
BAG3: Nature’s Quintessential Multi-Functional Protein Functions as a Ubiquitous Intra-Cellular Glue


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure-Function Relationship
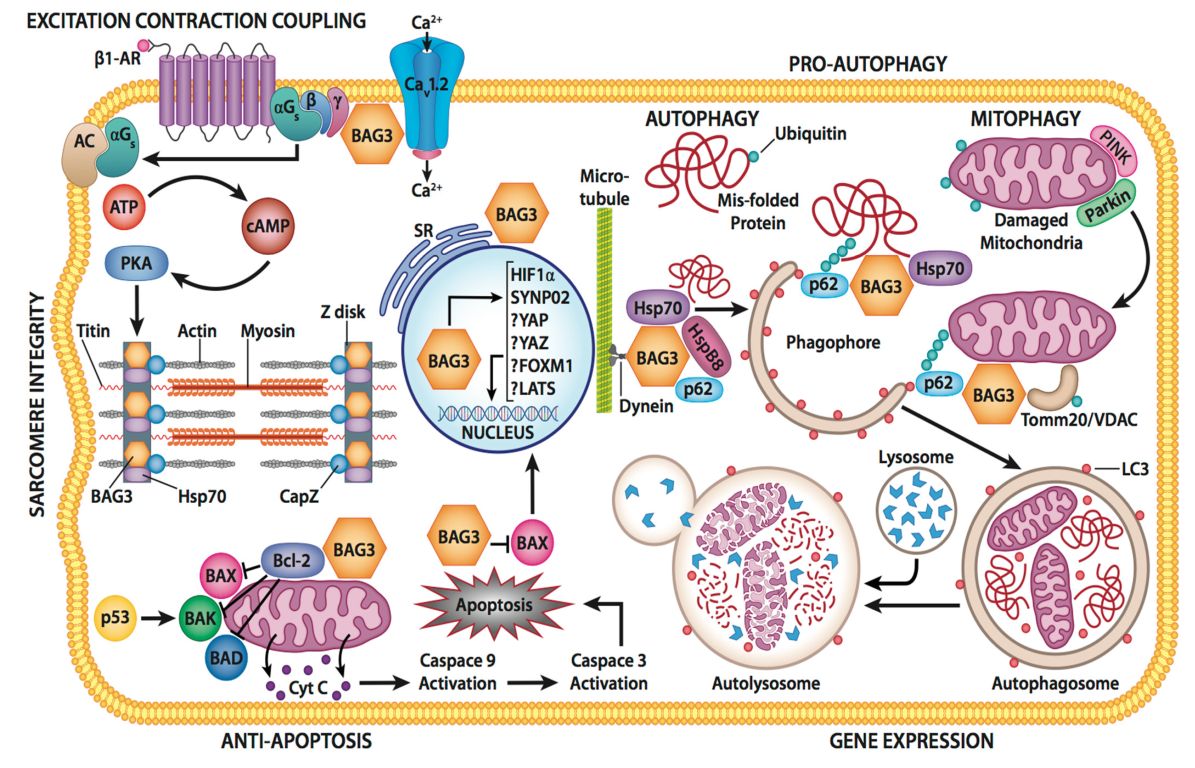
3. Cellular Function of BAG3
3.1. Protein Quality Control
3.2. BAG3 and Autophagy
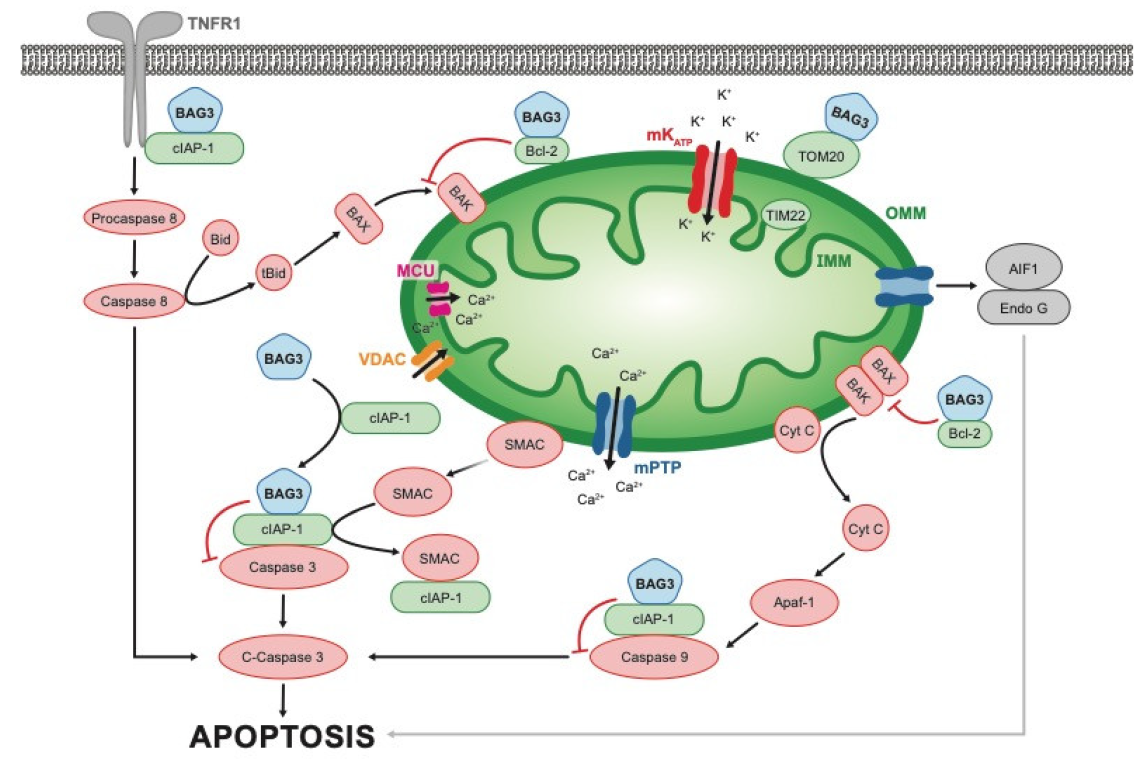
3.3. Apoptosis and Oxidative Stress
3.4. BAG3 and the Sarcomere
3.5. BAG3 and Insulin Release
3.6. Cell Cycle
3.7. BAG3 and Cell-Cell Junctions
3.8. BAG3 and Proteasome Inhibitors
3.9. Effects of BAG3 Depletion on the Proteosome the Inflammasome and the Metabolome
3.10. BAG3 Secretion/Excretion/Leak
3.11. Cell Metabolism
3.12. Angiogenesis
4. BAG3 in Heart and Skeletal Muscle
4.1. BAG3 Genetic Variants and Dilated Cardiomyopathy
4.2. Cardiomyocyte Structural Integrity and Protein Quality Control
4.3. BAG3 as a Stress-Responsive Protein in the Heart
4.4. Role of BAG3 in Skeletal Muscle
5. Role of BAG3 in Cancer Biology
5.1. Fundamental Observations
5.2. Epithelial-Mesenchymal Transition/Stemness
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirk, J.A.; Cheung, J.Y.; Feldman, A.M. Therapeutic targeting of BAG3: Considering its complexity in cancer and heart disease. J. Clin. Investig. 2021, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Carra, S.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Graziano, V.; De Laurenzi, V.; Pascale, M.; Turco, M.C. BAG3: A multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011, 2, e141. [Google Scholar] [CrossRef]
- Lee, J.H.; Takahashi, T.; Yasuhara, N.; Inazawa, J.; Kamada, S.; Tsujimoto, Y. Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene 1999, 18, 6183–6190. [Google Scholar] [CrossRef]
- Qu, H.Q.; Feldman, A.M.; Hakonarson, H. Genetics of BAG3: A Paradigm for Developing Precision Therapies for Dilated Cardiomyopathies. J. Am. Heart Assoc. 2022, 11, e027373. [Google Scholar] [CrossRef]
- Meister-Broekema, M.; Freilich, R.; Jagadeesan, C.; Rauch, J.N.; Bengoechea, R.; Motley, W.W.; Kuiper, E.F.E.; Minoia, M.; Furtado, G.V.; van Waarde, M.; et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat. Commun. 2018, 9, 5342. [Google Scholar] [CrossRef]
- Chakraborty, D.; Felzen, V.; Hiebel, C.; Sturner, E.; Perumal, N.; Manicam, C.; Sehn, E.; Grus, F.; Wolfrum, U.; Behl, C. Enhanced autophagic-lysosomal activity and increased BAG3-mediated selective macroautophagy as adaptive response of neuronal cells to chronic oxidative stress. Redox Biol. 2019, 24, 101181. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Bultynck, G.; Savvides, S.N. Pore-forming proteins as drivers of membrane permeabilization in cell death pathways. Nat. Rev. Mol. Cell Biol. 2022. (online ahead of print). [Google Scholar] [CrossRef]
- Baeken, M.W.; Behl, C. On the origin of BAG(3) and its consequences for an expansion of BAG3’s role in protein homeostasis. J. Cell Biochem. 2022, 123, 102–114. [Google Scholar] [CrossRef]
- Wang, J.; Tomar, D.; Martin, T.G.; Dubey, S.; Dubey, P.K.; Song, J.; Landensberg, B.S.; Myers, V.D.; Merali, S.; Merali, C.; et al. BAG3 regulates mitochondrial function and the inflammasome through canonical and non-canonicall pathways in the heart. J. Am. Coll. Cardiol. Basic Trans. Sci. 2023, in press. [Google Scholar]
- Huang, S.C.; Vu, L.V.; Yu, F.H.; Nguyen, D.T.; Benz, E.J., Jr. Multifunctional protein 4.1R regulates the asymmetric segregation of Numb during terminal erythroid maturation. J. Biol. Chem. 2021, 297, 101051. [Google Scholar] [CrossRef]
- Takayama, S.; Xie, Z.; Reed, J.C. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef]
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 2009, 425, 245–255. [Google Scholar] [CrossRef]
- McCollum, A.K.; Casagrande, G.; Kohn, E.C. Caught in the middle: The role of Bag3 in disease. Biochem. J. 2009, 425, e1–e3. [Google Scholar] [CrossRef]
- Zhang, R.; Varela, M.; Forn-Cuni, G.; Torraca, V.; van der Vaart, M.; Meijer, A.H. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria-infected macrophages. Cell Death Dis. 2020, 11, 277. [Google Scholar] [CrossRef]
- Kogel, D.; Linder, B.; Brunschweiger, A.; Chines, S.; Behl, C. At the Crossroads of Apoptosis and Autophagy: Multiple Roles of the Co-Chaperone BAG3 in Stress and Therapy Resistance of Cancer. Cells 2020, 9, 574. [Google Scholar] [CrossRef]
- Marzullo, L.; Turco, M.C.; De Marco, M. The multiple activities of BAG3 protein: Mechanisms. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129628. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef]
- Ni, E.; Zhao, L.; Yao, N.; Zhu, X.; Cao, H.; Sun, S.; Zhu, W. The PXXP domain is critical for the protective effect of BAG3 in cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 2019, 46, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Li, L.; Cui, B.; Men, S.; Shen, Y.; Yang, X. Structural insight into plant programmed cell death mediated by BAG proteins in Arabidopsis thaliana. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, B.; Vendredy, L.; Timmerman, V.; Poletti, A. The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy 2023, 3, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Tawfik, S.; Moravec, C.S.; Pak, T.R.; Kirk, J.A. BAG3 expression and sarcomere localization in the human heart are linked to HSF-1 and are differentially affected by sex and disease. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2339–H2350. [Google Scholar] [CrossRef]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Millan, J.A.; Reed, J.C. Cloning and functional analysis of BAG-1: A novel Bcl-2-binding protein with anti-cell death activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef]
- Moribe, Y.; Niimi, T.; Yamashita, O.; Yaginuma, T. Samui, a novel cold-inducible gene, encoding a protein with a BAG domain similar to silencer of death domains (SODD/BAG-4), isolated from Bombyx diapause eggs. Eur. J. Biochem. 2001, 268, 3432–3442. [Google Scholar] [CrossRef]
- Briknarova, K.; Takayama, S.; Brive, L.; Havert, M.L.; Knee, D.A.; Velasco, J.; Homma, S.; Cabezas, E.; Stuart, J.; Hoyt, D.W.; et al. Structural analysis of BAG1 cochaperone and its interactions with Hsc70 heat shock protein. Nat. Struct. Biol. 2001, 8, 349–352. [Google Scholar] [CrossRef]
- Franceschelli, S.; Rosati, A.; Lerose, R.; De Nicola, S.; Turco, M.C.; Pascale, M. Bag3 gene expression is regulated by heat shock factor 1. J. Cell Physiol. 2008, 215, 575–577. [Google Scholar] [CrossRef]
- Qi, H.; Fillion, C.; Labrie, Y.; Grenier, J.; Fournier, A.; Berger, L.; El-Alfy, M.; Labrie, C. AIbZIP, a novel bZIP gene located on chromosome 1q21.3 that is highly expressed in prostate tumors and of which the expression is up-regulated by androgens in LNCaP human prostate cancer cells. Cancer Res. 2002, 62, 721–733. [Google Scholar]
- Gentilella, A.; Khalili, K. BAG3 expression is sustained by FGF2 in neural progenitor cells and impacts cell proliferation. Cell Cycle 2010, 9, 4245–4247. [Google Scholar] [CrossRef]
- Gout, E.; Gutkowska, M.; Takayama, S.; Reed, J.C.; Chroboczek, J. Co-chaperone BAG3 and adenovirus penton base protein partnership. J. Cell. Biochem. 2010, 111, 699–708. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, Q.; Xia, H.; Xu, H.; Bi, F. PKM2 compensates for proteasome dysfunction by mediating the formation of the CHIP-HSP70-BAG3 complex and the aggregation of ubiquitinated proteins. FASEB J. 2022, 36, e22121. [Google Scholar] [CrossRef]
- Kassis, J.N.; Guancial, E.A.; Doong, H.; Virador, V.; Kohn, E.C. CAIR-1/BAG-3 modulates cell adhesion and migration by downregulating activity of focal adhesion proteins. Exp. Cell Res. 2006, 312, 2962–2971. [Google Scholar] [CrossRef]
- Martin, T.G.; Myers, V.D.; Dubey, P.; Dubey, S.; Perez, E.; Moravec, C.S.; Willis, M.S.; Feldman, A.M.; Kirk, J.A. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat. Commun. 2021, 12, 2942. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef]
- Bryantsev, A.L.; Kurchashova, S.Y.; Golyshev, S.A.; Polyakov, V.Y.; Wunderink, H.F.; Kanon, B.; Budagova, K.R.; Kabakov, A.E.; Kampinga, H.H. Regulation of stress-induced intracellular sorting and chaperone function of Hsp27 (HspB1) in mammalian cells. Biochem. J. 2007, 407, 407–417. [Google Scholar] [CrossRef]
- Martin, J.L.; Dawson, S.J.; Gale, J.E. An emerging role for stress granules in neurodegenerative disease and hearing loss. Hear. Res. 2022, 426, 108634. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; Orth, K.; O’Rourke, K.; Duan, H.; Poirier, G.G.; Dixit, V.M. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J. Biol. Chem. 1996, 271, 4573–4576. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Y.; Miao, W.; Shi, C.; Chen, Z.; Wu, B.; Zou, Y.; Ma, Q.; You, S.; Lu, S.; et al. An unexpected role for BAG3 in regulating PARP1 ubiquitination in oxidative stress-related endothelial damage. Redox Biol. 2022, 50, 102238. [Google Scholar] [CrossRef]
- Chen, H.; Moreno-Moral, A.; Pesce, F.; Devapragash, N.; Mancini, M.; Heng, E.L.; Rotival, M.; Srivastava, P.K.; Harmston, N.; Shkura, K.; et al. Author Correction: WWP2 regulates pathological cardiac fibrosis by modulating SMAD2 signaling. Nat. Commun. 2019, 10, 4085. [Google Scholar] [CrossRef]
- Ganassi, M.; Badodi, S.; Ortuste Quiroga, H.P.; Zammit, P.S.; Hinits, Y.; Hughes, S.M. Myogenin promotes myocyte fusion to balance fibre number and size. Nat. Commun. 2018, 9, 4232. [Google Scholar] [CrossRef]
- Ghozlan, H.; Cox, A.; Nierenberg, D.; King, S.; Khaled, A.R. The TRiCky Business of Protein Folding in Health and Disease. Front. Cell Dev. Biol. 2022, 10, 906530. [Google Scholar] [CrossRef]
- De Marco, M.; Turco, M.C.; Rosati, A. BAG3 protein is induced during cardiomyoblast differentiation and modulates myogenin expression. Cell Cycle 2011, 10, 850–852. [Google Scholar] [CrossRef][Green Version]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef]
- Iorio, V.; Festa, M.; Rosati, A.; Hahne, M.; Tiberti, C.; Capunzo, M.; De Laurenzi, V.; Turco, M.C. BAG3 regulates formation of the SNARE complex and insulin secretion. Cell Death Dis. 2015, 6, e1684. [Google Scholar] [CrossRef]
- Falco, A.; Festa, M.; Basile, A.; Rosati, A.; Pascale, M.; Florenzano, F.; Nori, S.L.; Nicolin, V.; Di Benedetto, M.; Vecchione, M.L.; et al. BAG3 controls angiogenesis through regulation of ERK phosphorylation. Oncogene 2012, 31, 5153–5161. [Google Scholar] [CrossRef]
- Saffitz, J.E.; Laing, J.G.; Yamada, K.A. Connexin expression and turnover: Implications for cardiac excitability. Circ. Res. 2000, 86, 723–728. [Google Scholar] [CrossRef]
- Wahl, C.M.; Schmidt, C.; Hecker, M.; Ullrich, N.D. Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases. Int. J. Mol. Sci. 2022, 23, 10147. [Google Scholar] [CrossRef]
- Saffitz, J.E.; Schuessler, R.B. Connexin-40, bundle-branch block, and propagation at the Purkinje-myocyte junction. Circ. Res. 2000, 87, 835–836. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef]
- Arena, A.; Romeo, M.A.; Benedetti, R.; Montani, M.S.G.; Cirone, M. JQ-1/Bortezomib combination strongly impairs MM and PEL survival by inhibiting c-Myc and mTOR despite the activation of pro-survival mechanisms. Exp. Hematol. 2023. [Google Scholar] [CrossRef]
- Engelhardt, M.; Wasch, R. Carfilzomib, lenalidomide, and dexamethasone maintenance for multiple myeloma: When and for whom? Lancet Oncol. 2023, 24, 118–119. [Google Scholar] [CrossRef]
- Judge, L.M.; Perez-Bermejo, J.A.; Truong, A.; Ribeiro, A.J.; Yoo, J.C.; Jensen, C.L.; Mandegar, M.A.; Huebsch, N.; Kaake, R.M.; So, P.L.; et al. A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight 2017, e94362. [Google Scholar] [CrossRef]
- Haudek, S.B.; Taffet, G.E.; Schneider, M.D.; Mann, D.L. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J. Clin. Investig. 2007, 117, 2692–2701. [Google Scholar] [CrossRef]
- Zhang, J.; Webster, J.D.; Dugger, D.L.; Goncharov, T.; Roose-Girma, M.; Hung, J.; Kwon, Y.C.; Vucic, D.; Newton, K.; Dixit, V.M. Ubiquitin Ligases cIAP1 and cIAP2 Limit Cell Death to Prevent Inflammation. Cell Rep. 2019, 27, 2679–2689.e2673. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ashurst, L.H.; Barrow, S.L.; Chvanov, M.A.; Gillies, S.; Criddle, D.N.; Tepikin, A.V.; Petersen, O.H.; et al. Caspase-8-mediated apoptosis induced by oxidative stress is independent of the intrinsic pathway and dependent on cathepsins. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 293, G296–G307. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Evan, G.I. Caspase-8 in apoptosis: The beginning of “the end”? IUBMB Life 2000, 50, 85–90. [Google Scholar] [CrossRef]
- Kearney, C.J.; Sheridan, C.; Cullen, S.P.; Tynan, G.A.; Logue, S.E.; Afonina, I.S.; Vucic, D.; Lavelle, E.C.; Martin, S.J. Inhibitor of apoptosis proteins (IAPs) and their antagonists regulate spontaneous and tumor necrosis factor (TNF)-induced proinflammatory cytokine and chemokine production. J. Biol. Chem. 2013, 288, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca(2+) Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, Y. Decreased ATP production during mitochondrial calcium uniporter inhibition enhances autophagy and mitophagy to provide cardioprotection in cardiac failure. Int. J. Cardiol. 2019, 282, 67. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, R.; Li, M.; Yu, Y.; Liang, Y.; Han, F.; Qin, S.; Chen, X.; Su, Y.; Ge, J. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 2018, 271, 161–168. [Google Scholar] [CrossRef]
- Garbincius, J.F.; Elrod, J.W. Is the Failing Heart Starved of Mitochondrial Calcium? Circ. Res. 2021, 128, 1205–1207. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Rosati, A.; Bersani, S.; Tavano, F.; Dalla Pozza, E.; De Marco, M.; Palmieri, M.; De Laurenzi, V.; Franco, R.; Scognamiglio, G.; Palaia, R.; et al. Expression of the antiapoptotic protein BAG3 is a feature of pancreatic adenocarcinoma and its overexpression is associated with poorer survival. Am. J. Pathol. 2012, 181, 1524–1529. [Google Scholar] [CrossRef]
- Falco, A.; Rosati, A.; Festa, M.; Basile, A.; De Marco, M.; d’Avenia, M.; Pascale, M.; Dal Piaz, F.; Tavano, F.; Di Mola, F.F.; et al. BAG3 is a novel serum biomarker for pancreatic adenocarcinomas. Am. J. Gastroenterol. 2013, 108, 1178–1180. [Google Scholar] [CrossRef]
- Rosati, A.; Basile, A.; D’Auria, R.; d’Avenia, M.; De Marco, M.; Falco, A.; Festa, M.; Guerriero, L.; Iorio, V.; Parente, R.; et al. BAG3 promotes pancreatic ductal adenocarcinoma growth by activating stromal macrophages. Nat. Commun. 2015, 6, 8695. [Google Scholar] [CrossRef]
- Basile, A.; De Marco, M.; Festa, M.; Falco, A.; Iorio, V.; Guerriero, L.; Eletto, D.; Rea, D.; Arra, C.; Lamolinara, A.; et al. Development of an anti-BAG3 humanized antibody for treatment of pancreatic cancer. Mol. Oncol. 2019, 13, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- An, M.X.; Li, S.; Yao, H.B.; Li, C.; Wang, J.M.; Sun, J.; Li, X.Y.; Meng, X.N.; Wang, H.Q. BAG3 directly stabilizes Hexokinase 2 mRNA and promotes aerobic glycolysis in pancreatic cancer cells. J. Cell Biol. 2017, 216, 4091–4105. [Google Scholar] [CrossRef]
- Lin, H.; Koren, S.A.; Cvetojevic, G.; Girardi, P.; Johnson, G.V.W. The role of BAG3 in health and disease: A “Magic BAG of Tricks”. J. Cell Biochem. 2022, 123, 4–21. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, J.M.; Yan, J.; Zhang, D.L.; Liu, B.Q.; Jiang, J.Y.; Li, C.; Li, S.; Meng, X.N.; Wang, H.Q. BAG3 promotes autophagy and glutaminolysis via stabilizing glutaminase. Cell Death Dis. 2019, 10, 284. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef]
- De Marco, M.; Basile, A.; Iorio, V.; Festa, M.; Falco, A.; Ranieri, B.; Pascale, M.; Sala, G.; Remondelli, P.; Capunzo, M.; et al. Role of BAG3 in cancer progression: A therapeutic opportunity. Semin. Cell Dev. Biol. 2018, 78, 85–92. [Google Scholar] [CrossRef]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Tedesco, B.; Mediani, L.; Asselbergh, B.; Crippa, V.; Antoniani, F.; Carra, S.; Poletti, A.; Timmerman, V. BAG3 Pro209 mutants associated with myopathy and neuropathy relocate chaperones of the CASA-complex to aggresomes. Sci. Rep. 2020, 10, 8755. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ooms, A.; Graf-Riesen, K.; Kuppusamy, M.; Unger, A.; Schuld, J.; Daerr, J.; Lother, A.; Geisen, C.; Hein, L.; et al. Overexpression of human BAG3(P209L) in mice causes restrictive cardiomyopathy. Nat. Commun. 2021, 12, 3575. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.L.; Sacconi, S.; Bach, J.E.; Liebe, C.; Burmann, J.; Kley, R.A.; Ferbert, A.; Anderheiden, R.; Van den Bergh, P.; Martin, J.J.; et al. Unusual multisystemic involvement and a novel BAG3 mutation revealed by NGS screening in a large cohort of myofibrillar myopathies. Orphanet J. Rare Dis. 2014, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Ellinor, P.T.; Sasse-Klaassen, S.; Probst, S.; Gerull, B.; Shin, J.T.; Toeppel, A.; Heuser, A.; Michely, B.; Yoerger, D.M.; Song, B.S.; et al. A novel locus for dilated cardiomyopathy, diffuse myocardial fibrosis, and sudden death on chromosome 10q25-26. J. Am. Coll. Cardiol. 2006, 48, 106–111. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]
- Chami, N.; Tadros, R.; Lemarbre, F.; Lo, K.S.; Beaudoin, M.; Robb, L.; Labuda, D.; Tardif, J.C.; Racine, N.; Talajic, M.; et al. Nonsense mutations in BAG3 are associated with early-onset dilated cardiomyopathy in French Canadians. Can. J. Cardiol. 2014, 30, 1655–1661. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Bilinska, Z.T.; Sobieszczanska-Malek, M.; Michalak, E.; Sleszycka, J.; Sioma, A.; Malek, L.A.; Kaczmarska, D.; Walczak, E.; Wlodarski, P.; et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: Four novel mutations and a genotype-phenotype correlation. J. Transl. Med. 2014, 12, 192. [Google Scholar] [CrossRef]
- Feldman, A.M.; Begay, R.L.; Knezevic, T.; Myers, V.D.; Slavov, D.B.; Zhu, W.; Gowan, K.; Graw, S.L.; Jones, K.L.; Tilley, D.G.; et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J. Cell Physiol. 2014, 229, 1697–1702. [Google Scholar] [CrossRef]
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.Q.; Gupta, M.K.; Tahrir, F.G.; Gordon, J.; McClung, J.M.; et al. Haplo-insufficiency of Bcl2-associated athanogene 3 in mice results in progressive left ventricular dysfunction, beta-adrenergic insensitivity, and increased apoptosis. J. Cell Physiol. 2018, 233, 6319–6326. [Google Scholar] [CrossRef]
- Dominguez, F.; Cuenca, S.; Bilinska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef]
- Shah, S.; Henry, A.; Roselli, C.; Lin, H.; Sveinbjornsson, G.; Fatemifar, G.; Hedman, A.K.; Wilk, J.B.; Morley, M.P.; Chaffin, M.D.; et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020, 11, 163. [Google Scholar] [CrossRef]
- Myers, V.D.; Gerhard, G.S.; McNamara, D.M.; Tomar, D.; Madesh, M.; Kaniper, S.; Ramsey, F.V.; Fisher, S.G.; Ingersoll, R.G.; Kasch-Semenza, L.; et al. Association of Variants in BAG3 With Cardiomyopathy Outcomes in African American Individuals. JAMA Cardiol. 2018, 3, 929–938. [Google Scholar] [CrossRef]
- Feldman, A.M.; Gordon, J.; Wang, J.; Song, J.; Zhang, X.Q.; Myers, V.D.; Tomar, D.; Gerhard, G.S.; Khalili, K.; Cheung, J.Y. Novel BAG3 Variants in African American Patients With Cardiomyopathy: Reduced beta-Adrenergic Responsiveness in Excitation-Contraction. J. Card Fail. 2020, 26, 1075–1085. [Google Scholar] [CrossRef]
- Cheung, J.Y.; Gordon, J.; Wang, J.; Song, J.; Zhang, X.Q.; Tilley, D.G.; Gao, E.; Koch, W.J.; Rabinowitz, J.; Klotman, P.E.; et al. Cardiac Dysfunction in HIV-1 Transgenic Mouse: Role of Stress and BAG3. Clin. Transl. Sci. 2015, 8, 305–310. [Google Scholar] [CrossRef]
- Feldman, A.M.; Gordon, J.; Wang, J.; Song, J.; Zhang, X.Q.; Myers, V.D.; Tilley, D.G.; Gao, E.; Hoffman, N.E.; Tomar, D.; et al. BAG3 regulates contractility and Ca(2+) homeostasis in adult mouse ventricular myocytes. J. Mol. Cell Cardiol. 2016, 92, 10–20. [Google Scholar] [CrossRef]
- Su, F.; Myers, V.D.; Knezevic, T.; Wang, J.; Gao, E.; Madesh, M.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Rabinowitz, J.; et al. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight 2016, 1, e90931. [Google Scholar] [CrossRef]
- Pagliuca, M.G.; Lerose, R.; Cigliano, S.; Leone, A. Regulation by heavy metals and temperature of the human BAG-3 gene, a modulator of Hsp70 activity. FEBS Lett. 2003, 541, 11–15. [Google Scholar] [CrossRef]
- Liao, Q.; Ozawa, F.; Friess, H.; Zimmermann, A.; Takayama, S.; Reed, J.C.; Kleeff, J.; Buchler, M.W. The anti-apoptotic protein BAG-3 is overexpressed in pancreatic cancer and induced by heat stress in pancreatic cancer cell lines. FEBS Lett. 2001, 503, 151–157. [Google Scholar] [CrossRef]
- Bonelli, P.; Petrella, A.; Rosati, A.; Romano, M.F.; Lerose, R.; Pagliuca, M.G.; Amelio, T.; Festa, M.; Martire, G.; Venuta, S.; et al. BAG3 protein regulates stress-induced apoptosis in normal and neoplastic leukocytes. Leukemia 2004, 18, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Kim, Y.K.; Youn, D.Y.; Lim, M.H.; Ko, J.H.; Ahn, Y.S.; Lee, J.H. Down-modulation of Bis sensitizes cell death in C6 glioma cells induced by oxygen-glucose deprivation. Brain Res. 2010, 1349, 1–10. [Google Scholar] [CrossRef] [PubMed]
- d’Avenia, M.; Citro, R.; De Marco, M.; Veronese, A.; Rosati, A.; Visone, R.; Leptidis, S.; Philippen, L.; Vitale, G.; Cavallo, A.; et al. A novel miR-371a-5p-mediated pathway, leading to BAG3 upregulation in cardiomyocytes in response to epinephrine, is lost in Takotsubo cardiomyopathy. Cell Death Dis. 2015, 6, e1948. [Google Scholar] [CrossRef] [PubMed]
- Liewluck, T.; Kintarak, J.; Sangruchi, T.; Selcen, D.; Kulkantrakorn, K. Myofibrillar myopathy with limb-girdle phenotype in a Thai patient. J. Med. Assoc. Thai. 2009, 92, 290–295. [Google Scholar]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. What have we learned from the congenital myasthenic syndromes. J. Mol. Neurosci. 2010, 40, 143–153. [Google Scholar] [CrossRef]
- Olive, M.; Goldfarb, L.G.; Shatunov, A.; Fischer, D.; Ferrer, I. Myotilinopathy: Refining the clinical and myopathological phenotype. Brain 2005, 128, 2315–2326. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Myofibrillar myopathies. Handb. Clin. Neurol. 2011, 101, 143–154. [Google Scholar] [CrossRef]
- McClung, J.M.; McCord, T.J.; Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Southerland, K.W.; Reinardy, J.L.; Mueller, S.B.; Venkatraman, T.N.; Lascola, C.D.; et al. BAG3 (Bcl-2-Associated Athanogene-3) Coding Variant in Mice Determines Susceptibility to Ischemic Limb Muscle Myopathy by Directing Autophagy. Circulation 2017, 136, 281–296. [Google Scholar] [CrossRef]
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther. 2015, 14, 642–648. [Google Scholar] [CrossRef]
- Ammirante, M.; Rosati, A.; Arra, C.; Basile, A.; Falco, A.; Festa, M.; Pascale, M.; d’Avenia, M.; Marzullo, L.; Belisario, M.A.; et al. IKKgamma protein is a target of BAG3 regulatory activity in human tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 7497–7502. [Google Scholar] [CrossRef]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef]
- Cesaro, E.; Montano, G.; Rosati, A.; Crescitelli, R.; Izzo, P.; Turco, M.C.; Costanzo, P. WT1 protein is a transcriptional activator of the antiapoptotic bag3 gene. Leukemia 2010, 24, 1204–1206. [Google Scholar] [CrossRef][Green Version]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef]
- Habata, S.; Iwasaki, M.; Sugio, A.; Suzuki, M.; Tamate, M.; Satohisa, S.; Tanaka, R.; Saito, T. BAG3 increases the invasiveness of uterine corpus carcinoma cells by suppressing miR-29b and enhancing MMP2 expression. Oncol. Rep. 2015, 33, 2613–2621. [Google Scholar] [CrossRef]
- Im, C.N.; Yun, H.H.; Song, B.; Youn, D.Y.; Cui, M.N.; Kim, H.S.; Park, G.S.; Lee, J.H. BIS-mediated STAT3 stabilization regulates glioblastoma stem cell-like phenotypes. Oncotarget 2016, 7, 35056–35070. [Google Scholar] [CrossRef]
- Liu, B.Q.; Zhang, S.; Li, S.; An, M.X.; Li, C.; Yan, J.; Wang, J.M.; Wang, H.Q. BAG3 promotes stem cell-like phenotype in breast cancer by upregulation of CXCR4 via interaction with its transcript. Cell Death Dis. 2017, 8, e2933. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, C.M.; Choudhary, M.; McCormick, M.G.; Cheung, D.; Landesberg, G.P.; Wang, J.-F.; Song, J.; Martin, T.G.; Cheung, J.Y.; Qu, H.-Q.; et al. BAG3: Nature’s Quintessential Multi-Functional Protein Functions as a Ubiquitous Intra-Cellular Glue. Cells 2023, 12, 937. https://doi.org/10.3390/cells12060937
Brenner CM, Choudhary M, McCormick MG, Cheung D, Landesberg GP, Wang J-F, Song J, Martin TG, Cheung JY, Qu H-Q, et al. BAG3: Nature’s Quintessential Multi-Functional Protein Functions as a Ubiquitous Intra-Cellular Glue. Cells. 2023; 12(6):937. https://doi.org/10.3390/cells12060937
Chicago/Turabian StyleBrenner, Caitlyn M., Muaaz Choudhary, Michael G. McCormick, David Cheung, Gavin P. Landesberg, Ju-Fang Wang, Jianliang Song, Thomas G. Martin, Joseph Y. Cheung, Hui-Qi Qu, and et al. 2023. "BAG3: Nature’s Quintessential Multi-Functional Protein Functions as a Ubiquitous Intra-Cellular Glue" Cells 12, no. 6: 937. https://doi.org/10.3390/cells12060937
APA StyleBrenner, C. M., Choudhary, M., McCormick, M. G., Cheung, D., Landesberg, G. P., Wang, J.-F., Song, J., Martin, T. G., Cheung, J. Y., Qu, H.-Q., Hakonarson, H., & Feldman, A. M. (2023). BAG3: Nature’s Quintessential Multi-Functional Protein Functions as a Ubiquitous Intra-Cellular Glue. Cells, 12(6), 937. https://doi.org/10.3390/cells12060937