
Quadra-Stable Dynamics of p53 and PTEN in the DNA Damage Response

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Simulations of the Boolean Network Model and Encoding of PubMed Articles into Boolean Rules
2.2. Gene Regulatory Network Evolution, as Well as Public Database and Tool Sets
3. Results
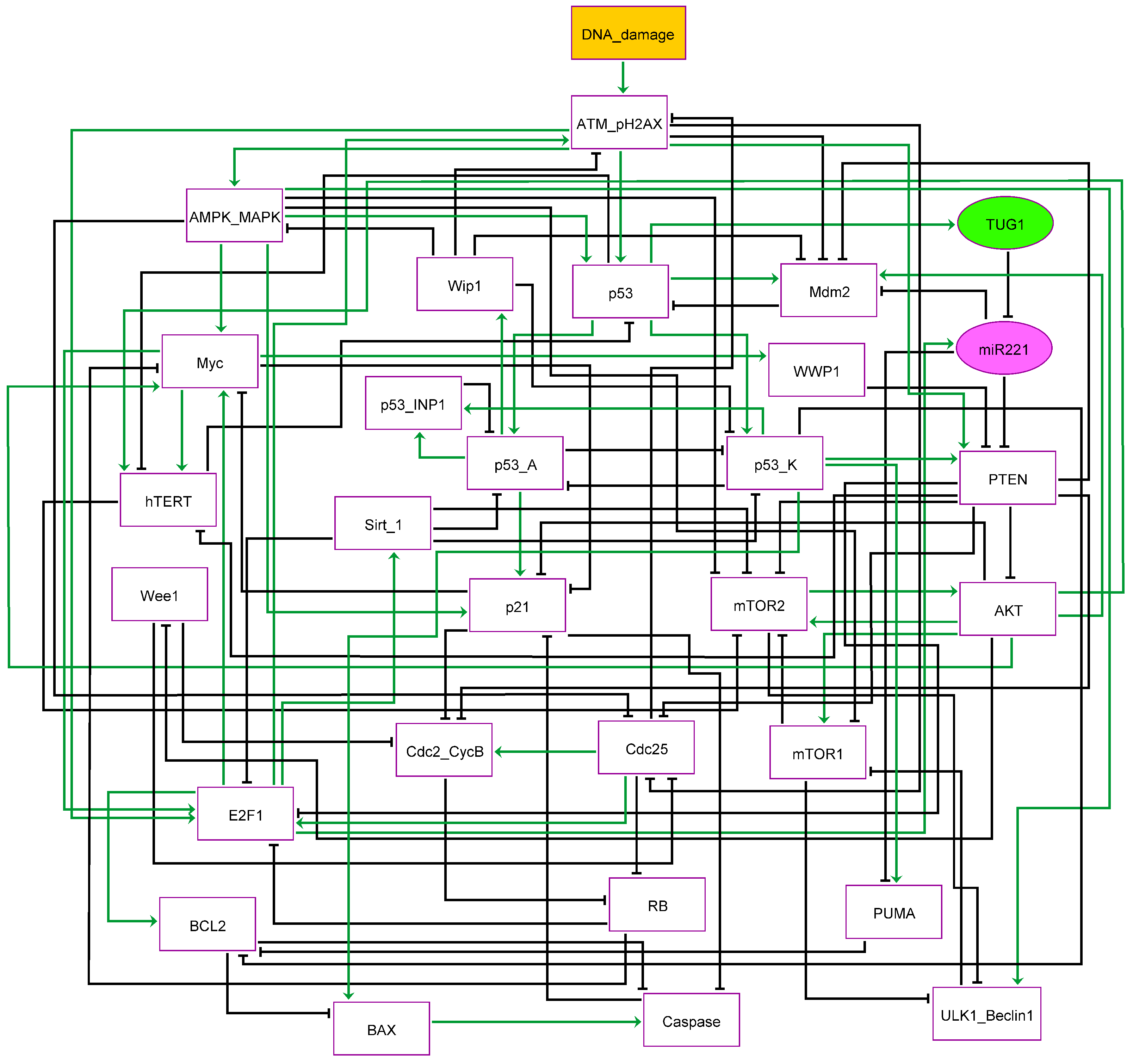
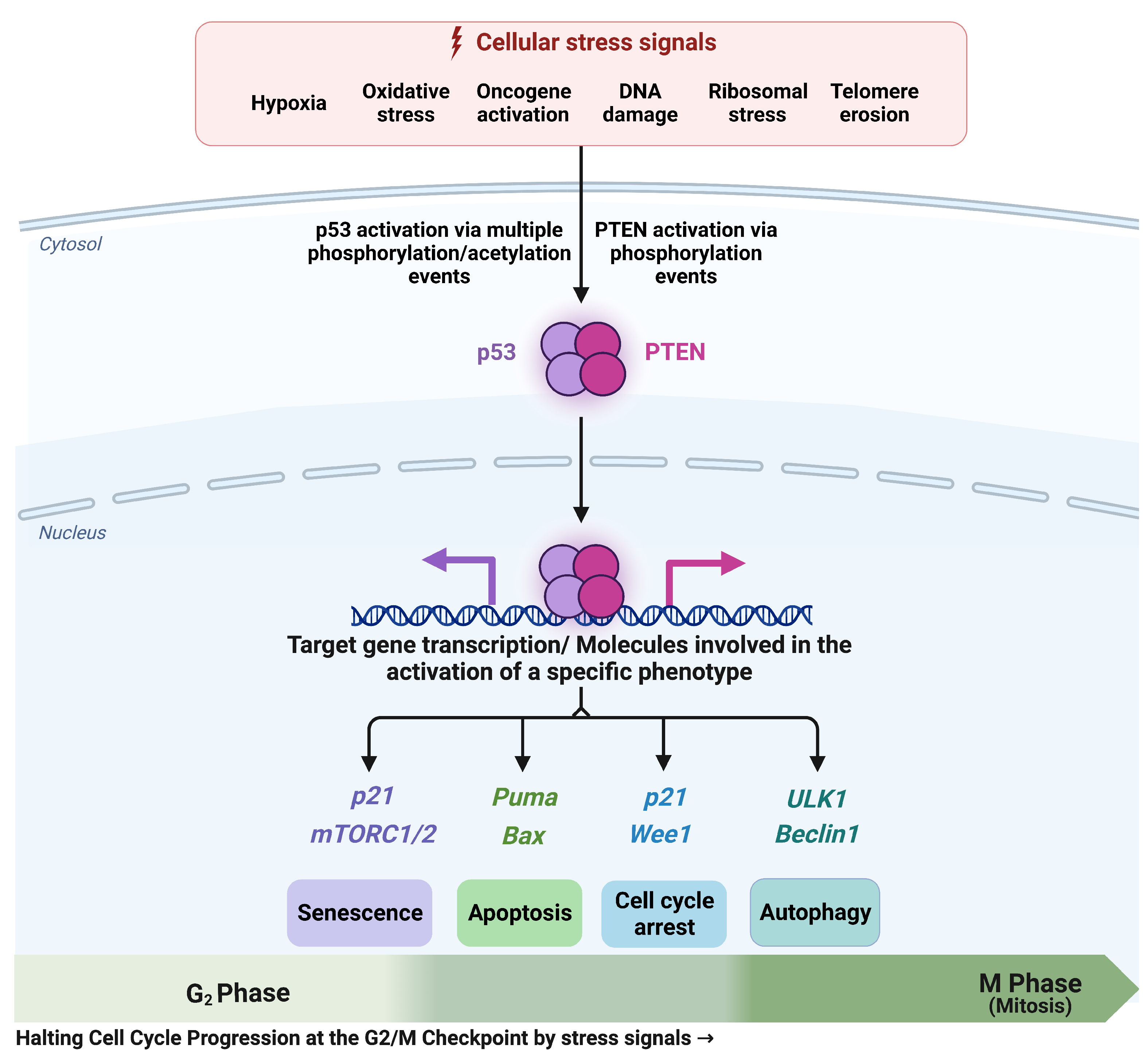
3.1. The G2/M Checkpoint’s Proposed Molecular Mechanism
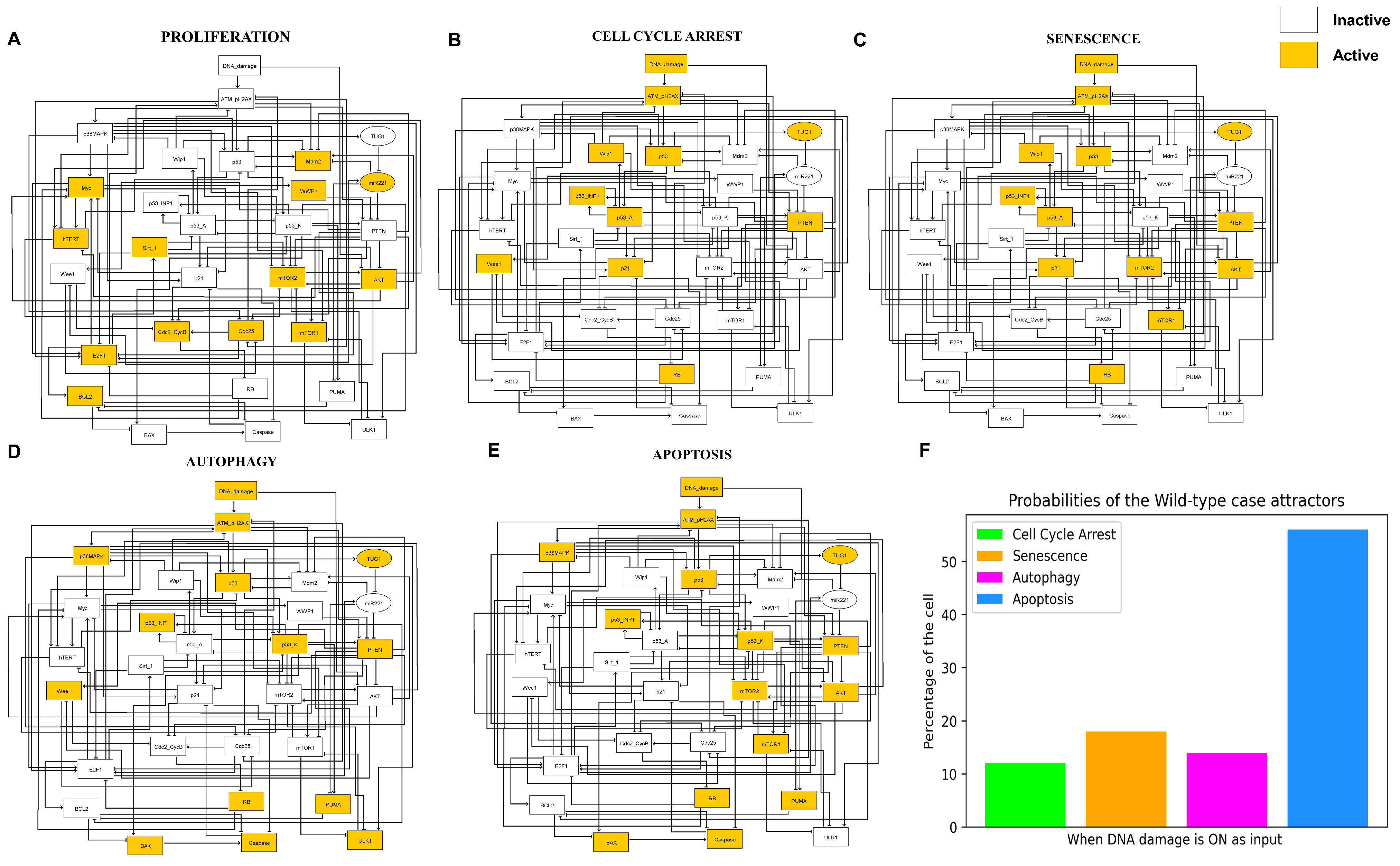
3.2. Simulations Indicate Quadra-Stable Dynamics of the Model
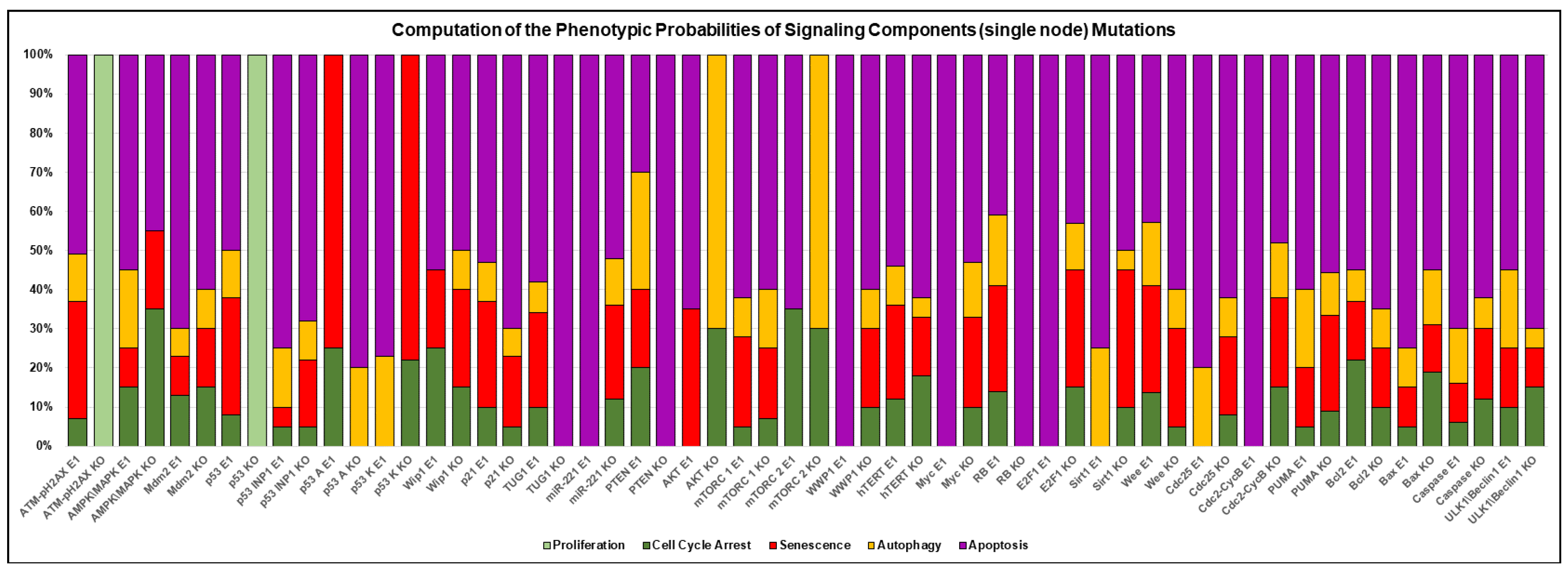
3.3. A Network-Guided Quadra Stability Correlation with Experimental Outcomes in DDR
3.4. Comparison of Experimental Observations with Phenotype Probabilities
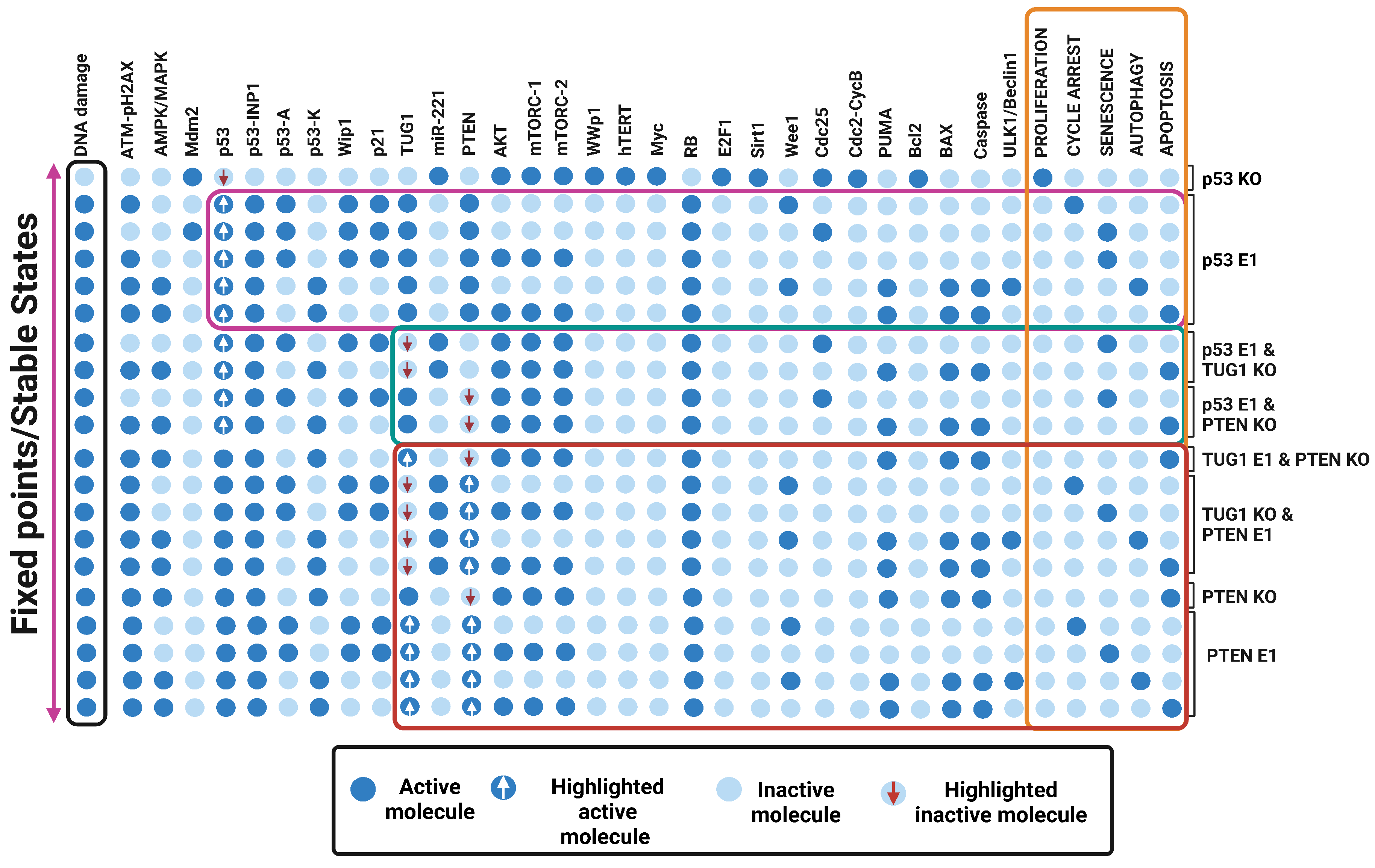
3.5. Quadra Stable Dynamics Is Regulated by p53 in DDR
3.6. Perturbations of TUG 1 and/or PTEN Can Break p53-Induced Quadratic Dynamics in DDR
3.7. “A New Guardian of Genome-PTEN” Is Required to Maintain the Quadra-Stable Dynamics of p53 in DDR
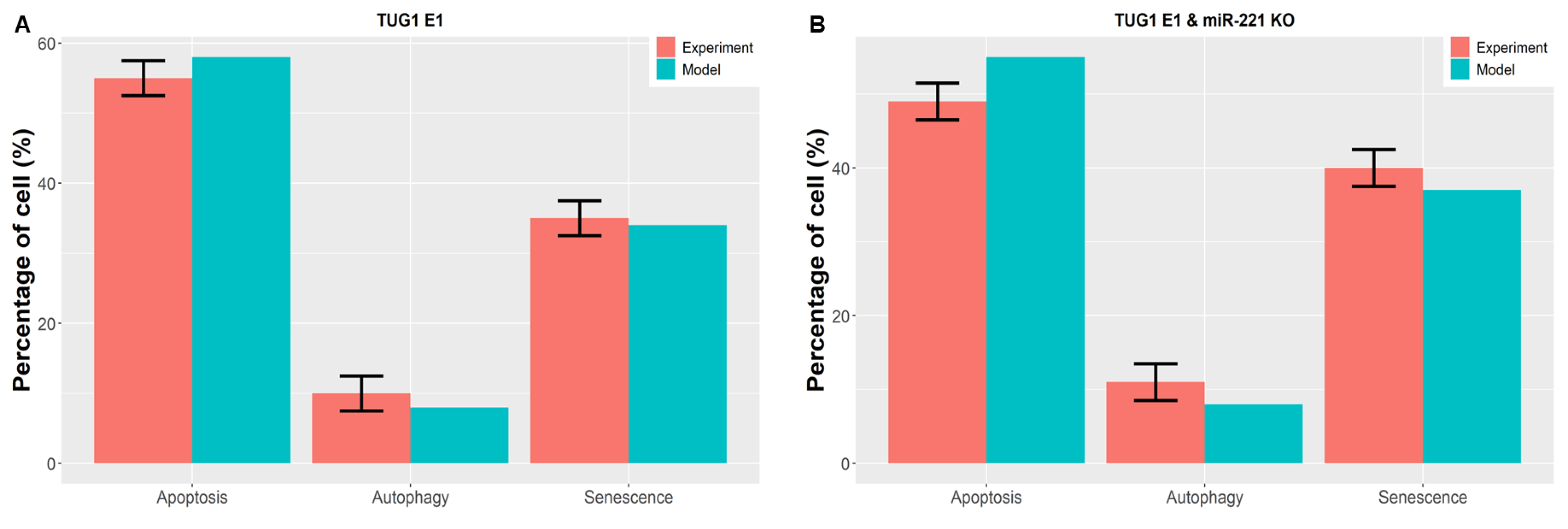
3.8. Consistency among Model Attractors and Other Experimental Investigations
4. Discussion
4.1. Influence of p53 on TUG1/miR-221 Axis and Its Consequences on PTEN Impulsivity in DDR
4.2. PTEN and Quadra-Stable Dynamics of p53 in DDR
4.2.1. Senescence and Apoptosis Induced by p53
4.2.2. Cell Cycle Arrest and Autophagy Is Regulated by PTEN
4.3. Implications of Quadra-Stable Dynamics of p53 and PTEN in Cancer Treatment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: Terminology for TOR-driven aging. Aging 2012, 4, 159–165. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.B., III; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21Cip1/Waf1 at both the G1/S and the G2/M cell cycle transitions: PRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar] [CrossRef]
- di Rorà, A.G.L.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in lung cancer: Dealing with the problem, building on new knowledge and turning the game around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef]
- Park, J.K.; Jung, H.Y.; Park, S.H.; Kang, S.Y.; Yi, M.R.; Um, H.D.; Hong, S.H. Combination of PTEN and γ-ionizing radiation enhances cell death and G2/M arrest through regulation of AKT activity and p21 induction in non–small-cell lung cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 1552–1560. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, S.; Yang, S.; Yang, Y.; Yang, T.; Yang, Y. Arsenic trioxide induces G2/M arrest in hepatocellular carcinoma cells by increasing the tumor suppressor PTEN expression. J. Cell. Biochem. 2012, 113, 3528–3535. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Dianatpour, A.; Ghafouri-Fard, S. The role of long non coding RNAs in the repair of DNA double strand breaks. Int. J. Mol. Cell. Med. 2017, 6, 1. [Google Scholar]
- Gebert, L.F.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 318030. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, L.; Zhang, Y.; Wu, Z.; He, D.; Li, X.; Wang, Z. Long non-coding RNA TUG1 enhances chemosensitivity in non-small cell lung cancer by impairing microRNA-221-dependent PTEN inhibition. Aging 2019, 11, 7553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yin, D.; Sun, M.; Kong, R.; Liu, X.; You, L.; Han, L.; Xia, R.; Wang, K.; Yang, J.; et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014, 5, e1243. [Google Scholar] [CrossRef]
- Schlatter, R.; Schmich, K.; Avalos Vizcarra, I.; Scheurich, P.; Sauter, T.; Borner, C.; Ederer, M.; Merfort, I.; Sawodny, O. ON/OFF and beyond-a Boolean model of apoptosis. PLoS Comput. Biol. 2009, 5, e1000595. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Panda, P.K.; Luo, W.; Hashimoto, R.F.; Ahuja, R. Network analysis reveals that the tumor suppressor lncRNA GAS5 acts as a double-edged sword in response to DNA damage in gastric cancer. Sci. Rep. 2022, 12, 18312. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hashimoto, R.F. Dynamical analysis of a boolean network model of the Oncogene role of lncRNA ANRIL and lncRNA UFC1 in non-small cell lung cancer. Biomolecules 2022, 12, 420. [Google Scholar] [CrossRef]
- Gupta, S.; Panda, P.K.; Hashimoto, R.F.; Samal, S.K.; Mishra, S.; Verma, S.K.; Mishra, Y.K.; Ahuja, R. Dynamical modeling of miR-34a, miR-449a, and miR-16 reveals numerous DDR signaling pathways regulating senescence, autophagy, and apoptosis in HeLa cells. Sci. Rep. 2022, 12, 4911. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Hashimoto, R.F.; Mombach, J.C.M. A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells. Biology 2022, 11, 480. [Google Scholar] [CrossRef]
- Silveira, D.A.; Gupta, S.; Sinigaglia, M.; Mombach, J.C.M. The Wnt pathway can stabilize hybrid phenotypes in the epithelial-mesenchymal transition: A logical modeling approach. Comput. Biol. Chem. 2022, 99, 107714. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.S.; Saadatpour, A.; Albert, R. Boolean modeling in systems biology: An overview of methodology and applications. Phys. Biol. 2012, 9, 055001. [Google Scholar] [CrossRef] [PubMed]
- Rozum, J.C.; Gómez Tejeda Zañudo, J.; Gan, X.; Deritei, D.; Albert, R. Parity and time reversal elucidate both decision-making in empirical models and attractor scaling in critical Boolean networks. Sci. Adv. 2021, 7, eabf8124. [Google Scholar] [CrossRef]
- Sizek, H.; Hamel, A.; Deritei, D.; Campbell, S.; Ravasz Regan, E. Boolean model of growth signaling, cell cycle and apoptosis predicts the molecular mechanism of aberrant cell cycle progression driven by hyperactive PI3K. PLoS Comput. Biol. 2019, 15, e1006402. [Google Scholar] [CrossRef] [PubMed]
- Silveira, D.A.; Gupta, S.; Mombach, J.C.M. Systems biology approach suggests new miRNAs as phenotypic stability factors in the epithelial–mesenchymal transition. J. R. Soc. Interface 2020, 17, 20200693. [Google Scholar] [CrossRef]
- Folkesson, E.; Sakshaug, C.; Hoel, A.D.; Klinkenberg, G.; Flobak, Å. Synergistic effects of complex drug combinations in colorectal cancer cells predicted by logical modelling. Front. Syst. Biol. 2023, 3, 12. [Google Scholar] [CrossRef]
- Abou-Jaoudé, W.; Traynard, P.; Monteiro, P.T.; Saez-Rodriguez, J.; Helikar, T.; Thieffry, D.; Chaouiya, C. Logical modeling and dynamical analysis of cellular networks. Front. Genet. 2016, 7, 94. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Hamberg, M.; Backes, C.; Fehlmann, T.; Hart, M.; Meder, B.; Meese, E.; Keller, A. MiRTargetLink—miRNAs, genes and interaction networks. Int. J. Mol. Sci. 2016, 17, 564. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Naldi, A.; Hernandez, C.; Abou-Jaoudé, W.; Monteiro, P.T.; Chaouiya, C.; Thieffry, D. Logical modeling and analysis of cellular regulatory networks with ginsim 3.0. Front. Physiol. 2018, 9, 646. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. ATM/miR-34a-5p axis regulates a p21-dependent senescence-apoptosis switch in non-small cell lung cancer: A Boolean model of G1/S checkpoint regulation. FEBS Lett. 2020, 594, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Barbé-Tuana, F.M.; Mombach, J.C.M. Integrative data modeling from lung and lymphatic cancer predicts functional roles for miR-34a and miR-16 in cell fate regulation. Sci. Rep. 2020, 10, 2511. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Modeling the role of microRNA-449a in the regulation of the G2/M cell cycle checkpoint in prostate LNCaP cells under ionizing radiation. PLoS ONE 2018, 13, e0200768. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef]
- Chène, P. Inhibiting the p53–MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef]
- Lu, X.; Ma, O.; Nguyen, T.A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: P53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Charrier-Savournin, F.B.; Château, M.T.; Gire, V.; Sedivy, J.; Piette, J.; Dulic, V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol. Biol. Cell 2004, 15, 3965–3976. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Eichbaum, Q.; Silberman, E.; Mayer, B.J.; Dutta, A. p21CIP1 and Cdc25A: Competition between an inhibitor and an activator of cyclin-dependent kinases. Mol. Cell. Biol. 1997, 17, 4338–4345. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G2/M cell cycle checkpoint induced by DNA damage. Mol. Cell. Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, L.; Zhang, L.; Xu, A.; Li, Z.; Xu, Y.; He, P.; Wu, M.; Wei, F.; Wang, C. PTEN enhances G2/M arrest in etoposide-treated MCF-7 cells through activation of the ATM pathway. Oncol. Rep. 2016, 35, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Qu, J.; Gao, Z.; Qi, Q.; Yin, H.; Zhu, L.; Wu, Y.; Liu, W.; Yang, J.; Huang, X. Timosaponin AIII induces G2/M arrest and apoptosis in breast cancer by activating the ATM/Chk2 and p38 MAPK signaling pathways. Front. Pharmacol. 2021, 11, 601468. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, Q.; Li, B.; Xie, B.; Wang, W. Temozolomide induces autophagy via ATM-AMPK-ULK1 pathways in glioma. Mol. Med. Rep. 2014, 10, 411–416. [Google Scholar] [CrossRef]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef]
- De, S.; Campbell, C.; Venkitaraman, A.R.; Esposito, A. Pulsatile MAPK signaling modulates p53 activity to control cell fate decisions at the G2 checkpoint for DNA damage. Cell Rep. 2020, 30, 2083–2093. [Google Scholar] [CrossRef]
- He, P.; Li, Z.; Xu, F.; Ru, G.; Huang, Y.; Lin, E.; Peng, S. AMPK activity contributes to G2 arrest and DNA damage decrease via p53/p21 pathways in oxidatively damaged mouse zygotes. Front. Cell Dev. Biol. 2020, 8, 539485. [Google Scholar] [CrossRef]
- Shen, Y.; Sherman, J.W.; Chen, X.; Wang, R. Phosphorylation of CDC25C by AMP-activated protein kinase mediates a metabolic checkpoint during cell-cycle G2/M-phase transition. J. Biol. Chem. 2018, 293, 5185–5199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sin, K.W.T.; Ding, H.; Doan, H.A.; Gao, S.; Miao, H.; Wei, Y.; Wang, Y.; Zhang, G.; Li, Y.P. p38β MAPK mediates ULK1-dependent induction of autophagy in skeletal muscle of tumor-bearing mice. Cell Stress 2018, 2, 311. [Google Scholar] [CrossRef]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. Fine-tuning of AMPK–ULK1–mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020, 10, 17803. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. OncoTargets Ther. 2018, 11, 7777. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Bak, Y.; Kim, M.J.; Srinivasrao, G.; Hwang, J.; Sung, N.K.; Kim, B.Y.; Yu, J.H.; Hong, J.T.; Yoon, D.Y. The novel small molecule STK899704 promotes senescence of the human A549 NSCLC cells by inducing DNA damage responses and cell cycle arrest. Front. Pharmacol. 2018, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Yong, Y.; Kim, C.G.; Lee, Y.H.; Lim, Y. Deoxypodophyllotoxin induces G2/M cell cycle arrest and apoptosis in HeLa cells. Cancer Lett. 2010, 287, 231–239. [Google Scholar] [CrossRef]
- Arafa, E.S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. Mol. Mech. Mutagen. 2011, 706, 28–35. [Google Scholar] [CrossRef]
- Huang, M.; Whang, P.; Lewicki, P.; Mitchell, B.S. Cyclopentenyl cytosine induces senescence in breast cancer cells through the nucleolar stress response and activation of p53. Mol. Pharmacol. 2011, 80, 40–48. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Vizza, D.; Plastina, P.; Barone, I.; Casaburi, I.; Lanzino, M.; De Amicis, F.; Sisci, D.; Mauro, L.; et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J. Cell. Physiol. 2013, 228, 1314–1322. [Google Scholar] [CrossRef]
- Pawar, J.S.; Mustafa, S.; Ghosh, I. Chrysin and Capsaicin induces premature senescence and apoptosis via mitochondrial dysfunction and p53 elevation in Cervical cancer cells. Saudi J. Biol. Sci. 2022, 29, 3838–3847. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H.Y.; Gao, X.J.; Zhu, H.X.; Zhang, X.P.; Liu, F.; Wang, W. Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses. Cells 2023, 12, 490. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Towards DNA-damage induced autophagy: A Boolean model of p53-induced cell fate mechanisms. DNA Repair 2020, 96, 102971. [Google Scholar] [CrossRef]
- Feroz, W.; Sheikh, A.M.A. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, W. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Chen, R.J.; Wu, P.H.; Ho, C.T.; Way, T.D.; Pan, M.H.; Chen, H.M.; Ho, Y.S.; Wang, Y.J. P53-dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment. Cell Death Dis. 2017, 8, e2985. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Jee, H.J.; Um, J.H.; Kim, Y.M.; Bae, S.S.; Yun, J. Cooperation between p21 and Akt is required for p53-dependent cellular senescence. Aging Cell 2017, 16, 1094–1103. [Google Scholar] [CrossRef]
- PORĘBSKA, I.; Wyrodek, E.; Kosacka, M.; Adamiak, J.; Jankowska, R.; Harłozińska-Szmyrka, A. Apoptotic markers p53, Bcl-2 and Bax in primary lung cancer. In Vivo 2006, 20, 599–604. [Google Scholar]
- de Gooijer, M.C.; van den Top, A.; Bockaj, I.; Beijnen, J.H.; Würdinger, T.; van Tellingen, O. The G2 checkpoint—A node-based molecular switch. FEBS Open Bio 2017, 7, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, H.; Baek, J.; Cho, Y.; Kang, H.; Jeong, J.; Song, J.; Park, H.; Chun, K. Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene 2016, 35, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell cycle control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stimulus/Perturbations | Method (In Vivo and In Vitro) | Model Response/Phenotype | References |
|---|---|---|---|
| Cervical Cancer (HeLa Cells) | |||
| p53 E1 and PTEN KO in DDR | In-vitro | Senescence and apoptosis at G2/M-phase checkpoint | [61] |
| PTEN E1 in DDR | In-vitro | Cell cycle arrest and apoptosis at G2/M-phase checkpoint | [57] |
| In-vitro | Autophagy and Apoptosis | [11] | |
| Breast Cancer (MCF-7 Cells) | |||
| p53 E1 and PTEN KO in DDR | In-vitro | Senescence and apoptosis at G2/M-phase checkpoint | [59] |
| PTEN E1 in DDR | In-vitro | Cell cycle arrest at the G2/M chcekpoint | [45] |
| In-vitro | Cell cycle arrest and apoptosis at G2/M-phase checkpoint | [58] | |
| In-vitro | Autophagy and Apoptosis | [60] | |
| Network Predictions for In-vitro/In-vivo experiments | |||
| PTEN E1 together with ATM E1 in DDR | In-vivo/In-vitro | Inhibition of proliferation | ? |
| PTEN E1 together with KO Cdc25c in DDR | In-vivo/In-vitro | Inhibition of proliferation and activation of DNA damage repair pathways | ? |
| PTEN E1 together with NEDD4-like E3 ubiquitin-protein ligase WWP1 (WWP1) KO in DDR | In-vivo/In-vitro | Inhibition of proliferation and Induction of cell cycle arrest, senescence, autophagy and apoptosis | ? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Panda, P.K.; Silveira, D.A.; Ahuja, R.; Hashimoto, R.F. Quadra-Stable Dynamics of p53 and PTEN in the DNA Damage Response. Cells 2023, 12, 1085. https://doi.org/10.3390/cells12071085
Gupta S, Panda PK, Silveira DA, Ahuja R, Hashimoto RF. Quadra-Stable Dynamics of p53 and PTEN in the DNA Damage Response. Cells. 2023; 12(7):1085. https://doi.org/10.3390/cells12071085
Chicago/Turabian StyleGupta, Shantanu, Pritam Kumar Panda, Daner A. Silveira, Rajeev Ahuja, and Ronaldo F. Hashimoto. 2023. "Quadra-Stable Dynamics of p53 and PTEN in the DNA Damage Response" Cells 12, no. 7: 1085. https://doi.org/10.3390/cells12071085
APA StyleGupta, S., Panda, P. K., Silveira, D. A., Ahuja, R., & Hashimoto, R. F. (2023). Quadra-Stable Dynamics of p53 and PTEN in the DNA Damage Response. Cells, 12(7), 1085. https://doi.org/10.3390/cells12071085