
DPP8 Selective Inhibitor Tominostat as a Novel and Broad-Spectrum Anticancer Agent against Hematological Malignancies

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentivirus and Transduction
2.3. Reagents
2.4. Cellular Cytotoxicity
2.5. Western Blot Analyses
2.6. Microarray Analysis
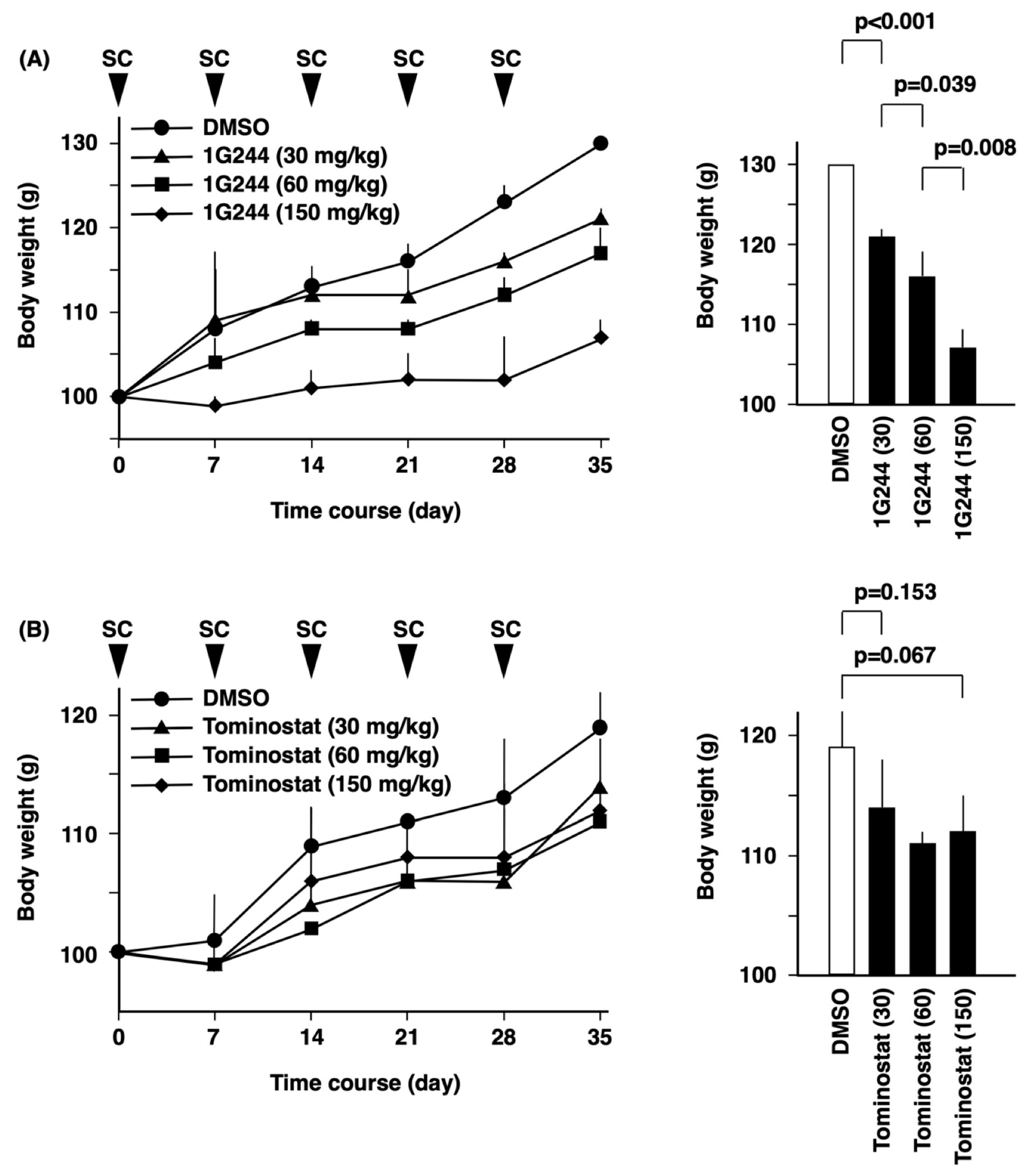
2.7. In Vivo Studies
3. Results
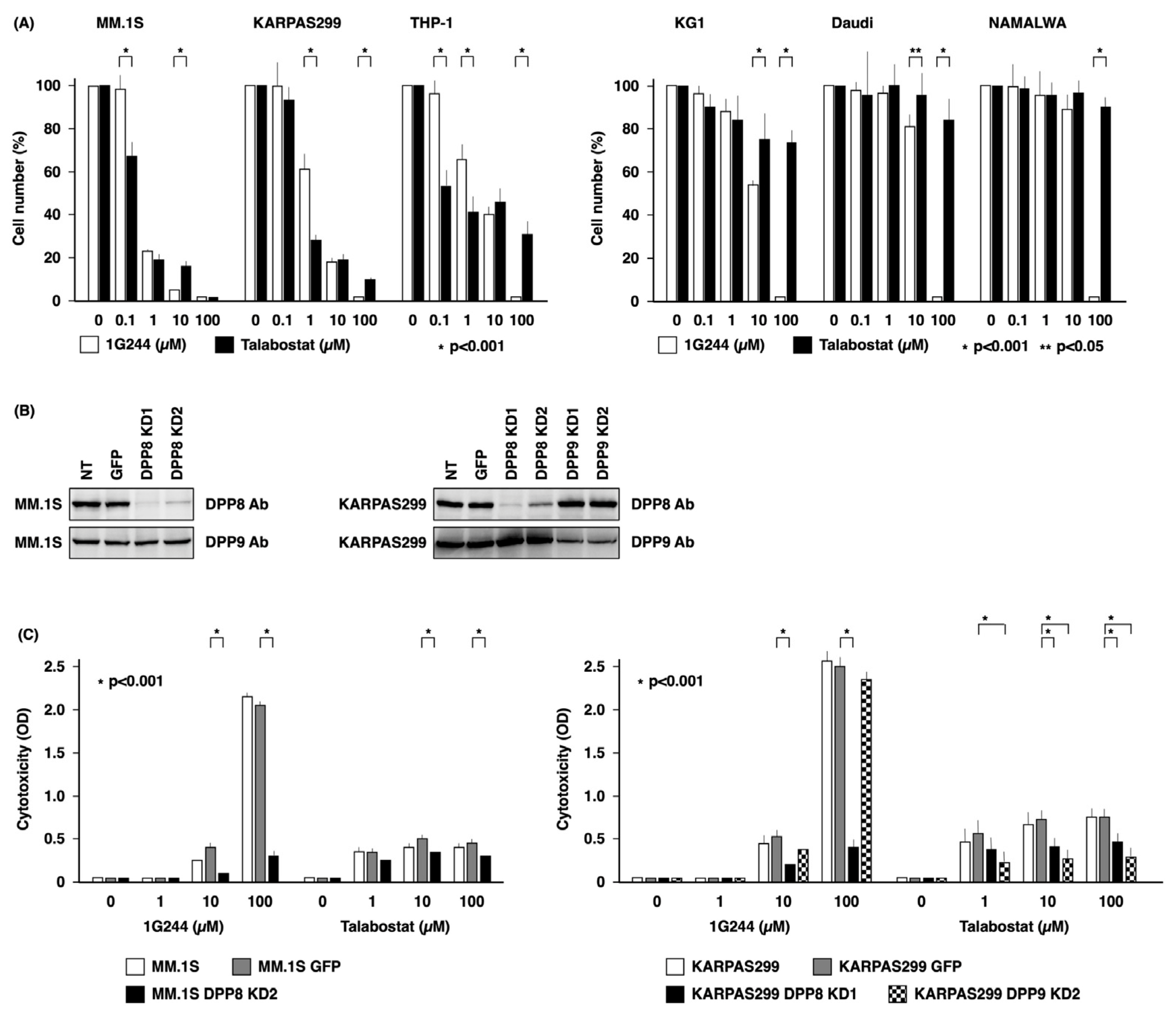
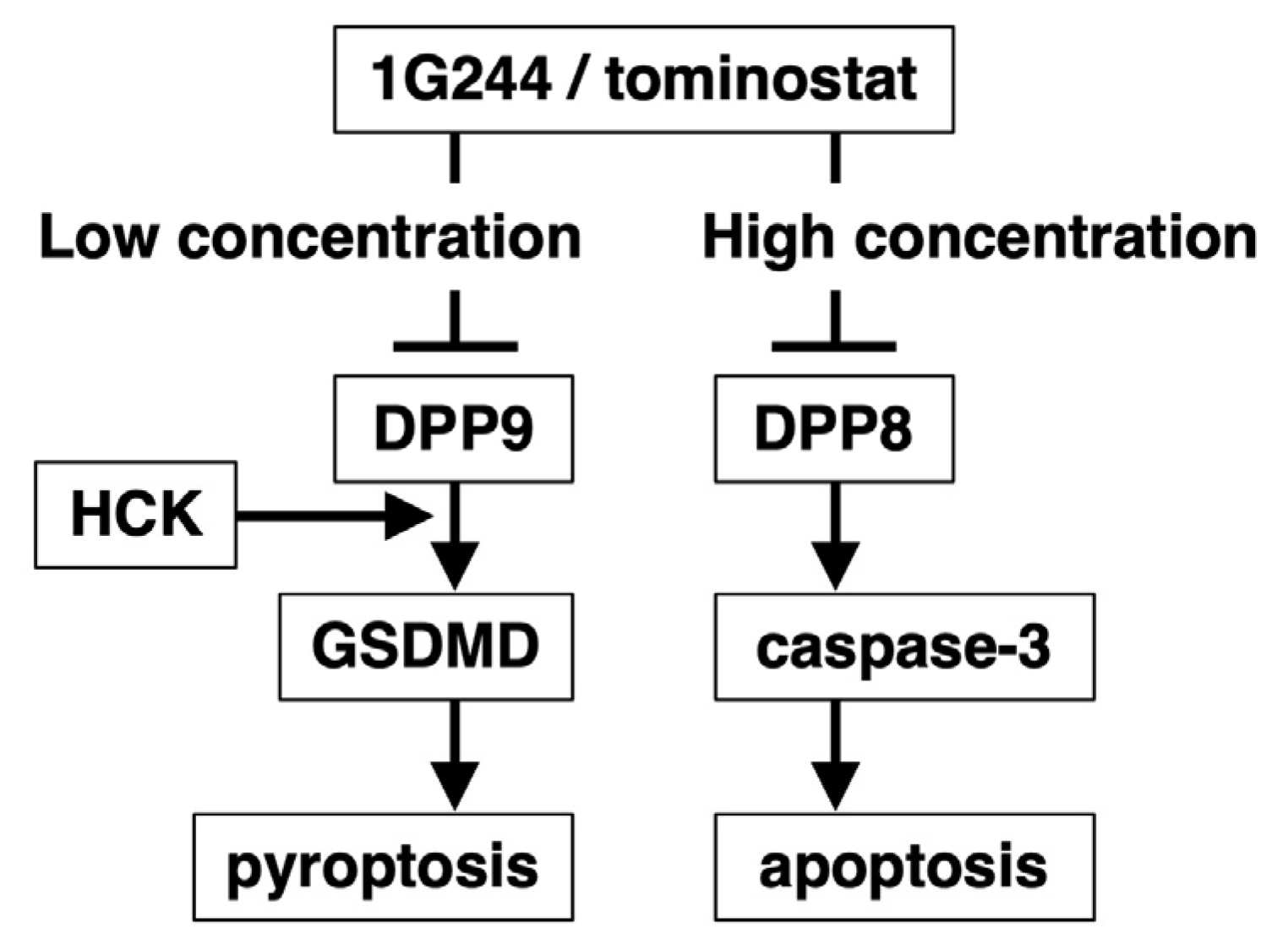
3.1. DPP8-Dependent Antineoplastic Effect of High-Dose 1G244
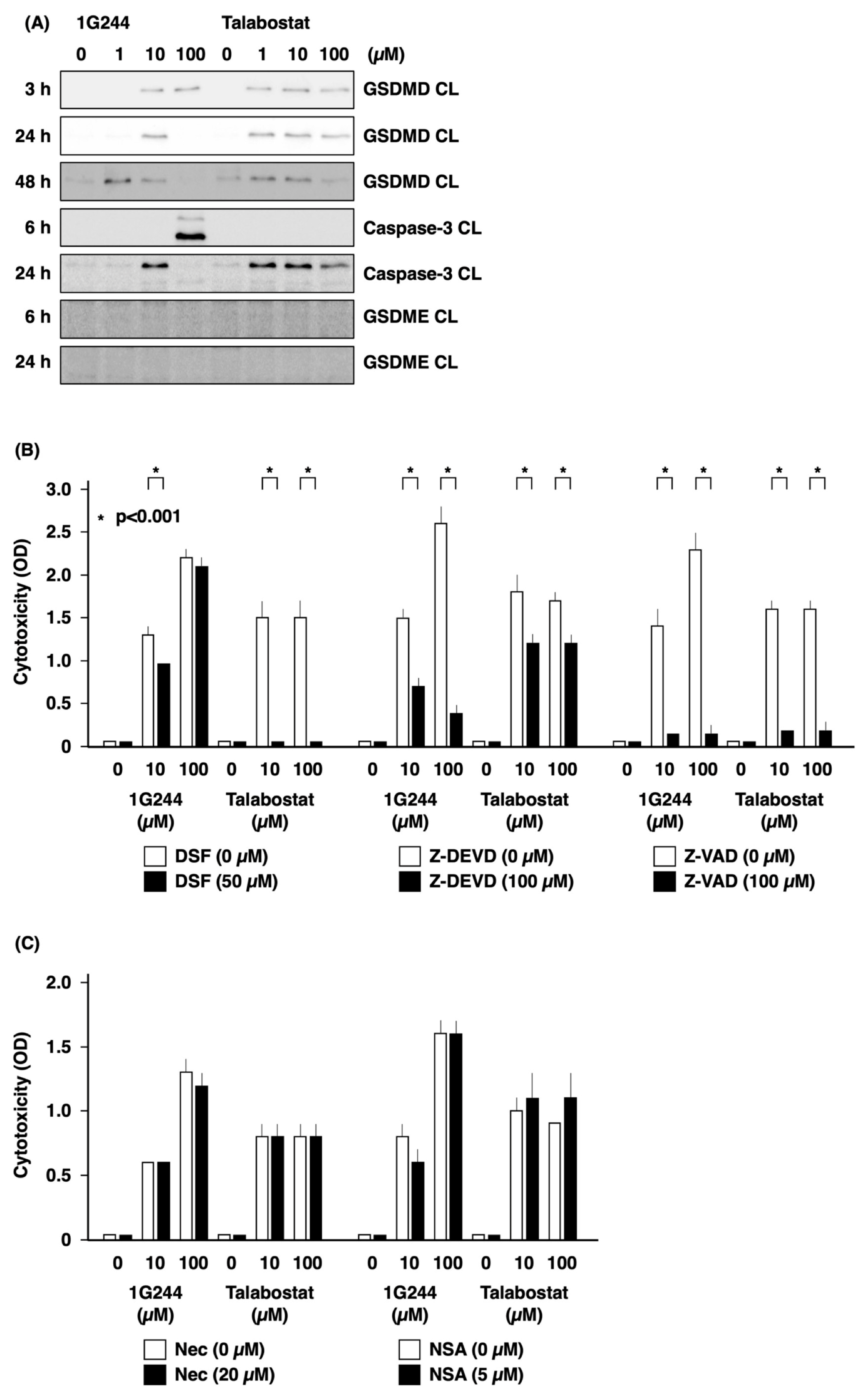
3.2. Caspase-3-Mediated Apoptosis as Anticancer Effect by High-Dose 1G244
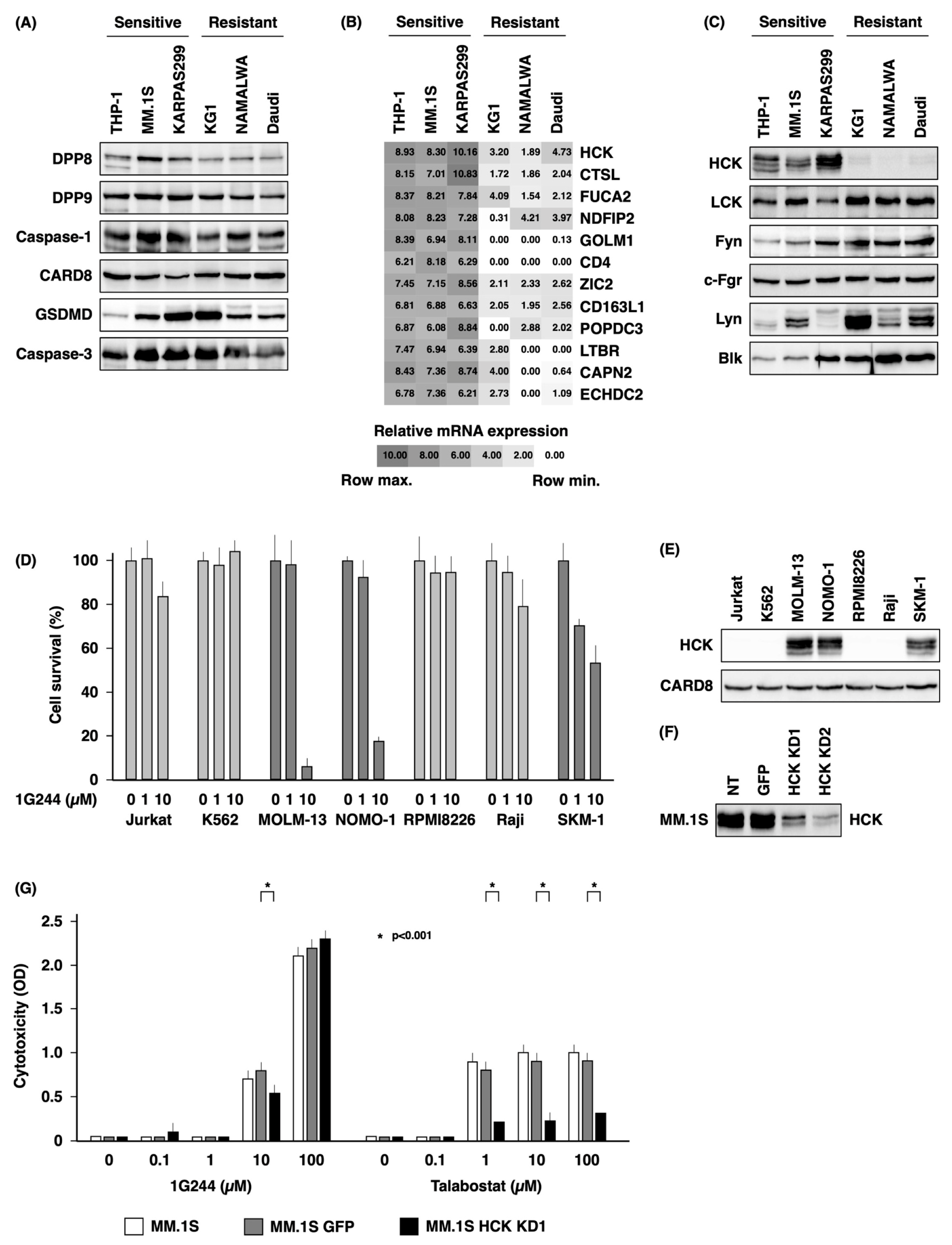
3.3. Dependence on HCK for DPP8/9 Inhibitor-Induced Pyroptosis
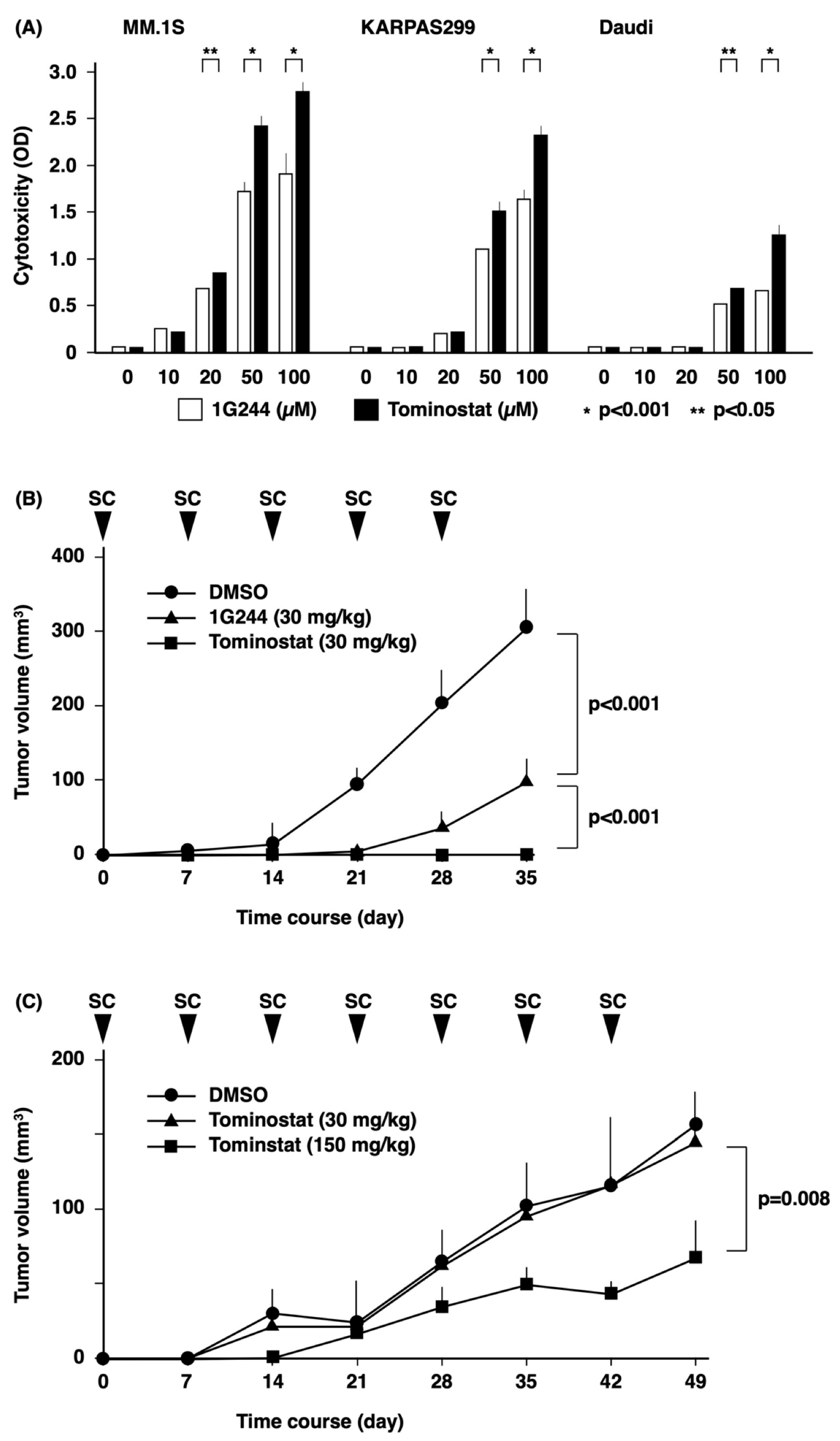
3.4. Antitumor Effects of Tominostat
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, C.H.; Zhang, H.E.; Gorrell, M.D.; Abbott, C.A. Dipeptidyl Peptidase 9 Substrates and Their Discovery: Current Progress and the Application of Mass Spectrometry-Based Approaches. Biol. Chem. 2016, 397, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M.; Proost, P.; Durinx, C.; Bal, G.; Senten, K.; Augustyns, K.; Scharpé, S.; van Dammes, J.; de Meester, I. Kinetic Investigation of Chemokine Truncation by CD26/Dipeptidyl Peptidase IV Reveals a Striking Selectivity within the Chemokine Family. J. Biol. Chem. 2001, 276, 29839–29845. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, C.; Schlossman, S.F. The Structure and Function of CD26 in the T-Cell Immune Response. Immunol. Rev. 1998, 161, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Yu, D.M.T.; O’Connor, S.P.; Gorrell, M.D. Inhibitor Selectivity in the Clinical Application of Dipeptidyl Peptidase-4 Inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef]
- Bjelke, J.R.; Christensen, J.; Nielsen, P.F.; Branner, S.; Kanstrup, A.B.; Wagtmann, N.; Rasmussen, H.B. Dipeptidyl Peptidases 8 and 9: Specificity and Molecular Characterization Compared with Dipeptidyl Peptidase IV. Biochem. J. 2006, 396, 391–399. [Google Scholar] [CrossRef]
- Bolgi, O.; Silva-Garcia, M.; Ross, B.; Pilla, E.; Kari, V.; Killisch, M.; Spitzner, M.; Stark, N.; Lenz, C.; Weiss, K.; et al. Dipeptidyl Peptidase 9 Triggers BRCA2 Degradation and Promotes DNA Damage Repair. EMBO Rep. 2022, 23, e54136. [Google Scholar] [CrossRef]
- Freeman, M.; Justa-Schuch, D.; Silva-Garcia, M.; Pilla, E.; Engelke, M.; Kilisch, M.; Lenz, C.; Mö ller, U.; Nakamura, F.; Urlaub, H.; et al. DPP9 Is a Novel Component of the N-End Rule Pathway Targeting the Tyrosine Kinase Syk. Elife 2016, 5, e16370. [Google Scholar] [CrossRef]
- Zhang, H.; Maqsudi, S.; Rainczuk, A.; Duffield, N.; Lawrence, J.; Keane, F.M.; Justa-Schuch, D.; Geiss-Friedlander, R.; Gorrell, M.D.; Stephens, A.N. Identification of Novel Dipeptidyl Peptidase 9 Substrates by Two-Dimensional Differential in-Gel Electrophoresis. FEBS J. 2015, 282, 3737–3757. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Parmentier, N.; Möller, U.; Urlaub, H.; van den Eynde, B.J.; Melchoir, F. The Cytoplasmic Peptidase DPP9 Is Rate-Limiting for Degradation of Proline-Containing Peptides. J. Biol. Chem. 2009, 284, 27211–27219. [Google Scholar] [CrossRef]
- Ajami, K.; Pitman, M.R.; Wilson, C.H.; Park, J.; Menz, R.I.; Starr, A.E.; Cox, J.H.; Abbott, C.A.; Overall, C.M.; Gorrell, M.D. Stromal Cell-Derived Factors 1α and 1β, Inflammatory Protein-10 and Interferon-Inducible T Cell Chemo-Attractant Are Novel Substrates of Dipeptidyl Peptidase 8. FEBS Lett. 2008, 582, 819–825. [Google Scholar] [CrossRef]
- Cui, C.; Tian, X.; Wei, L.; Wang, Y.; Wang, K.; Fu, R. New Insights into the Role of Dipeptidyl Peptidase 8 and Dipeptidyl Peptidase 9 and Their Inhibitors. Front. Pharmacol. 2022, 13, 1002871. [Google Scholar] [CrossRef]
- Ajami, K.; Abbott, C.A.; McCaughan, G.W.; Gorrell, M.D. Dipeptidyl Peptidase 9 Has Two Forms, a Broad Tissue Distribution, Cytoplasmic Localization and DPIV-like Peptidase Activity. Biochim. Et Biophys. Acta-Gene Struct. Expr. 2004, 1679, 18–28. [Google Scholar] [CrossRef]
- Johnson, D.C.; Taabazuing, C.Y.; Okondo, M.C.; Chui, A.J.; Rao, S.D.; Brown, F.C.; Reed, C.; Peguero, E.; de Stanchina, E.; Kentsis, A.; et al. DPP8/DPP9 Inhibitor-Induced Pyroptosis for Treatment of Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1151–1156. [Google Scholar] [CrossRef]
- Okondo, M.C.; Johnson, D.C.; Sridharan, R.; Go, E.B.; Chui, A.J.; Wang, M.S.; Poplawski, S.E.; Wu, W.; Liu, Y.; Lai, J.H.; et al. DPP8 and DPP9 Inhibition Induces Pro-Caspase-1-Dependent Monocyte and Macrophage Pyroptosis. Nat. Chem. Biol. 2017, 13, 46–53. [Google Scholar] [CrossRef]
- Okondo, M.C.; Rao, S.D.; Taabazuing, C.Y.; Chui, A.J.; Poplawski, S.E.; Johnson, D.C.; Bachovchin, D.A. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell. Chem. Biol. 2018, 25, 262–267.e5. [Google Scholar] [CrossRef]
- Sato, T.; Tatekoshi, A.; Takada, K.; Iyama, S.; Kamihara, Y.; Jawaid, P.; Rehman, M.U.; Noguchi, K.; Kondo, T.; Kajikawa, S.; et al. DPP8 Is a Novel Therapeutic Target for Multiple Myeloma. Sci. Rep. 2019, 9, 18094. [Google Scholar] [CrossRef]
- van Goethem, S.; Matheeussen, V.; Joossens, J.; Lambeir, A.M.; Chen, X.; de Meester, I.; Haemers, A.; Augustyns, K.; van der Veken, P. Structure-Activity Relationship Studies on Isoindoline Inhibitors of Dipeptidyl Peptidases 8 and 9 (DPP8, DPP9): Is DPP8-Selectivity an Attainable Goal? J. Med. Chem. 2011, 54, 5737–5746. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The Caspase-3/GSDME Signal Pathway as a Switch between Apoptosis and Pyroptosis in Cancer. Cell. Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Poh, A.R.; O’Donoghue, R.J.J.; Ernst, M. Hematopoietic Cell Kinase (HCK) as a Therapeutic Target in Immune and Cancer Cells. Oncotarget 2015, 6, 15752–15771. [Google Scholar] [CrossRef]
- Radha, V.; Sudhakar, C.; Ray, P.; Swarup, G. Induction of Cytochrome c Release and Apoptosis by Hck-SH3 Domain-Mediated Signalling Requires Caspase-3. Apoptosis 2002, 7, 195–207. [Google Scholar] [CrossRef]
- Wu, J.J.; Tang, H.K.; Yeh, T.K.; Chen, C.M.; Shy, H.S.; Chu, Y.R.; Chien, C.H.; Tsai, T.Y.; Huang, Y.C.; Huang, Y.L.; et al. Biochemistry, Pharmacokinetics, and Toxicology of a Potent and Selective DPP8/9 Inhibitor. Biochem. Pharmacol. 2009, 78, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.; Krapp, S.; Augustin, M.; Kierfersauer, R.; Arciniega, M.; Geiss-Friedlander, R.; Huber, R. Structures and Mechanism of Dipeptidyl Peptidases 8 and 9, Important Players in Cellular Homeostasis and Cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E1437–E1445. [Google Scholar] [CrossRef] [PubMed]
- Gall, M.G.; Chen, Y.; de Ribeiro, A.J.V.; Zhang, H.; Bailey, C.G.; Spielman, D.S.; Yu, D.M.T.; Gorrell, M.D. Targeted Inactivation of Dipeptidyl Peptidase 9 Enzymatic Activity Causes Mouse Neonate Lethality. PLoS ONE 2013, 8, e78378. [Google Scholar] [CrossRef]
- Kim, M.; Minoux, M.; Piaia, A.; Kueng, B.; Gapp, B.; Weber, D.; Haller, C.; Barbieri, S.; Namoto, K.; Lorenz, T.; et al. DPP9 Enzyme Activity Controls Survival of Mouse Migratory Tongue Muscle Progenitors and Its Absence Leads to Neonatal Lethality Due to Suckling Defect. Dev. Biol. 2017, 431, 297–308. [Google Scholar] [CrossRef]
- Kong, X.; Liao, Y.; Zhou, L.; Zhang, Y.; Cheng, J.; Yuan, Z.; Wang, S. Hematopoietic Cell Kinase (HCK) Is Essential for NLRP3 Inflammasome Activation and Lipopolysaccharide-Induced Inflammatory Response In Vivo. Front. Pharmacol. 2020, 11, 581011. [Google Scholar] [CrossRef]
- Kim, H.; Lee, H.J.; Oh, Y.; Choi, S.G.; Hong, S.H.; Kim, H.J.; Lee, S.Y.; Choi, J.W.; Su Hwang, D.; Kim, K.S.; et al. The DUSP26 Phosphatase Activator Adenylate Kinase 2 Regulates FADD Phosphorylation and Cell Growth. Nat. Commun. 2014, 5, 3351. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Matsuyoshi, S.; Nakamura, M.; Ishida, E.; Kishi, M.; Konishi, N. Phosphorylation of FADD Is Critical for Sensitivity to Anticancer Drug-Induced Apoptosis. Carcinogenesis 2004, 25, 1089–1097. [Google Scholar] [CrossRef]
- Wilson, C.H.; Indarto, D.; Doucet, A.; Pogson, L.D.; Pitman, M.R.; McNicholas, K.; Menz, R.I.; Overall, C.M.; Abbott, C.A. Identifying Natural Substrates for Dipeptidyl Peptidases 8 and 9 Using Terminal Amine Isotopic Labeling of Substrates (TAILS) Reveals in Vivo Roles in Cellular Homeostasis and Energy Metabolism. J. Biol. Chem. 2013, 288, 13936–13949. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikuchi, S.; Wada, A.; Kamihara, Y.; Okazaki, K.; Jawaid, P.; Rehman, M.U.; Kobayashi, E.; Susukida, T.; Minemura, T.; Nabe, Y.; et al. DPP8 Selective Inhibitor Tominostat as a Novel and Broad-Spectrum Anticancer Agent against Hematological Malignancies. Cells 2023, 12, 1100. https://doi.org/10.3390/cells12071100
Kikuchi S, Wada A, Kamihara Y, Okazaki K, Jawaid P, Rehman MU, Kobayashi E, Susukida T, Minemura T, Nabe Y, et al. DPP8 Selective Inhibitor Tominostat as a Novel and Broad-Spectrum Anticancer Agent against Hematological Malignancies. Cells. 2023; 12(7):1100. https://doi.org/10.3390/cells12071100
Chicago/Turabian StyleKikuchi, Shohei, Akinori Wada, Yusuke Kamihara, Kosuke Okazaki, Paras Jawaid, Mati Ur Rehman, Eiji Kobayashi, Takeshi Susukida, Tomoki Minemura, Yoshimi Nabe, and et al. 2023. "DPP8 Selective Inhibitor Tominostat as a Novel and Broad-Spectrum Anticancer Agent against Hematological Malignancies" Cells 12, no. 7: 1100. https://doi.org/10.3390/cells12071100
APA StyleKikuchi, S., Wada, A., Kamihara, Y., Okazaki, K., Jawaid, P., Rehman, M. U., Kobayashi, E., Susukida, T., Minemura, T., Nabe, Y., Iwao, N., Ozawa, T., Hatano, R., Yamada, M., Kishi, H., Matsuya, Y., Mizuguchi, M., Hayakawa, Y., Dang, N. H., ... Sato, T. (2023). DPP8 Selective Inhibitor Tominostat as a Novel and Broad-Spectrum Anticancer Agent against Hematological Malignancies. Cells, 12(7), 1100. https://doi.org/10.3390/cells12071100