

Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
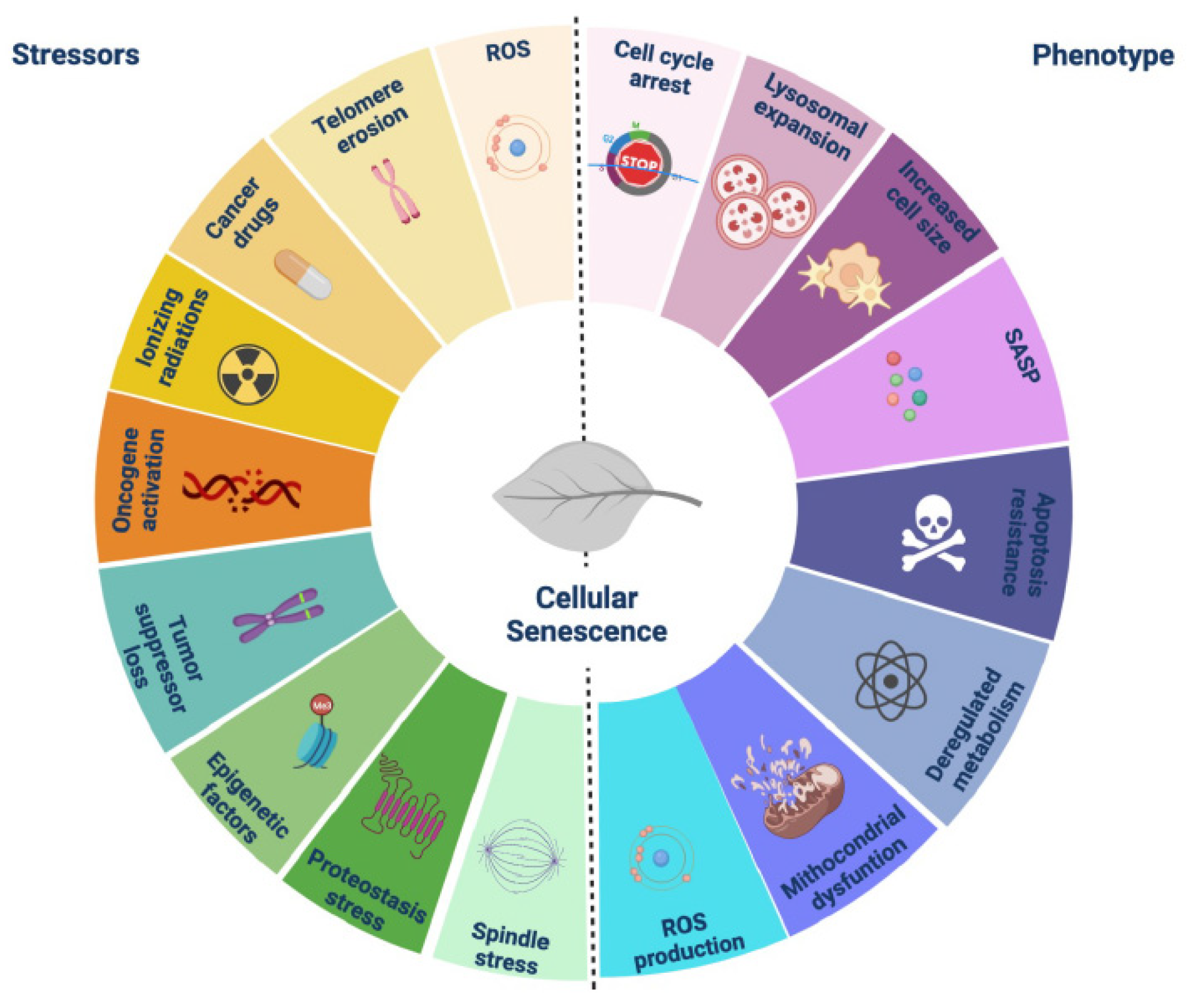
1. Introduction
2. Invasion: Lessons from Mammalian Placenta
3. Common Features Required for Cell Invasion and Colonization
3.1. Fusogenic Potential
3.2. Polyploidy
3.3. EMT–MET Stemness
3.4. Angiogenic and ECM Remodeling Properties
3.5. Immune-Suppressive Modulation
3.6. Pro-Survival Intrinsic and External Anoikis Signals
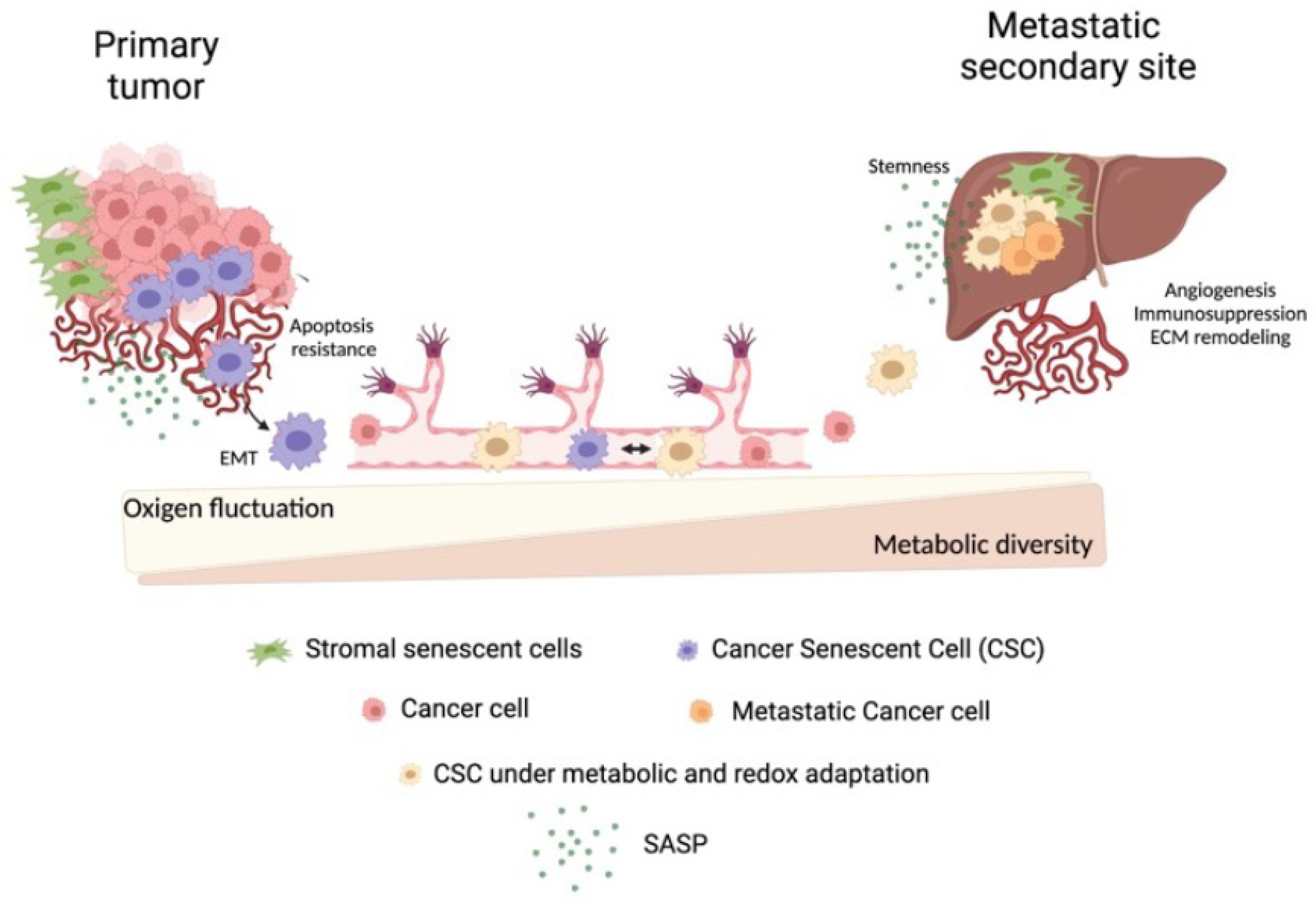
4. Metabolic Plasticity
4.1. Pyruvate and Lactate
4.2. NAD+
4.3. Lipids
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Celià-Terrassa, T.; Kang, Y. Distinctive Properties of Metastasis-Initiating Cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Metastatic Niche Functions and Therapeutic Opportunities. Nat. Cell Biol. 2018, 20, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative Evidence for Early Metastatic Seeding in Colorectal Cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The Seed and Soil Hypothesis Revisited-The Role of Tumor-Stroma Interactions in Metastasis to Different Organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-Dependent Effects of Cellular Senescence in Cancer Development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-Associated Senescence-like Cell Cycle Arrest of Human Naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in Premalignant Tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial Role of P53-Dependent Cellular Senescence in Suppression of Pten-Deficient Tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent Tumor Cells Lead the Collective Invasion in Thyroid Cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Komoda, K.; Mikawa, R.; Asai, A.; Sugimoto, M. Cellular Senescence Promotes Cancer Metastasis by Enhancing Soluble E-Cadherin Production. iScience 2021, 24, 103022. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Kim, Y.H.; Oh, S.Y.; Suh, K.W.; Kim, Y.S.; Lee, G.Y.; Yoon, J.E.; Park, S.S.; Lee, Y.K.; Park, Y.J.; et al. Senescent Tumor Cells Build a Cytokine Shield in Colorectal Cancer. Adv. Sci. 2021, 8, 2002497. [Google Scholar] [CrossRef]
- Xiao, Z.; Yan, L.; Liang, X.; Wang, H. Progress in Deciphering Trophoblast Cell Differentiation during Human Placentation. Curr. Opin. Cell Biol. 2020, 67, 86–91. [Google Scholar] [CrossRef]
- D’Souza, A.W.; Wagner, G.P. Malignant Cancer and Invasive Placentation: A Case for Positive Pleiotropy between Endometrial and Malignancy Phenotypes. Evol. Med. Public Health 2014, 2014, 136–145. [Google Scholar] [CrossRef]
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef]
- Dupressoir, A.; Vernochet, C.; Harper, F.; Guégan, J.; Dessen., P.; Pierron., G.; Heidmann., T. A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast. Proc. Natl. Acad. Sci. USA 2011, 108, E1164–E1173. [Google Scholar] [CrossRef]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell Fusion Induced by ERVWE1 or Measles Virus Causes Cellular Senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef]
- Gal, H.; Lysenko, M.; Stroganov, S.; Vadai, E.; Youssef, S.A.; Tzadikevitch-Geffen, K.; Rotkopf, R.; Biron-Shental, T.; Bruin, A.; Neeman, M.; et al. Molecular Pathways of Senescence Regulate Placental Structure and Function. EMBO J. 2019, 38, e100849. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; McKinney, S.; Gerton, J.L. Persistent DNA Damage and Senescence in the Placenta Impacts Developmental Outcomes of Embryos. Dev. Cell 2020, 54, 333–347.e7. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent Insights into Targeting the IL-6 Cytokine Family in Inflammatory Diseases and Cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/MiR-34a Feedback Loop Promotes EMT-Mediated Colorectal Cancer Invasion and Metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef]
- Wang, L.-H.; Lin, C.-Y.; Liu, S.-C.; Liu, G.-T.; Chen, Y.-L.; Chen, J.-J.; Chan, C.-H.; Lin, T.-Y.; Chen, C.-K.; Xu, G.-H.; et al. CCL5 Promotes VEGF-C Production and Induces Lymphangiogenesis by Suppressing MiR-507 in Human Chondrosarcoma Cells. Oncotarget 2016, 7, 36896–36908. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; DuBois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Sacco, M.G.; Susani, L.; Montagna, C.; Vezzoni, P. Cell Fusion Is a Physiological Process in Mouse Liver. Hepatology 2008, 48, 1655–1664. [Google Scholar] [CrossRef]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common Signalling Pathways in Macrophage and Osteoclast Multinucleation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Petrany, M.J.; Millay, D.P. Cell Fusion: Merging Membranes and Making Muscle. Trends Cell Biol. 2019, 29, 964–973. [Google Scholar] [CrossRef]
- Mortensen, K.; Lichtenberg, J.; Thomsen, P.D.; Larsson, L.I. Spontaneous Fusion between Cancer Cells and Endothelial Cells. Cell. Mol. Life Sci. 2004, 61, 2125–2131. [Google Scholar] [CrossRef]
- Jacobsen, B.M.; Harrell, J.C.; Jedlicka, P.; Borges, V.F.; Varella-Garcia, M.; Horwitz, K.B. Spontaneous Fusion with, and Transformation of Mouse Stroma by, Malignant Human Breast Cancer Epithelium. Cancer Res. 2006, 66, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of Tumour Cells with Bone Marrow-Derived Cells: A Unifying Explanation for Metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Miller, O.J.; Klein, G.; Worst, P.; Tachibana, T. Suppression of Malignancy by Cell Fusion. Nature 1969, 223, 363–368. [Google Scholar] [CrossRef]
- Duelli, D.M.; Hearn, S.; Myers, M.P.; Lazebnik, Y. A Primate Virus Generates Transformed Human Cells by Fusion. J. Cell Biol. 2005, 171, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Reddel, R.R. Senescence: An Antiviral Defense That Is Tumor Suppressive? Carcinogenesis 2010, 31, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M.; Christensen, I.J.; Nielsen, H.J.; Hansen, U.; Bjerregaard, B.; Talts, J.F.; Larsson, L.I. Syncytin Immunoreactivity in Colorectal Cancer: Potential Prognostic Impact. Cancer Lett. 2009, 280, 44–49. [Google Scholar] [CrossRef]
- Larsson, L.-I.; Holck, S.; Christensen, I.J. Prognostic Role of Syncytin Expression in Breast Cancer. Human Pathol. 2007, 38, 726–731. [Google Scholar] [CrossRef]
- Strick, R.; Ackermann, S.; Langbein, M.; Swiatek, J.; Schubert, S.W.; Hashemolhosseini, S.; Koscheck, T.; Fasching, P.A.; Schild, R.L.; Beckmann, M.W.; et al. Proliferation and Cell-Cell Fusion of Endometrial Carcinoma Are Induced by the Human Endogenous Retroviral Syncytin-1 and Regulated by TGF-β. J. Mol. Med. 2007, 85, 23–38. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell-to-Cell Fusion as a Link between Viruses and Cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell Fusion: A Hidden Enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes. Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, B.N.; Kudryavtseva, M.V.; Sakuta, G.A.; Stein, G.I. Human Hepatocyte Polyploidization Kinetics in the Course of Life Cycle. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 64, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Vezzoni, P.; Montagna, C. Single-Cell Analysis of Ploidy and Centrosomes Underscores the Peculiarity of Normal Hepatocytes. PLoS ONE 2011, 6, e26080. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Pang, L.; Poncz, M.; Dowdy, S.F.; Blobel, G.A.; Crispino, J.D. Cyclin D–Cdk4 Is Regulated by GATA-1 and Required for Megakaryocyte Growth and Polyploidization. Blood 2007, 109, 5199–5207. [Google Scholar] [CrossRef]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Falette, N.; Tabone, E.; Levrat, C.; Britten, R.; Voorzanger-Rousselot, N.; Roesch-Gateau, O.; Vanier-Viornery, A.; Puisieux, A.; Dumontet, C. Inactivation of Wild-Type P53 by a Dominant Negative Mutant Renders MCF-7 Cells Resistant to Tubulin-Binding Agent Cytotoxicity. Br. J. Cancer 2001, 85, 902–908. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing Radiation-Induced Responses in Human Cells with Differing TP53 Status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid Giant Cancer Cells: Unrecognized Actuators of Tumorigenesis, Metastasis, and Resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell 2018, 44, 447–459.e5. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kang, Y. Probing the Fifty Shades of EMT in Metastasis. Trends Cancer 2016, 2, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-Cell Analysis Reveals a Stem-Cell Program in Human Metastatic Breast Cancer Cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Christofori, G. Epithelial-Mesenchymal Transition (EMT) and Metastasis: Yes, No, Maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone Vascular Niche E-Selectin Induces Mesenchymal–Epithelial Transition and Wnt Activation in Cancer Cells to Promote Bone Metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef]
- Ombrato, L.; Malanchi, I. The EMT Universe: Space between Cancer Cell Dissemination and Metastasis Initiation. Crit. Rev. Oncog. 2014, 19, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Klejman, A.; Piwocka, K.; Kaminska, B.; et al. Bafilomycin A1 Triggers Proliferative Potential of Senescent Cancer Cells in Vitro and in NOD/SCID Mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J. Release of mitotic descendants by giant cells from irradiated burkitt’s lymphoma cell lines. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into Blastomere-like Cancer Stem Cells via Formation of Polyploid Giant Cancer Cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.Z.; Battistini, L.; Blandino, R.; et al. SASP Mediates Chemoresistance and Tumor-Initiating-Activity of Mesothelioma Cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the Aneuploidy and Senescence Paradoxes: A Study of Triploid Breast Cancers Non-Responsive to Neoadjuvant Therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal. Cancer 2016, 15, 170–178.e3. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link between Cancer and Aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S.; Coppe, J.P.; Krtolica, A.; Campisi, J. Stromal-Epithelial Interactions in Aging and Cancer: Senescent Fibroblasts Alter Epithelial Cell Differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beauséjour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.Y.; Campisi, J. A Human-like Senescence-Associated Secretory Phenotype Is Conserved in Mouse Cells Dependent on Physiological Oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Pazolli, E.; Luo, X.; Brehm, S.; Carbery, K.; Chung, J.J.; Prior, J.L.; Doherty, J.; Demehri, S.; Salavaggione, L.; Piwnica-Worms, D.; et al. Senescent Stromal-Derived Osteopontin Promotes Preneoplastic Cell Growth. Cancer Res. 2009, 69, 1230–1239. [Google Scholar] [CrossRef]
- Jia, R.; Liang, Y.; Chen, R.; Liu, G.; Wang, H.; Tang, M.; Zhou, X.; Wang, H.; Yang, Y.; Wei, H.; et al. Osteopontin Facilitates Tumor Metastasis by Regulating Epithelial–Mesenchymal Plasticity. Cell Death Dis. 2016, 7, e2564. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C Produced by S100A4 + Stromal Cells Are Important for Metastatic Colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between Cancer Stem Cells and Their Niche Govern Metastatic Colonization. Nature 2012, 481, 85–91. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.F.; et al. Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, A.A.; Chothani, S.; Viswanathan, S.; Goh, J.W.T.; Lim, W.-W.; Cook, S.A. IL11 Stimulates IL33 Expression and Proinflammatory Fibroblast Activation across Tissues. Int. J. Mol. Sci. 2022, 23, 8900. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Angelini, P.D.; Zacarias Fluck, M.F.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Morales, C.B.; Vicario, R.; Luque-Garciá, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 Signaling Promotes Breast Cancer Metastasis through Cellular Senescence. Cancer Res. 2013, 73, 450–458. [Google Scholar] [CrossRef]
- Sahai, E. Illuminating the Metastatic Process. Nat. Rev. Cancer 2007, 7, 737–749. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and Cancer: Regulators of Cancer Stemness, Metastasis, and Drug Resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic Cancer Exosomes Initiate Pre-Metastatic Niche Formation in the Liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Murgai, M.; Ju, W.; Eason, M.; Kline, J.; Beury, D.W.; Kaczanowska, S.; Miettinen, M.M.; Kruhlak, M.; Lei, H.; Shern, J.F.; et al. KLF4-Dependent Perivascular Cell Plasticity Mediates Pre-Metastatic Niche Formation and Metastasis. Nat. Med. 2017, 23, 1176–1190. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef]
- Tsai, K.K.C.; Yao-, E.; Chuang, Y.; Little, J.B.; Yuan, Z.-M. Cellular Mechanisms for Low-Dose Ionizing Radiation-Induced Perturbation of the Breast Tissue Microenvironment. Cancer Res. 2005, 65, 6734–6744. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vercamer, C.; Bouali, F.; Martien, S.; Deruy, E.; Wernert, N.; Chwastyniak, M.; Pinet, F.; Abbadie, C.; Pourtier, A. Senescent Fibroblasts Enhance Early Skin Carcinogenic Events via a Paracrine MMP-PAR-1 Axis. PLoS ONE 2013, 8, e63607. [Google Scholar] [CrossRef] [PubMed]
- Tonini, T.; Rossi, F.; Claudio, P.P. Molecular Basis of Angiogenesis and Cancer. Oncogene 2003, 22, 6549–6556. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tuxhorn, J.A.; Ressler, S.J.; McAlhany, S.J.; Dang, T.D.; Rowley, D.R. Stromal Expression of Connective Tissue Growth Factor Promotes Angiogenesis and Prostate Cancer Tumorigenesis. Cancer Res. 2005, 65, 8887–8895. [Google Scholar] [CrossRef] [PubMed]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of Il-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Expect the Unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Liu, Y.; Kosaka, A.; Ikeura, M.; Kohanbash, G.; Fellows-Mayle, W.; Snyder, L.A.; Okada, H. Premetastatic Soil and Prevention of Breast Cancer Brain Metastasis. Neuro. Oncol. 2013, 15, 891–903. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin Shapes Immunosuppression in the Metastatic Niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; DiMitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten-Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-Infiltrating Gr-1 + Myeloid Cells Antagonize Senescence in Cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through Pge2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule Exocytosis Mediates Immune Surveillance of Senescent Cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular Metabolic Energetics Can Promote Cancer Progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F. Metabolism in Cancer Metastasis. Cancer Lett. 2008, 270, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Nabi, K.; Le, A. The Intratumoral Heterogeneity of Cancer Metabolism. Adv. Exp. Med. Biol. 2018, 1063, 131–145. [Google Scholar] [PubMed]
- Tarragó-Celada, J.; Cascante, M. Targeting the Metabolic Adaptation of Metastatic Cancer. Cancers 2021, 13, 1641. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized Activities of the Pyruvate Dehydrogenase Complex Sustain Lipogenesis in Prostate Cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic Lethal Metabolic Targeting of Cellular Senescence in Cancer Therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; MacKay, G.; van der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A Key Role for Mitochondrial Gatekeeper Pyruvate Dehydrogenase in Oncogene-Induced Senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Takebayashi, S.I.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma Protein Promotes Oxidative Phosphorylation through Upregulation of Glycolytic Genes in Oncogene-Induced Senescent Cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef]
- Olenchock, B.A.; vander Heiden, M.G. XPyruvate as a Pivot Point for Oncogene-Induced Senescence. Cell 2013, 153, 1429. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. The Metabolic Roots of Senescence: Mechanisms and Opportunities for Intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.Y.; Liu, J.M.; Chen, Q.K.; Chen, H.Z.; Ye, C.J.; Xue, J.; Yang, H.Q.; Li, J.L.; Liu, X.F.; Kuang, S.J. Inhibition of LDHA Suppresses Tumor Progression in Prostate Cancer. Tumor. Biology 2015, 36, 8093–8100. [Google Scholar] [CrossRef] [PubMed]
- He, T.-L.; Zhang, Y.-J.; Jiang, H.; Li, X.-H.; Zhu, H.; Zheng, K.-L. The C-Myc–LDHA Axis Positively Regulates Aerobic Glycolysis and Promotes Tumor Progression in Pancreatic Cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, X.; Xu, Z.; Dai, J.; He, H.; Zhu, Y.; Wang, H. LDHA Promotes Tumor Metastasis by Facilitating Epithelial-Mesenchymal Transition in Renal Cell Carcinoma. Mol. Med. Rep. 2017, 16, 8335–8344. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F.; Sun, L. MCT1 Regulates Aggressive and Metabolic Phenotypes in Bladder Cancer. J. Cancer 2018, 9, 2492–2501. [Google Scholar] [CrossRef]
- Liao, E.C.; Hsu, Y.T.; Chuah, Q.Y.; Lee, Y.J.; Hu, J.Y.; Huang, T.C.; Yang, P.M.; Chiu, S.J. Radiation Induces Senescence and a Bystander Effect through Metabolic Alterations. Cell Death Dis. 2014, 5, e1255. [Google Scholar] [CrossRef]
- Payen, V.L.; Hsu, M.Y.; Rädecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporter MCT1 Promotes Tumor Metastasis Independently of Its Activity as a Lactate Transporter. Cancer Res. 2017, 77, 5591–5601. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ Metabolism, Stemness, the Immune Response, and Cancer. Signal. Transduct. Target. Ther. 2021, 6. [Google Scholar] [CrossRef]
- da Veiga Moreira, J.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Pérès, S.; Paulevé, L.; Nogueira, M.L.; Steyaert, J.M.; Schwartz, L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef]
- Bonuccelli, G.; de Francesco, E.M.; de Boer, R.; Tanowitz, H.B.; Lisanti, M.P. NADH Autofluorescence, a New Metabolic Biomarker for Cancer Stem Cells: Identification of Vitamin C and CAPE as Natural Products Targeting “Stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD + Metabolism Governs the Proinflammatory Senescence-Associated Secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent Cells Promote Tissue NAD+ Decline during Ageing via the Activation of CD38+ Macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Nath, A.; Chan, C. Genetic Alterations in Fatty Acid Transport and Metabolism Genes Are Associated with Metastatic Progression and Poor Prognosis of Human Cancers. Sci. Rep. 2016, 6, srep18669. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated Free Fatty Acid Uptake via CD36 Promotes Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef]
- Deng, M.; Cai, X.; Long, L.; Xie, L.; Ma, H.; Zhou, Y.; Liu, S.; Zeng, C. CD36 Promotes the Epithelial-Mesenchymal Transition and Metastasis in Cervical Cancer by Interacting with TGF-β. J. Transl. Med. 2019, 17, 352. [Google Scholar] [CrossRef]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.; et al. CD 36 Initiates the Secretory Phenotype during the Establishment of Cellular Senescence. EMBO Rep. 2018, 19, e45274. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-Regulated FASN Expression Promotes Transcoelomic Metastasis of Ovarian Cancer Cell through Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef] [PubMed]
- Seguin, F.; Carvalho, M.A.; Bastos, D.C.; Agostini, M.; Zecchin, K.G.; Alvarez-Flores, M.P.; Chudzinski-Tavassi, A.M.; Coletta, R.D.; Graner, E. The Fatty Acid Synthase Inhibitor Orlistat Reduces Experimental Metastases and Angiogenesis in B16-F10 Melanomas. Br. J. Cancer 2012, 107, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Rychahou, P.G.; Gulhati, P.; Elliott, V.A.; Mustain, W.C.; O’Connor, K.; Morris, A.J.; Sunkara, M.; Weiss, H.L.; Lee, E.Y.; et al. Inhibition of Fatty Acid Synthase Attenuates CD44-Associated Signaling and Reduces Metastasis in Colorectal Cancer. Cancer Res. 2012, 72, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Fafián-Labora, J.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN Activity Is Important for the Initial Stages of the Induction of Senescence. Cell Death Dis. 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A Signature of Enhanced Lipid Metabolism, Lipid Peroxidation and Aldehyde Stress in Therapy-Induced Senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. “Cell Membrane Theory of Senescence” and the Role of Bioactive Lipids in Aging, and Aging Associated Diseases and Their Therapeutic Implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef]
- Wiley, C.D.; Sharma, R.; Davis, S.S.; Lopez-Dominguez, J.A.; Mitchell, K.P.; Wiley, S.; Alimirah, F.; Kim, D.E.; Payne, T.; Rosko, A.; et al. Oxylipin Biosynthesis Reinforces Cellular Senescence and Allows Detection of Senolysis. Cell Metab. 2021, 33, 1124–1136.e5. [Google Scholar] [CrossRef]
- Wisastra, R.; Dekker, F.J. Inflammation, Cancer and Oxidative Lipoxygenase Activity Are Intimately Linked. Cancers 2014, 6, 1500–1521. [Google Scholar] [CrossRef]
- Catalano, A.; Rodilossi, S.; Caprari, P.; Coppola, V.; Procopio, A. 5-Lipoxygenase Regulates Senescence-like Growth Arrest by Promoting ROS-Dependent P53 Activation. EMBO J. 2005, 24, 170–179. [Google Scholar] [CrossRef]
- Azrad, M.; Turgeon, C.; Demark-Wahnefried, W. Current Evidence Linking Polyunsaturated Fatty Acids with Cancer Risk and Progression. Front. Oncol. 2013, 3, 224. [Google Scholar] [CrossRef]
- Montecillo-Aguado, M.; Tirado-Rodriguez, B.; Antonio-Andres, G.; Morales-Martinez, M.; Tong, Z.; Yang, J.; Hammock, B.D.; Hernandez-Pando, R.; Huerta-Yepez, S. Omega-6 Polyunsaturated Fatty Acids Enhance Tumor Aggressiveness in Experimental Lung Cancer Model: Important Role of Oxylipins. Int. J. Mol. Sci. 2022, 23, 6179. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Guryleva, M.V.; Stepanova, E.S.; Makarenkova, L.M.; Ptitsyna, E.V.; Goriainov, S.V.; Nikolskaya, A.I.; Astakhova, A.A.; Klimenko, A.S.; Bezborodova, O.A.; et al. Multi-Omics Approach Points to the Importance of Oxylipins Metabolism in Early-Stage Breast Cancer. Cancers 2022, 14, 2041. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting Senescence for the Treatment of Cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. MTOR Regulates MAPKAPK2 Translation to Control the Senescence-Associated Secretory Phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, J.; Jiang, L.; Li, W.; Su, Q.; Zhang, J.; Li, H.; Song, M.; Cheng, B.; Xia, J.; et al. Metformin as a Senostatic Drug Enhances the Anticancer Efficacy of CDK4/6 Inhibitor in Head and Neck Squamous Cell Carcinoma. Cell Death Dis. 2020, 11, 925. [Google Scholar] [CrossRef] [PubMed]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting Interconnected Synthetic Lethal Interactions between PARP Inhibition and Cancer Cell Reversible Senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and Exploiting Vulnerabilities for the Treatment of Liver Cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Poblocka, M.; Bassey, A.L.; Smith, V.M.; Falcicchio, M.; Manso, A.S.; Althubiti, M.; Sheng, X.; Kyle, A.; Barber, R.; Frigerio, M.; et al. Targeted Clearance of Senescent Cells Using an Antibody-Drug Conjugate against a Specific Membrane Marker. Sci. Rep. 2021, 11, 20358. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faggioli, F.; Velarde, M.C.; Wiley, C.D. Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases. Cells 2023, 12, 860. https://doi.org/10.3390/cells12060860
Faggioli F, Velarde MC, Wiley CD. Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases. Cells. 2023; 12(6):860. https://doi.org/10.3390/cells12060860
Chicago/Turabian StyleFaggioli, Francesca, Michael C. Velarde, and Christopher D. Wiley. 2023. "Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases" Cells 12, no. 6: 860. https://doi.org/10.3390/cells12060860
APA StyleFaggioli, F., Velarde, M. C., & Wiley, C. D. (2023). Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases. Cells, 12(6), 860. https://doi.org/10.3390/cells12060860