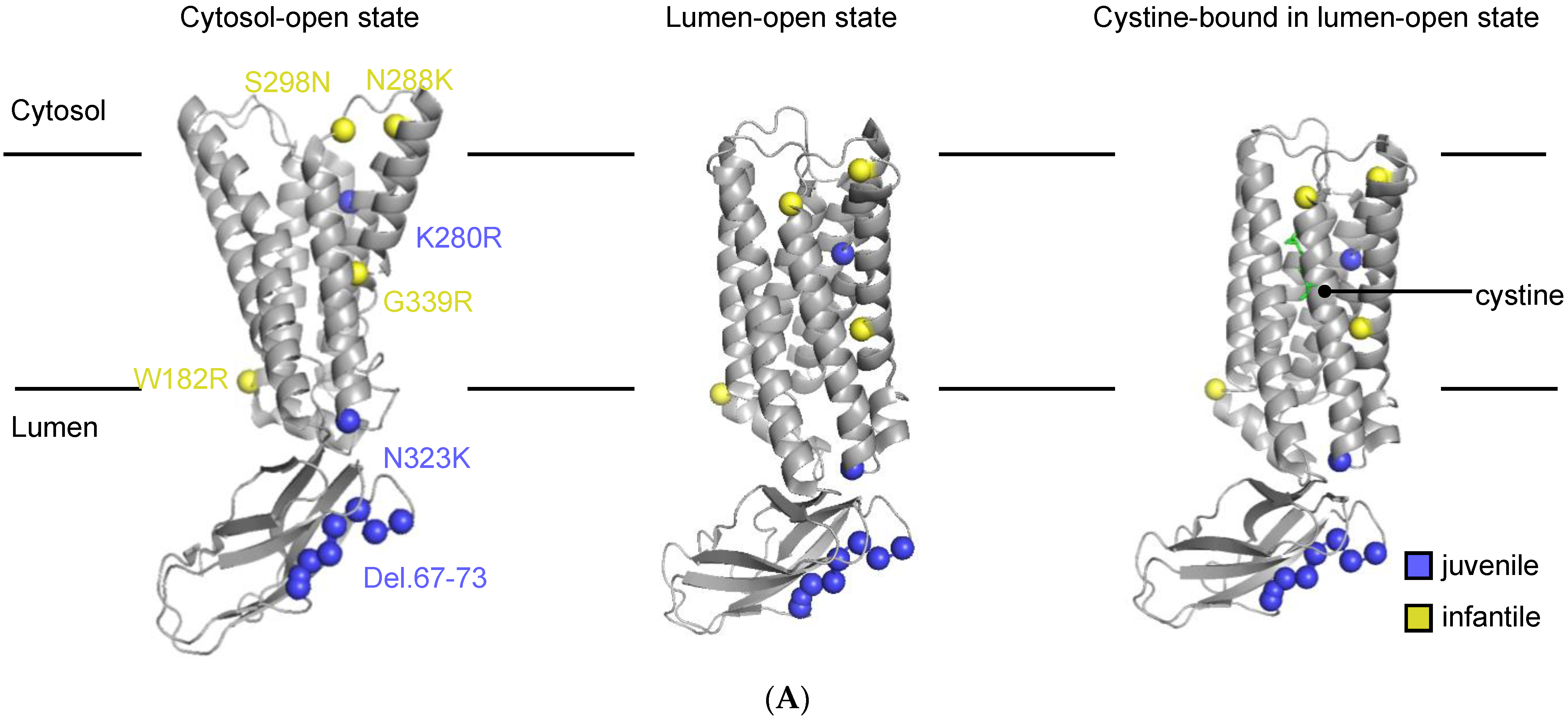
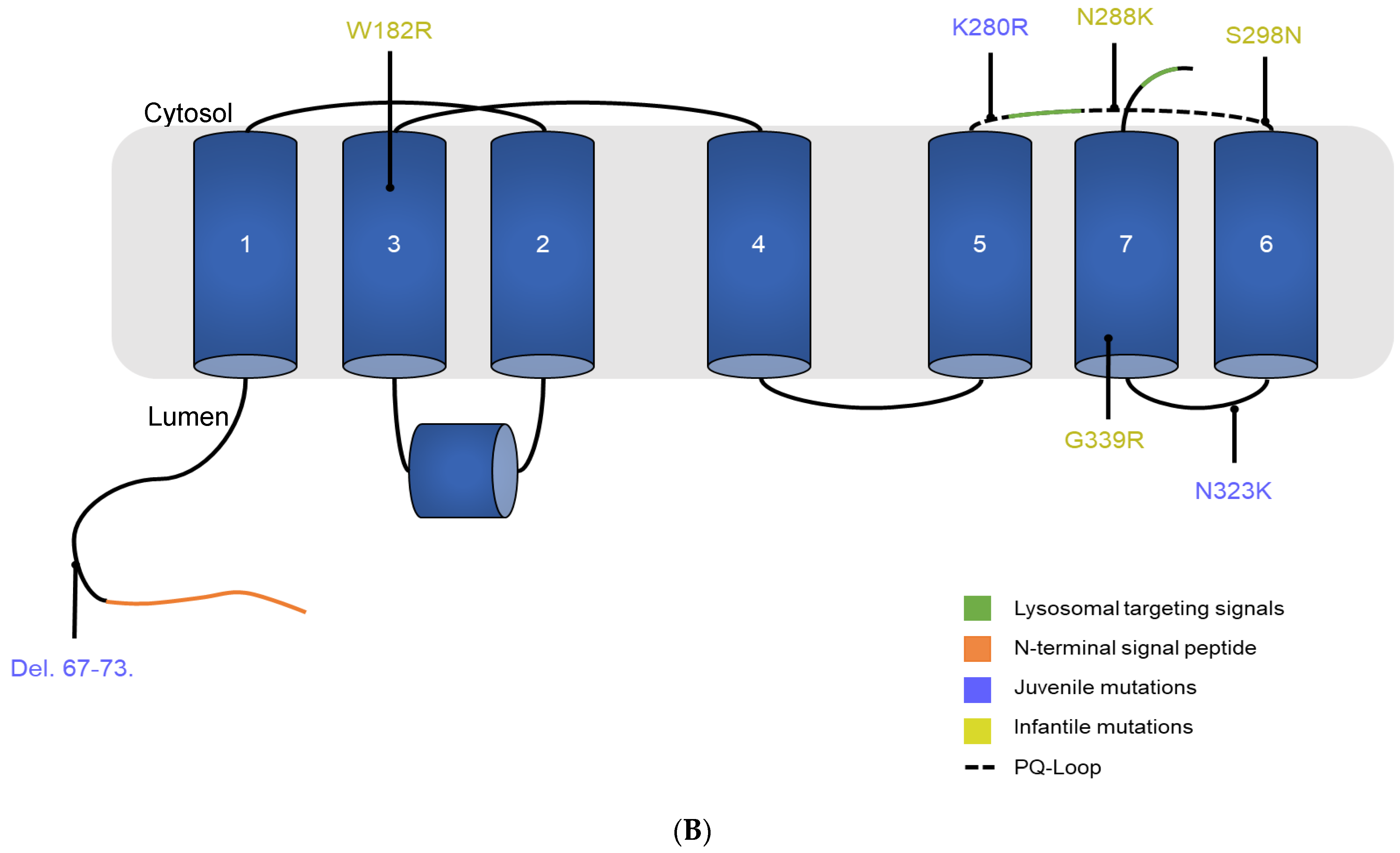
3.1. Description of the Selected CTNS Mutations
3.2. Both CTNSWT and CTNSmutants Restore Cystine Content in Cystinosis Cell Models upon Overexpression
3.3. Physiologically More Relevant CTNSWT Expression Levels Rescue Cystine Accumulation
3.4. CTNSmutant Expression Driven by EFS- and CTNS Promoter Still Reverts Cystine Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jean, G.; Fuchshuber, A.; Town, M.M.; Gribouval, O.; Schneider, J.A.; Broyer, M.; Hoff, W.V.N.T.; Niaudet, P.; Antignac, C. High-Resolution Mapping of the Gene for Cystinosis, Using Combined Biochemical and Linkage Analysis. Am. J. Hum. Genet. 1996, 58, 535–543. [Google Scholar] [PubMed]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Gallen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A Novel Gene Encoding an Integral Membrane Protein Is Mutated in Nephropathic Cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, F.; Roberds, S.L.; Jeanpierre, M.; Leturcq, F.; Azibi, K.; Beldjord, C.; Carrié, A.; Récan, D.; Chaouch, M.; Reghis, A.; et al. Linkage of the Gene for Cystinosis to Markers on the Short Arm of Chromosome 17. Nat. Genet. 1995, 10, 246–248. [Google Scholar] [CrossRef]
- Gahl, W.A.; Tietze, F.; Bashan, N.; Bernardini, I.; Raiford, D.; Schulman, J.D. Characteristics of Cystine Counter-Transport in Normal and Cystinotic Lysosome-Rich Leucocyte Granular Fractions. Biochem. J. 1983, 216, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Antignac, C.; Gasnier, B.; Cherqui, S.S.; Antignac, C.; Gasnier, B.; Cherqui, Â.; Kalatzis, V.; Antignac, C.; Gasnier, B.; et al. Cystinosin, the Protein Defective in Cystinosis, Is a H+-Driven Lysosomal Cystine Transporter. EMBO J. 2001, 20, 5940–5949. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Bashan, N.; Tietze, F.; Bernardini, I.; Schulman, J.D. Cystine Transport Is Defective in Isolated Leukocyte Lysosomes from Patients with Cystinosis. Science 1982, 217, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.D.; Bradley, K.H.; Seegmiller, J.E. Cystine: Compartmentalization within Lysosomes in Cystinotic Leukocytes. Science 1969, 166, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Bradley, K.; Seegmiller, J.E. Increased Cystine in Leukocytes from Individuals Homozygous and Heterozygous for Cystinosis. Science 1967, 157, 1321–1322. [Google Scholar] [CrossRef] [PubMed]
- Jonas, A.J.; Greene, A.A.; Smith, M.L.; Schneider, J.A. Cystine Accumulation and Loss in Normal, Heterozygous, and Cystinotic Fibroblasts. Proc. Natl. Acad. Sci. USA 1982, 79, 4442–4445. [Google Scholar] [CrossRef] [PubMed]
- Goldman, H.; Scriver, C.R.; Aaron, K.; Sc, M. Adolescent Cystinosis: Comparisons with Infantile and Adult Forms. Pediatrics 1971, 47, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Gahl, W. Nephropathic Cystinosis: Late Complications of a Multisystemic Disease. Pediartric Nephrol. 2008, 23, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Wong, V.; Bradley, K.; Seegmiller, J.E. Biochemical Comparisons of the Adult and Childhood Forms of Cystinosis. N. Engl. J. Med. 1968, 279, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Schoeber, J.P.; Van Den Heuvel, L.P.; Levtchenko, E.N. Cystinosis: Practical Tools for Diagnosis and Treatment. Pediatr. Nephrol. 2011, 26, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Medical Progress: Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Morinière, V.; Grünfeld, J.P.; Noël, L.H.; Goujon, J.M.; Chadefaux-Vekemans, B.; Antignac, C. Late-Onset Nephropathic Cystinosis: Clinical Presentation, Outcome, and Genotyping. Clin. J. Am. Soc. Nephrol. 2008, 3, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Anikster, Y.; Lucero, C.; Guo, J.; Huizing, M.; Shotelersuk, V.; Bernardini, I.; McDowell, G.; Iwata, F.; Kaiser-Kupfer, M.I.; Jaffe, R.; et al. Ocular Nonnephropathic Cystinosis: Clinical, Biochemical, and Molecular Correlations. Pediatr. Res. 2000, 47, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Brodin-sartorius, A.; Tête, M.-J.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine Therapy Delays the Progression of Nephropathic Cystinosis in Late Adolescents and Adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; Nesterova, G.; Langman, C.; Labbé, A.; Cherqui, S.; Goodyer, P.; Janssen, M.C.; Greco, M.; Topaloglu, R.; Elenberg, E.; et al. Full Review Nephropathic Cystinosis: An International Consensus Document. Nephrol. Dial. Transplant. 2014, 29, 87–94. [Google Scholar] [CrossRef] [PubMed]
- HGMD® Gene Result. Available online: https://www.hgmd.cf.ac.uk/ac/gene.php?gene=CTNS (accessed on 4 October 2023).
- David, D.; Berlingerio, P.; Elmonem, A.; Princiero Berlingerio, S.; Elmonem, M.A.; Oliveira Arcolino, F.; Soliman, N.; Van Den Heuvel, B.; Gijsbers, R.; Levtchenko, E.; et al. Molecular Basis of Cystinosis: Geographic Distribution, Functional Consequences of Mutations in the CTNS Gene, and Potential for Repair. Nephron 2019, 141, 133–146. [Google Scholar] [CrossRef]
- Touchman, J.W.; Anikster, Y.; Dietrich, N.L.; Braden Maduro, V.V.; McDowell, G.; Shotelersuk, V.; Bouffard, G.G.; Beckstrom-Sternberg, S.M.; Gahl, W.A.; Green, E.D. The Genomic Region Encompassing the Nephropathic Cystinosis Gene (CTNS): Complete Sequencing of a 200-Kb Segment and Discovery of a Novel Gene within the Common Cystinosis-Causing Deletion. Genome Res. 2000, 10, 165–173. [Google Scholar] [CrossRef]
- Anikster, Y.; Lucero, C.; Touchman, J.W.; Huizing, M.; McDowell, G.; Shotelersuk, V.; Green, E.D.; Gahl, W.A. Identification and Detection of the Common 65-Kb Deletion Breakpoint in the Nephropathic Cystinosis Gene (CTNS). Mol. Genet. Metab. 1999, 66, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; van’t Hoff, W.; Hohenfellner, K.; Topaloglu, R.; Greco, M.; Ariceta, G.; Bettini, C.; Bockenhauer, D.; Veys, K.; Pape, L.; et al. An International Cohort Study Spanning Five Decades Assessed Outcomes of Nephropathic Cystinosis. Kidney Int. 2021, 100, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Nevo, N.; Cherqui, S.; Gasnier, B.; Antignac, C.; Kalatzis, V.; Nevo, N.; Antignac, C.; Cherqui, S.; Gasnier, B.; et al. Molecular Pathogenesis of Cystinosis: Effect of CTNS Mutations on the Transport Activity and Subcellular Localization of Cystinosin. Hum. Mol. Genet. 2004, 13, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Kalatzis, V.; Trugnan, G.; Antignac, C. The Targeting of Cystinosin to the Lysosomal Membrane Requires a Tyrosine-Based Signal and a Novel Sorting Motif*. J. Biol. Chem. 2001, 276, 13314–13321. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Cordier, A.; Cochat, P.; Broyer, M.; Antignac, C.; Malades, N.; Bernard, C. Cystinosin, the Protein Defective in Cystinosis, Is a H+-Driven Lysosomal Cystine Transporter. J. Am. Soc. Nephrol. 2001, 12, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Schmiege, P.; Assafa, T.E.; Wang, R.; Xu, Y.; Donnelly, L.; Fine, M.; Ni, X.; Jiang, J.; Millhauser, G.; et al. Structure and Mechanism of Human Cystine Exporter Cystinosin. Cell 2022, 185, 3739–3752.e18. [Google Scholar] [CrossRef] [PubMed]
- Löbel, M.; Salphati, S.P.; El Omari, K.; Wagner, A.; Tucker, S.J.; Parker, J.L.; Newstead, S. Structural Basis for Proton Coupled Cystine Transport by Cystinosin. Nat. Commun. 2022, 13, 4845. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulyte, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate Proteome-Wide Missense Variant Effect Prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Saleem, M.A.; Masereeuw, R.; Ni, L.; Van Der Velden, T.J.; Russel, F.G.; Mathieson, P.W.; Monnens, L.A.; Van Den Heuvel, L.P.; Levtchenko, E.N. Novel Conditionally Immortalized Human Proximal Tubule Cell Line Expressing Functional Influx and Efflux Transporters. Cell Tissue Res. 2010, 339, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Jamalpoor, A.; Van Gelder, C.A.G.H.; Yousef Yengej, F.A.; Zaal, E.A.; Berlingerio, S.P.; Veys, K.R.; Pou Casellas, C.; Voskuil, K.; Essa, K.; Ammerlaan, C.M.E.; et al. Cysteamine–Bicalutamide Combination Therapy Corrects Proximal Tubule Phenotype in Cystinosis. EMBO Mol. Med. 2021, 13, e13067. [Google Scholar] [CrossRef] [PubMed]
- Besouw, M.; Van Den Heuvel, L.; Van Eijsden, R.; Bongaers, I.; Kluijtmans, L.; Dewerchin, M.; Levtchenko, E. Increased Human Dermal Microvascular Endothelial Cell Survival Induced by Cysteamine. J. Inherit. Metab. Dis. 2013, 36, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.M.; El-Kares, R.; Taranta, A.; Bellomo, F.; Emma, F.; Besouw, M.; Levtchenko, E.; Toelen, J.; van den Heuvel, L.; Chu, L.L.; et al. Stem Cell Microvesicles Transfer Cystinosin to Human Cystinotic Cells and Reduce Cystine Accumulation in Vitro. PLoS ONE 2012, 7, e42840. [Google Scholar] [CrossRef] [PubMed]
- Corallini, S.; Taranta, A.; Bellomo, F.; Palma, A.; Pastore, A.; Emma, F. Transcriptional and Post-Transcriptional Regulation of the CTNS Gene. Pediatr. Res. 2011, 70, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Van Looveren, D.; Giacomazzi, G.; Thiry, I.; Sampaolesi, M.; Gijsbers, R. Improved Functionality and Potency of next Generation BinMLV Viral Vectors toward Safer Gene Therapy. Mol. Ther. Methods Clin. Dev. 2021, 23, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, A.; Vande Velde, G.; Reumers, V.; Toelen, J.; Thiry, I.; Vandeputte, C.; Vets, S.; Deroose, C.; Bormans, G.; Baekelandt, V.; et al. Highly Efficient Multicistronic Lentiviral Vectors with Peptide 2A Sequences. Hum. Gene Ther. 2009, 20, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Veys, K.; Berlingerio, S.P.; David, D.; Bondue, T.; Held, K.; Reda, A.; van den Broek, M.; Theunis, K.; Janssen, M.; Cornelissen, E.; et al. Urine-Derived Kidney Progenitor Cells in Cystinosis. Cells 2022, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Fehse, B.; Kustikova, O.S.; Bubenheim, M.; Baum, C. Pois(s)on—It’s a Question of Dose. Gene Ther. 2004, 11, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Tordai, H.; Torres, O.; Csepi, M.; Padányi, R.; Lukács, G.L.; Hegedűs, T. Lightway Access to AlphaMissense Data That Demonstrates a Balanced Performance of This Missense Mutation Predictor. bioRxiv 2023. [Google Scholar] [CrossRef]
- Minton, K. Predicting Variant Pathogenicity with AlphaMissense. Nat. Rev. Genet. 2023, 24, 804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Bigio, B.; Rapaport, F.; Zhang, S.Y.; Casanova, J.L.; Abel, L.; Boisson, B.; Itan, Y. PopViz: A Webserver for Visualizing Minor Allele Frequencies and Damage Prediction Scores of Human Genetic Variations. Bioinformatics 2018, 34, 4307–4309. [Google Scholar] [CrossRef] [PubMed]
- Shotelersuk, V.; Larson, D.; Anikster, Y.; Mcdowell, G.; Lemons, R.; Bernardini, I.; Guo, J.; Thoene, J.; Gahl, W.A. CTNS Mutations in an American-Based Population of Cystinosis Patients. Am. J. Hum. Genet. 1998, 63, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Midgley, J.P.; El-Kares, R.; Mathieu, F.; Goodyer, P. Natural History of Adolescent-Onset Cystinosis. Pediatr. Nephrol. 2011, 26, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Nevo, N.; Thomas, L.; Chhuon, C.; Andrzejewska, Z.; Lipecka, J.; Bailleux, A.; Edelman, A.; Antignac, C.; Guerrera, I.C.; Guillonneau, F.; et al. Impact of Cystinosin Glycosylation on Protein Stability by Differential Dynamic Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC). Mol. Cell. Proteomics 2017, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole Exome Sequencing Identifies Causative Mutations in the Majority of Consanguineous or Familial Cases with Childhood-Onset Increased Renal Echogenicity. Kidney Int. 2016, 89, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Serrano, M.; Junckerstorff, R.C.; Alisheri, A.; Pestronk, A.; Laing, N.G.; Weihl, C.C.; Lamont, P.J. Cystinosis Distal Myopathy, Novel Clinical, Pathological and Genetic Features. Neuromuscul. Disord. 2017, 27, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Savostyanov, K.V.; Pushkov, A.A.; Shchagina, O.A.; Maltseva, V.V.; Suleymanov, E.A.; Zhanin, I.S.; Mazanova, N.N.; Fisenko, A.P.; Mishakova, P.S.; Polyakov, A.V.; et al. Genetic Landscape of Nephropathic Cystinosis in Russian Children. Front. Genet. 2022, 13, 863157. [Google Scholar] [CrossRef] [PubMed]
- Venkatarangan, V.; Zhang, W.; Yang, X.; Thoene, J.G.; Hahn, S.H.; Li, M. ER-Associated Degradation in Cystinosis Pathogenesis and the Prospects of Precision Medicine. J. Clin. Investig. 2023, 133, e169551. [Google Scholar] [CrossRef] [PubMed]
- Thoene, J.; Lemons, R.; Anikster, Y.; Mullet, J.; Paelicke, K.; Lucero, C.; Gahl, W.; Schneider, J.; Shu, S.G.; Campbell, H.T. Mutations of CTNS Causing Intermediate Cystinosis. Mol. Genet. Metab. 1999, 67, 283–293. [Google Scholar] [CrossRef]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the Small GTPase Rab11, and the Rab7 Effector RILP Regulate Intracellular Trafficking of the Chaperone-Mediated Autophagy Receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Chiara Guerrera, I.; Antignac, C. Cystinosin Is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Cohen-Solal, L.; Cordier, B.; Frishberg, Y.; Kemper, M.; Nuutinen, E.M.; Legrand, E.; Cochat, P.; Antignac, C. Identification of 14 Novel CTNS Mutations and Characterization of Seven Splice Site Mutations Associated with Cystinosis. Hum. Mutat. 2002, 20, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Yeetong, P.; Tongkobpetch, S.; Kingwatanakul, P.; Deekajorndech, T.; Bernardini, I.M.; Suphapeetiporn, K.; Gahl, W.A.; Shotelersuk, V. Two Novel CTNS Mutations in Cystinosis Patients in Thailand. Gene 2012, 499, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Koudstaal, W.; Fletcher, L.; Costa, M.; van Winsen, M.; Siregar, B.; Inganäs, H.; Kim, J.; Keogh, E.; Macedo, J.; et al. Naturally Occurring Antibodies Isolated from PD Patients Inhibit Synuclein Seeding in Vitro and Recognize Lewy Pathology. Acta Neuropathol. 2019, 137, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Attard, M.; Jean, G.G.; Forestier, L.; Cherqui, S.S.; Van’t Hoff, W.; Broyer, M.; Antignac, C.; Town, M. Severity of Phenotype in Cystinosis Varies with Mutations in the CTNS Gene: Predicted Effect on the Model of Cystinosin. Hum. Mol. Genet. 1999, 8, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Rupar, C.A.; Matsell, D.; Surry, S.; Siu, V. A G339R Mutation in the CTNS Gene Is a Common Cause of Nephropathic Cystinosis in the South Western Ontario Amish Mennonite Population. J. Med. Genet. 2001, 38, 615–616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kiehntopf, M.; Schickel, J.; von der Gönne, B.; Koch, H.G.; Superti-Furga, A.; Steinmann, B.; Deufel, T.; Harms, E. Analysis of the CTNS Gene in Patients of German and Swiss Origin with Nephropathic Cystinosis. Hum. Mutat. 2002, 20, 237. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.; Pepe, G.; Dall’Amico, R.; Tartaglia, S.; Casciani, S.; Greco, M.; Bencivenga, P.; Murer, L.; Rizzoni, G.; Tenconi, R.; et al. Mutational Spectrum of the CTNS Gene in Italy. Eur. J. Hum. Genet. 2003, 11, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Macías-Vidal, J.; Rodés, M.; Hernández-Pérez, J.M.; Vilaseca, M.A.; Coll, M.J. Analysis of the CTNS Gene in 32 Cystinosis Patients from Spain. Clin. Genet. 2009, 76, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Soliman, N.A.; Elmonem, M.A.; van den Heuvel, L.; Abdel Hamid, R.H.; Gamal, M.; Bongaers, I.; Marie, S.; Levtchenko, E. Mutational Spectrum of the CTNS Gene in Egyptian Patients with Nephropathic Cystinosis. JIMD Rep. 2014, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, F.; Basiratnia, M.; Derakhshan, A.; Fardaei, M. Mutation Analysis of the CTNS Gene in Iranian Patients with Infantile Nephropathic Cystinosis: Identification of Two Novel Mutations. Hum. Genome Var. 2017, 4, 17038. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, R.; Gulhan, B.; İnözü, M.; Canpolat, N.; Yilmaz, A.; Noyan, A.; Dursun, İ.; Gökçe, İ.; Gürgöze, M.K.; Akinci, N.; et al. The Clinical and Mutational Spectrum of Turkish Patients with Cystinosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Emma, F.; Levtchenko, E.N. The Pathogenesis of Cystinosis: Mechanisms beyond Cystine Accumulation. Am. J. Physiol.-Ren. Physiol. 2010, 299, F905–F916. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, S.; Martin, S.; Motamedi, S.; van Veen, S.; Holemans, T.; Van den Haute, C.; Jordanova, A.; Baekelandt, V.; Vangheluwe, P.; Agostinis, P. ATP13A2/PARK9 Regulates Endo-/Lysosomal Cargo Sorting and Proteostasis through a Novel PI(3, 5)P2-Mediated Scaffolding Function. Hum. Mol. Genet. 2017, 26, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, F.; Corallini, S.; Pastore, A.; Palma, A.; Laurenzi, C.; Emma, F.; Taranta, A. Modulation of CTNS Gene Expression by Intracellular Thiols. Free Radic. Biol. Med. 2010, 48, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Donello, J.E.; Loeb, J.E.; Hope, T.J. Woodchuck Hepatitis Virus Contains a Tripartite Posttranscriptional Regulatory Element. J. Virol. 1998, 72, 5085–5092. [Google Scholar] [CrossRef] [PubMed]
- Ruivo, R.; Bellenchi, G.C.; Chen, X.; Zifarelli, G.; Sagneá, C.; Debacker, C.C.; Pusch, M.; Supplisson, S.S.; Gasnier, B.; Sagné, C.; et al. Mechanism of Proton/Substrate Coupling in the Heptahelical Lysosomal Transporter Cystinosin. Proc. Natl. Acad. Sci. USA 2012, 109, E210–E217. [Google Scholar] [CrossRef]
- Berquez, M.; Chen, Z.; Festa, B.P.; Krohn, P.; Keller, S.A.; Parolo, S.; Korzinkin, M.; Gaponova, A.; Laczko, E.; Domenici, E.; et al. Lysosomal Cystine Export Regulates MTORC1 Signaling to Guide Kidney Epithelial Cell Fate Specialization. Nat. Commun. 2023, 14, 3994. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.M.; Carlon, M.S. One Size Does Not Fit All: The Past, Present and Future of CF Causal Therapies. Cells 2022, 11, 1868. [Google Scholar] [CrossRef] [PubMed]
- Helip-Wooley, A.; Park, M.A.; Lemons, R.M.; Thoene, J.G. Expression of CTNS Alleles: Subcellular Localization and Aminoglycoside Correction in Vitro. Mol. Genet. Metab. 2002, 75, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Brasell, E.J.; Chu, L.L.; El Kares, R.; Seo, J.H.; Loesch, R.; Iglesias, D.M.; Goodyer, P. The Aminoglycoside Geneticin Permits Translational Readthrough of the CTNS W138X Nonsense Mutation in Fibroblasts from Patients with Nephropathic Cystinosis. Pediatr. Nephrol. 2019, 34, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Brasell, E.J.; Chu, L.L.; Akpa, M.M.; Eshkar-Oren, I.; Alroy, I.; Corsini, R.; Gilfix, B.M.; Yamanaka, Y.; Huertas, P.; Goodyer, P. The Novel Aminoglycoside, ELX-02, Permits CTNSW138X Translational Read-through and Restores Lysosomal Cystine Efflux in Cystinosis. PLoS ONE 2019, 14, e0223954. [Google Scholar] [CrossRef] [PubMed]
- European Rare Kidney Disease Reference Network. Available online: https://www.erknet.org/ (accessed on 26 January 2024).
- RaDiCo-French National Programme on Rare Disease Cohorts. Available online: https://radico.fr/en/ (accessed on 26 January 2024).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gDNA Mutation | Exon | Protein Mutation | Location | Phenotype | Cystine Transport Activity (%) A | AlphaMissense Score B | CADD Score C | References |
|---|---|---|---|---|---|---|---|---|
| c.198_218del21 | Exon 5 | ITILELP-Del.67–73 | N-terminal tail | Juvenile | 19 ± 6.1 | NA | NA | [24,43,44,45,46,47,48,49] |
| c.544T > C | Exon 8 | W182R | 2nd TM | Infantile | 34 ± 5 | 0.7778 | 26.9 | [24,43] |
| c.839A > G | Exon 10 | K280R | 5th inter-TM loop | Juvenile | 0.68 ± 0.9 | 0.6602 | 35 | [15,24,27,28,50,51,52] |
| c.864C > A | Exon 11 | N288K | 5th inter-TM loop | Infantile | 1.6 ± 1.2 | 0.9942 | 25 | [24,27,45,52,53] |
| c.893G > A | Exon 11 | S298N | 5th inter-TM loop | Infantile | 77 ± 21 | 0.5957 | 29.8 | [24,43] |
| c.969C > G | Exon 11 | N323K | 6th inter-TM loop | Juvenile | 0.14 ± 0.8 | 0.7791 | 22.6 | [24,45,50,52,54,55] |
| c.1354G > A | Exon 12 | G339R | 7th TM | Infantile | −0.8 ± 3.3 | 0.9803 | 31 | [24,43,56,57,58,59,60,61,62,63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medaer, L.; David, D.; Smits, M.; Levtchenko, E.; Sampaolesi, M.; Gijsbers, R. Residual Cystine Transport Activity for Specific Infantile and Juvenile CTNS Mutations in a PTEC-Based Addback Model. Cells 2024, 13, 646. https://doi.org/10.3390/cells13070646
Medaer L, David D, Smits M, Levtchenko E, Sampaolesi M, Gijsbers R. Residual Cystine Transport Activity for Specific Infantile and Juvenile CTNS Mutations in a PTEC-Based Addback Model. Cells. 2024; 13(7):646. https://doi.org/10.3390/cells13070646
Chicago/Turabian StyleMedaer, Louise, Dries David, Maxime Smits, Elena Levtchenko, Maurilio Sampaolesi, and Rik Gijsbers. 2024. "Residual Cystine Transport Activity for Specific Infantile and Juvenile CTNS Mutations in a PTEC-Based Addback Model" Cells 13, no. 7: 646. https://doi.org/10.3390/cells13070646
APA StyleMedaer, L., David, D., Smits, M., Levtchenko, E., Sampaolesi, M., & Gijsbers, R. (2024). Residual Cystine Transport Activity for Specific Infantile and Juvenile CTNS Mutations in a PTEC-Based Addback Model. Cells, 13(7), 646. https://doi.org/10.3390/cells13070646