
Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection


 ,
, 
 , and
, and 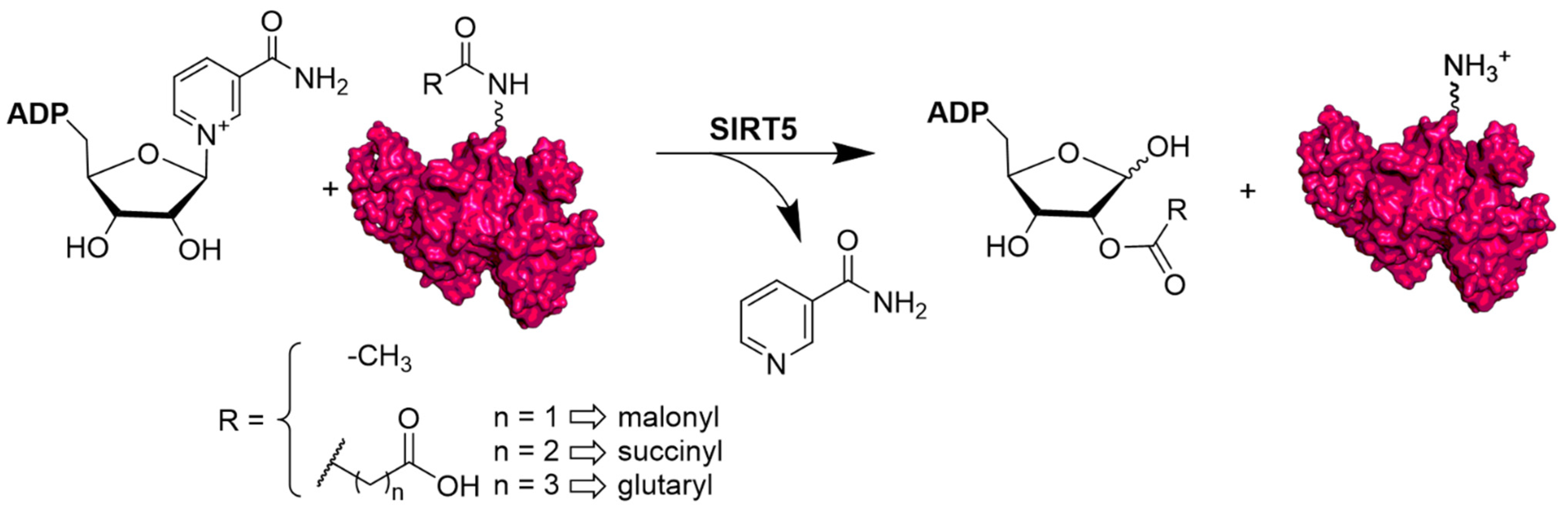{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
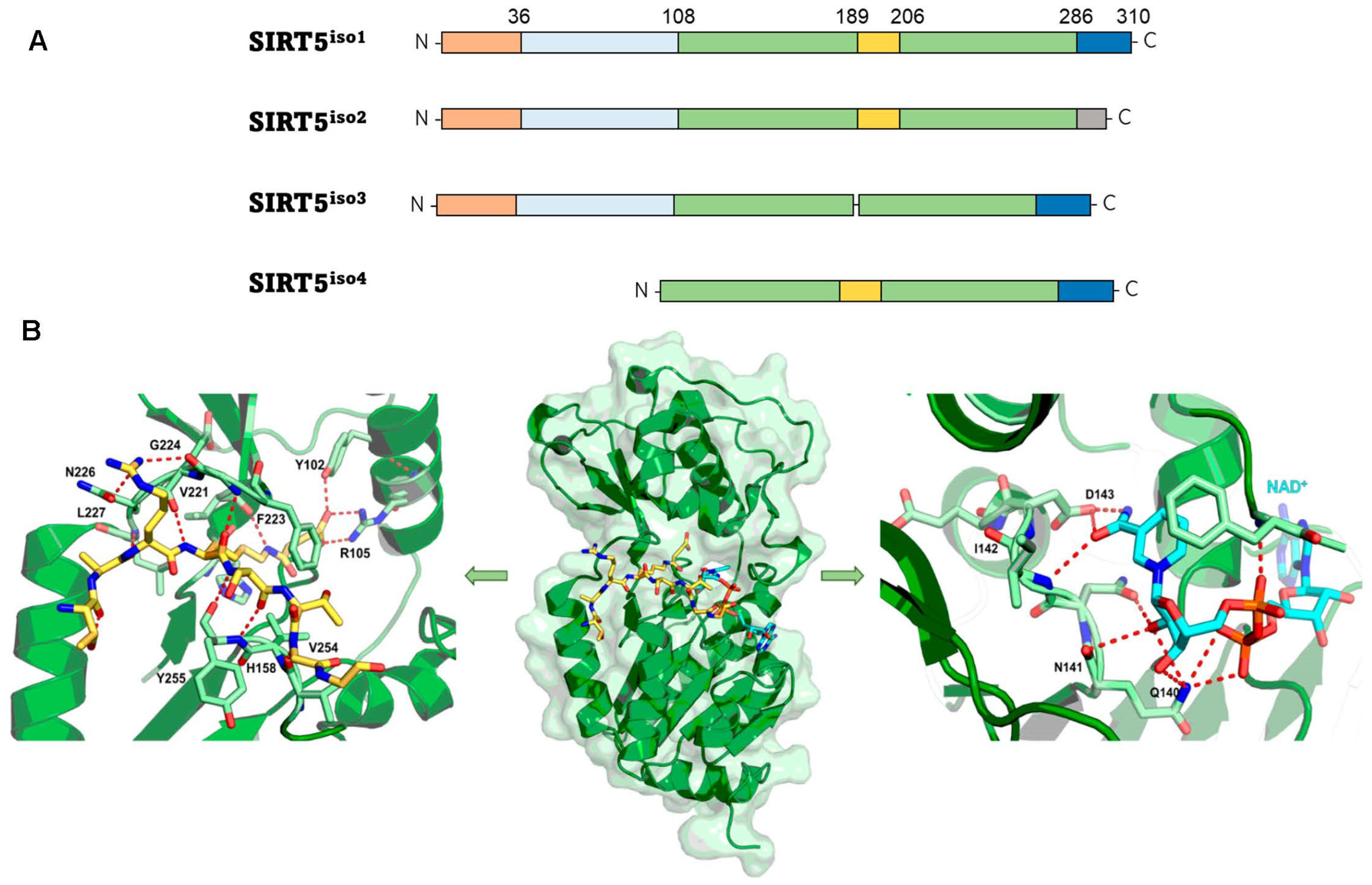
2. Structural and Functional Properties of SIRT5
3. Biological Activities and Disease Relevance of SIRT5
3.1. Oxidative Stress
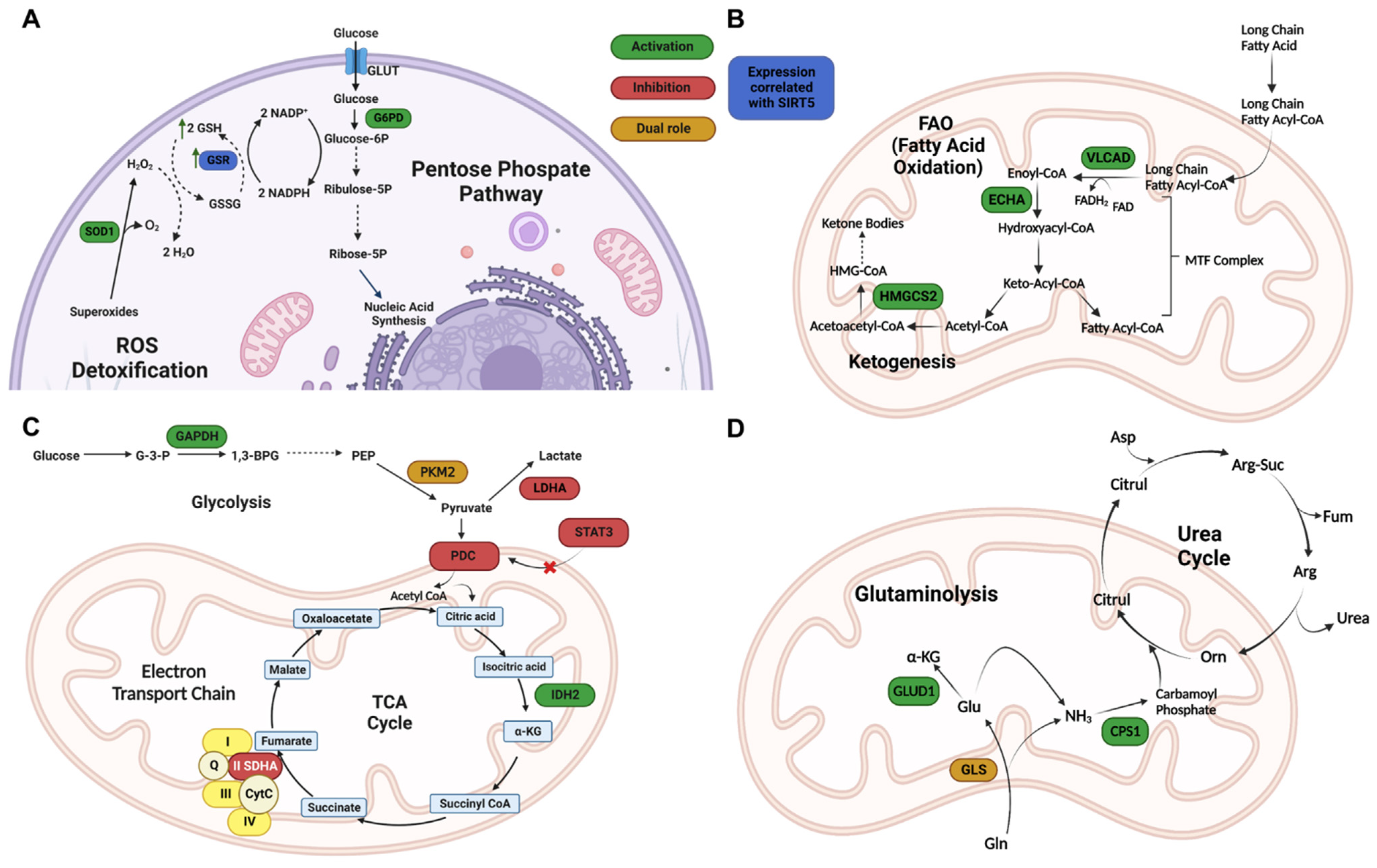
3.2. Metabolism and Mitochondrial Regulation
3.3. Cardiovascular Regulation
3.4. Neurodegeneration
3.5. Inflammation
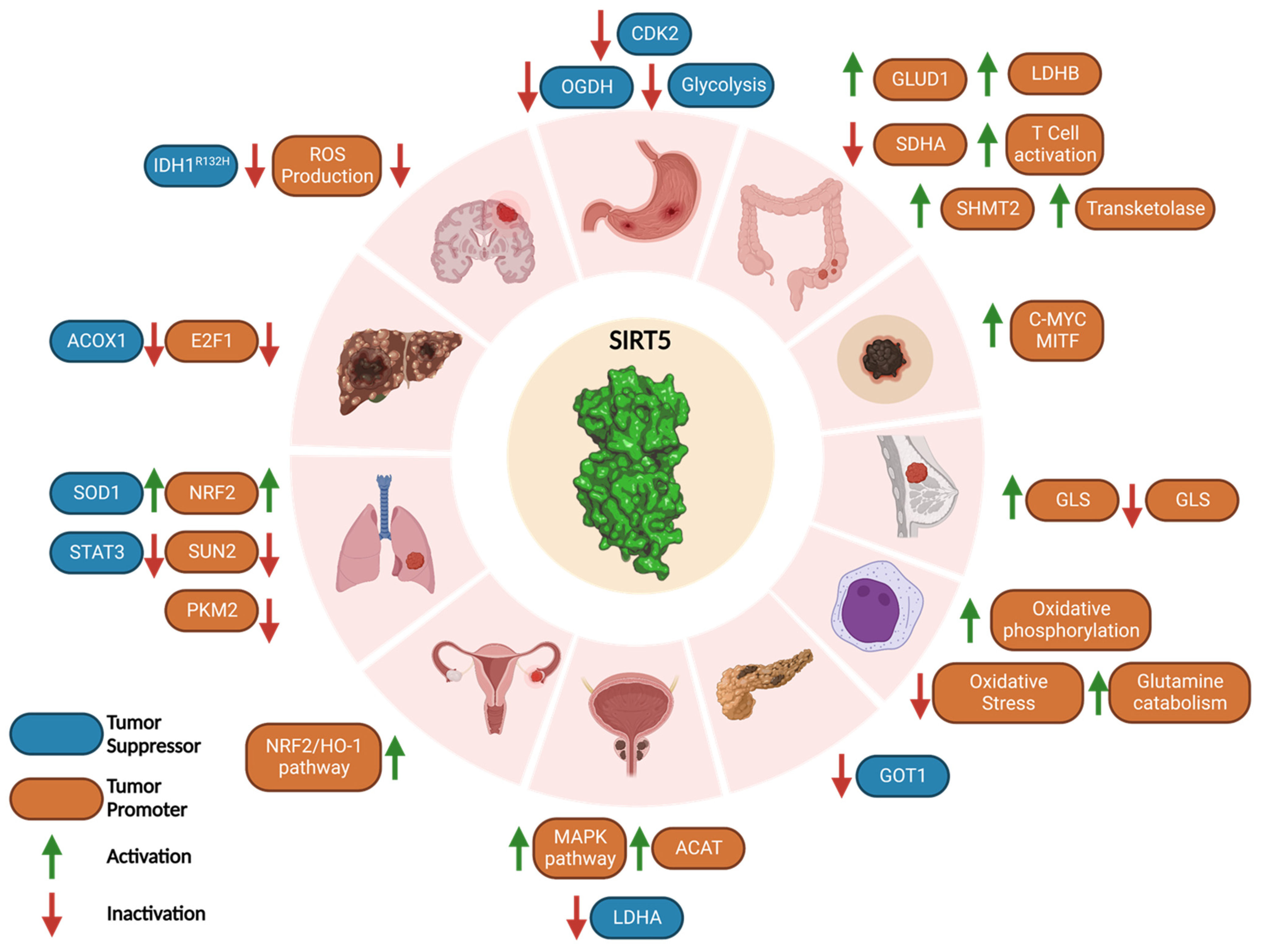
3.6. Cancer: A Janus-Faced Role
3.6.1. Tumor Promoter Role of SIRT5 in Cancer
3.6.2. Tumor Suppressor Role of SIRT5 in Cancer
3.7. SARS-CoV-2 Infection
4. Pharmacological Modulation of SIRT5
4.1. SIRT5 Activators
4.2. SIRT5 Inhibitors
4.2.1. Small Molecules
4.2.2. Peptide-Based and Amino Acid Mimetics Molecules
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,4-DHP | 1,4-dihydropyridine |
| α-KG | α-ketoglutarate |
| ACAD | Acyl-CoA dehydrogenase |
| ACAT1 | Acetyl-CoA acetyltransferase 1 |
| ACOX1 | Acyl-CoA oxidase 1 |
| AD | Alzheimer’s disease |
| ADP | Adenosine diphosphate |
| AIFM1 | Apoptosis inducing factor mitochondrion-associated 1 |
| AML | Acute myeloid leukemia |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ANXA1 | Annexin A1 |
| AP-1 | Activator protein 1 |
| ATP | Adenosine triphosphate |
| BAT | Brown adipose tissue |
| Bax | Bcl-2 associated X protein |
| Bcl-XL | B-cell lymphoma-extra large |
| cAMP | Cyclic adenosine monophosphate |
| CAPT1A | Carnitine palmitoyltransferase 1A |
| CoA | Coenzyme A |
| CDK2 | Cyclin dependent kinase 2 |
| CETSA | Cellular thermal shift assay |
| CPS1 | Carbamoyl phosphate synthetase |
| CR | Caloric restriction |
| CRC | Colorectal cancer |
| CTL | Cell toxicity T lymphocytes |
| DRP1 | Dynamin-related protein 1 |
| EC50 | Half maximal effective concentration |
| ECHA | Enoyl-CoA hydratase |
| EGFR | Epidermal growth factor receptor |
| ELOVL | Fatty acid elongase |
| ETC | Electron transport chain |
| FAD | Flavin adenine dinucleotide |
| FAO | Fatty acid oxidation |
| FFA | Free fatty acid |
| FOXO3A | Forkhead box O3 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| G6PD | Glucose-6-phosphate dehydrogenase G6PD |
| GLS | Glutaminase |
| GLUD1 | Glutamate dehydrogenase 1 |
| GOT1 | Glutamic-oxaloacetic transaminase |
| GSH | Reduced glutathione |
| GSR | Glutathione-disulfide reductase |
| GSSG | Oxidized glutathione |
| HCC | Hepatocellular carcinoma |
| Hda1 | Histone deacetylase 1 |
| HDAC | Histone deacetylase |
| HMG | 3-hydroxy-3-methyl-glutaryl |
| HMGCS2 | 3-hydroxy-3-methylglutaryl CoA synthase 2 |
| HNSCC | Head and neck squamous cell carcinoma |
| IC50 | Half maximal inhibitory concentration |
| IDD | Intervertebral disc degeneration |
| IDH | Isocitrate dehydrogenase |
| IFN-β | Interferon-β |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| ITDRF-CETSA | Isothermal dose-response fingerprinting cellular thermal shift assay |
| KDAC | Lysine deacetylase |
| LC | Liquid chromatography |
| LCAD | Long-chain acyl-CoA dehydrogenase |
| LDHA | Lactate dehydrogenase A |
| LDHB | Lactate dehydrogenase B |
| LINC | Linker of nucleoskeleton and cytoskeleton |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MCAD | Medium-chain acyl-CoA dehydrogenase |
| MITF | Microphthalmia family of transcription factors |
| MM | Multiple myeloma |
| MLS | Mitochondrial localization signal |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mRNA | Messenger RNA |
| MS | Mass spectrometry |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NADP+ | Nicotinamide adenine dinucleotide phosphate |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| NP | Nucleus pulposus |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NRF2/HO-1 | Nuclear factor erythroid 2-related factor 2-heme oxygenase 1 |
| NSCLC | Non-small cell lung cancer |
| NSP10 | Non-structural viral protein 10 |
| NSP14 | Non-structural viral protein 14 |
| OGDH | Oxoglutarate dehydrogenase |
| PARP1 | Poly (ADP-ribose) polymerase 1 |
| PD | Parkinson’s disease |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDC | Pyruvate dehydrogenase complex |
| PDX | Patient-derived xenograft |
| PGC-1α | Proliferator-activated receptor γ coactivator 1-α |
| PKA | Protein kinase A |
| PKM2 | Pyruvate kinase M2 |
| PTEN | Phosphatase and tensin homolog |
| PTMs | Post-translational modifications |
| ROS | Reactive oxygen species |
| Rpd3 | Reduced potassium dependency 3 |
| SDH | Succinate dehydrogenase |
| SHMT2 | Serine hydroxymethyltransferase 2 |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| SUN2 | SAD1/UNC84 domain protein 2 |
| TAC | Transverse aortic constriction |
| TCA | Tricarboxylic acids cycle |
| Th1 | T-helper 1 |
| THF | Tetrahydrofolate |
| TNF-α | Tumor necrosis factor α |
| TrxR2 | Thioredoxin reductase 2 |
| UCP-1 | Uncoupling protein 1 |
| VLCAD | Very long-chain acyl-CoA dehydrogenase |
| VDAC3 | Voltage dependent anion channel 3 |
| WAT | White adipose tissue |
References
- Audagnotto, M.; Dal Peraro, M. Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput. Struct. Biotechnol. J. 2017, 15, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lombard, D.B. Cycling around Lysine Modifications. Trends Biochem. Sci. 2017, 42, 501–503. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Paine, P.L.; Moore, L.C.; Horowitz, S.B. Nuclear envelope permeability. Nature 1975, 254, 109–114. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Lysine acetyltransferase inhibitors: Structure–activity relationships and potential therapeutic implications. Future Med. Chem. 2018, 10, 1067–1091. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Lysine Acetyltransferase Inhibitors From Natural Sources. Front. Pharmacol. 2020, 11, 1243. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Mautone, N.; Zwergel, C.; Mai, A.; Rotili, D. Sirtuin modulators: Where are we now? A review of patents from 2015 to 2019. Expert Opin. Ther. Pat. 2020, 30, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Mautone, N.; Menna, M.; D’Acunzo, F.; Mai, A.; Rotili, D. Sirtuin modulators: Past, present, and future perspectives. Future Med. Chem. 2022, 14, 915–939. [Google Scholar] [CrossRef]
- Levine, D.C.; Kuo, H.Y.; Hong, H.K.; Cedernaes, J.; Hepler, C.; Wright, A.G.; Sommars, M.A.; Kobayashi, Y.; Marcheva, B.; Gao, P.; et al. NADH inhibition of SIRT1 links energy state to transcription during time-restricted feeding. Nat. Metab. 2021, 3, 1621–1632. [Google Scholar] [CrossRef]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Yang, L.; Ma, X.; He, Y.; Yuan, C.; Chen, Q.; Li, G.; Chen, X. Sirtuin 5: A review of structure, known inhibitors and clues for developing new inhibitors. Sci. China Life Sci. 2017, 60, 249–256. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Carafa, V.; Lara, E.; Meade, S.; Botta, G.; Nebbioso, A.; Schemies, J.; Jung, M.; Kazantsev, A.G.; et al. Identification of tri- and tetracyclic pyrimidinediones as sirtuin inhibitors. ChemMedChem 2010, 5, 674–677. [Google Scholar] [CrossRef]
- Rack, J.G.; VanLinden, M.R.; Lutter, T.; Aasland, R.; Ziegler, M. Constitutive nuclear localization of an alternatively spliced sirtuin-2 isoform. J. Mol. Biol. 2014, 426, 1677–1691. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef]
- Tomaselli, D.; Steegborn, C.; Mai, A.; Rotili, D. Sirt4: A Multifaceted Enzyme at the Crossroads of Mitochondrial Metabolism and Cancer. Front. Oncol. 2020, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- Taurone, S.; De Ponte, C.; Rotili, D.; De Santis, E.; Mai, A.; Fiorentino, F.; Scarpa, S.; Artico, M.; Micera, A. Biochemical Functions and Clinical Characterizations of the Sirtuins in Diabetes-Induced Retinal Pathologies. Int. J. Mol. Sci. 2022, 23, 4048. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.M.; Skinner, M.E.; et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Hershberger, K.A.; Abraham, D.M.; Martin, A.S.; Mao, L.; Liu, J.; Gu, H.; Locasale, J.W.; Hirschey, M.D. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 2017, 292, 19767–19781. [Google Scholar] [CrossRef]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Weinert, B.T.; Choudhary, C.; Jackson, S.P. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010, 329, 1348–1353. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Emerging Therapeutic Potential of SIRT6 Modulators. J. Med. Chem. 2021, 64, 9732–9758. [Google Scholar] [CrossRef]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. The Two-Faced Role of SIRT6 in Cancer. Cancers 2021, 13, 1156. [Google Scholar] [CrossRef]
- Pannek, M.; Simic, Z.; Fuszard, M.; Meleshin, M.; Rotili, D.; Mai, A.; Schutkowski, M.; Steegborn, C. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun. 2017, 8, 1513. [Google Scholar] [CrossRef]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017, 25, 838–855.e15. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Chen, S.; Seiler, J.; Santiago-Reichelt, M.; Felbel, K.; Grummt, I.; Voit, R. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol. Cell 2013, 52, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Roessler, C.; Tuting, C.; Meleshin, M.; Steegborn, C.; Schutkowski, M. A Novel Continuous Assay for the Deacylase Sirtuin 5 and Other Deacetylases. J. Med. Chem. 2015, 58, 7217–7223. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Uppala, R.; Verdin, E.; Gibson, B.W.; Goetzman, E.S. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain Acyl-CoA dehydrogenase. PLoS ONE 2015, 10, e0122297. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.B.; Jin, W.; Huang, H.H.; Chen, X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef]
- Fiorentino, F.; Castiello, C.; Mai, A.; Rotili, D. Therapeutic Potential and Activity Modulation of the Protein Lysine Deacylase Sirtuin 5. J. Med. Chem. 2022, 65, 9580–9606. [Google Scholar] [CrossRef]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Ho, A.D.; Letzel, S.; Voelter-Mahlknecht, S. Assignment of the NAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band 6p23 by in situ hybridization. Cytogenet. Genome Res. 2006, 112, 208–212. [Google Scholar] [CrossRef]
- Matsushita, N.; Yonashiro, R.; Ogata, Y.; Sugiura, A.; Nagashima, S.; Fukuda, T.; Inatome, R.; Yanagi, S. Distinct regulation of mitochondrial localization and stability of two human Sirt5 isoforms. Genes Cells 2011, 16, 190–202. [Google Scholar] [CrossRef]
- Voelter-Mahlknecht, S.; Mahlknecht, U. Cloning, chromosomal characterization and FISH mapping of the NAD(+)-dependent histone deacetylase gene sirtuin 5 in the mouse. Int. J. Oncol. 2013, 43, 237–245. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Szczepankiewicz, B.G.; Dai, H.; Koppetsch, K.J.; Qian, D.; Jiang, F.; Mao, C.; Perni, R.B. Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5. J. Org. Chem. 2012, 77, 7319–7329. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; He, B.; Du, J.; Lin, H.; Cerione, R.A.; Hao, Q. The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5). J. Biol. Chem. 2012, 287, 28307–28314. [Google Scholar] [CrossRef]
- Yang, L.L.; Wang, H.L.; Yan, Y.H.; Liu, S.; Yu, Z.J.; Huang, M.Y.; Luo, Y.; Zheng, X.; Yu, Y.; Li, G.B. Sensitive fluorogenic substrates for sirtuin deacylase inhibitor discovery. Eur. J. Med. Chem. 2020, 192, 112201. [Google Scholar] [CrossRef]
- Gertz, M.; Nguyen, G.T.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A Molecular Mechanism for Direct Sirtuin Activation by Resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef]
- Davenport, A.M.; Huber, F.M.; Hoelz, A. Structural and functional analysis of human SIRT1. J. Mol. Biol. 2014, 426, 526–541. [Google Scholar] [CrossRef]
- Moniot, S.; Weyand, M.; Steegborn, C. Structures, substrates, and regulators of mammalian Sirtuins—Opportunities and challenges for drug development. Front. Pharmacol. 2012, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.L.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumor Biol. 2014, 35, 10699–10705. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, Y.; Xing, S.; Ma, P.; Lin, D. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3. Cell Stress Chaperones 2015, 20, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Greene, K.S.; Lukey, M.J.; Wang, X.; Blank, B.; Druso, J.E.; Lin, M.C.J.; Stalnecker, C.A.; Zhang, C.; Abril, Y.N.; Erickson, J.W.; et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 26625–26632. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Fould, B.; Garlatti, V.; Neumann, E.; Fenel, D.; Gaboriaud, C.; Arlaud, G.J. Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry 2010, 49, 8608–8617. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Mynatt, R.L.; Park, E.A.; Cook, G.A. Membrane microenvironment regulation of carnitine palmitoyltranferases I and II. Biochem. Soc. Trans. 2011, 39, 833–837. [Google Scholar] [CrossRef]
- Jukarainen, S.; Heinonen, S.; Rämö, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity is associated with low nad+/sirt pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Sas, K.; He, C.; Nair, V.; Giblin, W.; Inoki, A.; Zhang, H.; Yingbao, Y.; Hodgin, J.; Nelson, R.G.; et al. The Deacylase Sirtuin 5 Reduces Malonylation in Non-mitochondrial Metabolic Pathways in Diabetic Kidney Disease. J. Biol. Chem. 2023, 299, 102960. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate Kinase M2 Regulates Gene Transcription by Acting as a Protein Kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; Wei, C.; Guo, F.; Chen, Y.; et al. PKM2 Regulates Chromosome Segregation and Mitosis Progression of Tumor Cells. Mol. Cell 2014, 53, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef]
- Xiangyun, Y.; Xiaomin, N.; Linping, G.; Yunhua, X.; Ziming, L.; Yongfeng, Y.; Zhiwei, C.; Shun, L. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8, 6984–6993. [Google Scholar] [CrossRef]
- Qi, H.; Ning, X.; Yu, C.; Ji, X.; Jin, Y.; McNutt, M.A.; Yin, Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019, 10, 170. [Google Scholar] [CrossRef]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.Z.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting Isocitrate Dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- Marcon, E.; Jain, H.; Bhattacharya, A.; Guo, H.; Phanse, S.; Pu, S.; Byram, G.; Collins, B.C.; Dowdell, E.; Fenner, M.; et al. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. Methods 2015, 12, 725–731. [Google Scholar] [CrossRef]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Ogura, M.; Nakamura, Y.; Tanaka, D.; Zhuang, X.; Fujita, Y.; Obara, A.; Hamasaki, A.; Hosokawa, M.; Inagaki, N. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem. Biophys. Res. Commun. 2010, 393, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Frontini, A.; Cinti, S. Distribution and Development of Brown Adipocytes in the Murine and Human Adipose Organ. Cell Metab. 2010, 11, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.H. Brown fat fuel utilization and thermogenesis. Trends Endrocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e847. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Bio. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef]
- Shuai, L.; Zhang, L.N.; Li, B.H.; Tang, C.L.; Wu, L.Y.; Li, J.; Li, J.Y. SIRT5 regulates brown adipocyte differentiation and browning of subcutaneous white adipose tissue. Diabetes 2019, 68, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wei, Y.H. Roles of mitochondrial sirtuins in mitochondrial function, redox homeostasis, insulin resistance and type 2 diabetes. Int. J. Mol. Sci. 2020, 21, 5266. [Google Scholar] [CrossRef]
- Guedouari, H.; Daigle, T.; Scorrano, L.; Hebert-Chatelain, E. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 169–176. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Wang, D.; Wu, Q.; Shang, Q.; Gao, C.; Wang, H.; Wang, H.; Du, M.; Peng, P.; et al. SIRT5-related desuccinylation modification of AIFM1 protects against compression-induced intervertebral disc degeneration by regulating mitochondrial homeostasis. Exp. Mol. Med. 2023, 55, 253–268. [Google Scholar] [CrossRef]
- Guo, A.H.; Baliira, R.; Skinner, M.E.; Kumar, S.; Andren, A.; Zhang, L.; Goldsmith, R.S.; Michan, S.; Davis, N.J.; Maccani, M.W.; et al. Sirtuin 5 levels are limiting in preserving cardiac function and suppressing fibrosis in response to pressure overload. Sci. Rep. 2022, 12, 12258. [Google Scholar] [CrossRef] [PubMed]
- Boylston, J.A.; Sun, J.; Chen, Y.; Gucek, M.; Sack, M.N.; Murphy, E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 88, 73–81. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Che, W.; Zheng, C.; Liu, W.; Wen, J.; Fu, H.; Tang, K.; Zhang, J.; Xu, Y. SIRT5: A safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell. Physiol. Biochem. 2013, 32, 1050–1059. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef]
- Williams, A.C.; Ramsden, D.B. Autotoxicity, methylation and a road to the prevention of Parkinson’s disease. J. Clin. Neurosci. 2005, 12, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wei, Y.; Li, J.; Bai, Y.; Yin, P.; Wang, S. SIRT5 Represses Neurotrophic Pathways and Aβ Production in Alzheimer’s Disease by Targeting Autophagy. ACS Chem. Neurosci. 2021, 12, 4428–4437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, S.; Simonyi, A.; Sun, G.Y.; Sun, A.Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005, 31, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, L. SIRT5 deficiency enhances susceptibility to kainate-induced seizures and exacerbates hippocampal neurodegeneration not through mitochondrial antioxidant enzyme SOD2. Front. Cell. Neurosci. 2016, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, D.; Shi, C. SIRT5 reduces the inflammatory response and barrier dysfunction in IL-17A-induced epidermal keratinocytes. Allergol. Immunopathol. 2023, 51, 30–36. [Google Scholar] [CrossRef]
- Wang, T.; Lin, B.; Qiu, W.; Yu, B.; Li, J.; An, S.; Weng, L.; Li, Y.; Shi, M.; Chen, Z.; et al. AMP-activated protein kinase phosphorylation mediated by Sirtuin 5 alleviates septic acute kidney injury. Shock 2022, 59, 477–485. [Google Scholar] [CrossRef]
- Xia, Q.; Gao, S.; Han, T.; Mao, M.; Zhan, G.; Wang, Y.; Li, X. Sirtuin 5 aggravates microglia-induced neuroinflammation following ischaemic stroke by modulating the desuccinylation of Annexin-A1. J. Neuroinflammation 2022, 19, 301. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Bringman-Rodenbarger, L.R.; Guo, A.H.; Lyssiotis, C.A.; Lombard, D.B. Emerging Roles for SIRT5 in Metabolism and Cancer. Antioxid. Redox Signal. 2018, 28, 677–690. [Google Scholar] [CrossRef]
- Takeshita, A.; Naito, K.; Shinjo, K.; Sahara, N.; Matsui, H.; Ohnishi, K.; Beppu, H.; Ohtsubo, K.; Horii, T.; Maekawa, M.; et al. Deletion 6p23 and add(11)(p15) leading to NUP98 translocation in a case of therapy-related atypical chronic myelocytic leukemia transforming to acute myelocytic leukemia. Cancer Genet. Cytogenet. 2004, 152, 56–60. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Sun, J.Y.; Xu, L.; Tseng, H.; Ciccarelli, B.; Fulciniti, M.; Hunter, Z.R.; Maghsoudi, K.; Hatjiharissi, E.; Zhou, Y.; Yang, G.; et al. Histone deacetylase inhibitors demonstrate significant preclinical activity as single agents, and in combination with bortezomib in Waldenstrom’s macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 2011, 11, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Kalender, M.E.; Borazan, E.; Bozgeyik, I.; Bayraktar, R.; Bozgeyik, E.; Camci, C.; Arslan, A. High-throughput screening of Sirtuin family of genes in breast cancer. Gene 2016, 586, 123–128. [Google Scholar] [CrossRef]
- Bartosch, C.; Monteiro-Reis, S.; Almeida-Rios, D.; Vieira, R.; Castro, A.; Moutinho, M.; Rodrigues, M.; Graça, I.; Lopes, J.M.; Jerónimo, C. Assessing sirtuin expression in endometrial carcinoma and non-neoplastic endometrium. Oncotarget 2016, 7, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Lin, P.M.; Lin, S.F.; Hsu, C.H.; Lin, H.C.; Hu, M.L.; Hsu, C.M.; Yang, M.Y. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 2013, 34, 1847–1854. [Google Scholar] [CrossRef]
- Lv, X.B.; Liu, L.; Cheng, C.; Yu, B.; Xiong, L.; Hu, K.; Tang, J.; Zeng, L.; Sang, Y. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci. Rep. 2015, 5, 17940. [Google Scholar] [CrossRef]
- Wang, J.X.; Yi, Y.; Li, Y.W.; Cai, X.Y.; He, H.W.; Ni, X.C.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; Fan, J.; et al. Down-regulation of sirtuin 3 is associated with poor prognosis in hepatocellular carcinoma after resection. BMC Cancer 2014, 14, 297. [Google Scholar] [CrossRef]
- Jaiswal, A.; Xudong, Z.; Zhenyu, J.; Saretzki, G. Mitochondrial sirtuins in stem cells and cancer. FEBS J. 2022, 289, 3393–3415. [Google Scholar] [CrossRef]
- Chang, L.; Xi, L.; Liu, Y.; Liu, R.; Wu, Z.; Jian, Z. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol. Med. Rep. 2018, 17, 342–349. [Google Scholar] [CrossRef]
- Abril, Y.L.N.; Fernandez, I.R.; Hong, J.Y.; Chiang, Y.L.; Kutateladze, D.A.; Zhao, Q.; Yang, M.; Hu, J.; Sadhukhan, S.; Li, B.; et al. Pharmacological and genetic perturbation establish SIRT5 as a promising target in breast cancer. Oncogene 2021, 40, 1644–1658. [Google Scholar] [CrossRef]
- Giblin, W.; Bringman-Rodenbarger, L.; Guo, A.H.; Kumar, S.; Monovich, A.C.; Mostafa, A.M.; Skinner, M.E.; Azar, M.; Mady, A.S.A.; Chung, C.H.; et al. The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics. J. Clin. Investig. 2021, 131, e138926. [Google Scholar] [CrossRef]
- Liang, F.; Wang, X.; Ow, S.H.; Chen, W.; Ong, W.C. Sirtuin 5 is Anti-apoptotic and Anti-oxidative in Cultured SH-EP Neuroblastoma Cells. Neurotox. Res. 2017, 31, 63–76. [Google Scholar] [CrossRef]
- He, Q.; Chen, K.; Ye, R.; Dai, N.; Guo, P.; Wang, L. Associations of sirtuins with clinicopathological variables and prognosis in human ovarian cancer. Oncol. Lett. 2020, 19, 3278–3288. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.; Gai, J.; Guan, J.; Li, J.; Li, Y.; Zhao, J.; Zhao, C.; Fu, L.; Li, Q. SIRT5 promotes cisplatin resistance in ovarian cancer by suppressing dna damage in a ROS-dependent manner via regulation of the Nrf2/HO-1 pathway. Front. Oncol. 2019, 9, 754. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Li, X.; Liu, B.; Liu, M.; Liu, L.; Chen, S.; Ren, M.; Wang, Y.; Yu, M.; et al. SHMT2 Desuccinylation by SIRT5 Drives Cancer Cell Proliferation. Cancer Res. 2018, 78, 372–386. [Google Scholar] [CrossRef]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, H.L.; Xu, J.; Tan, J.; Fu, L.N.; Wang, J.L.; Zou, T.H.; Sun, D.F.; Gao, Q.Y.; Chen, Y.X.; et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018, 9, 545. [Google Scholar] [CrossRef]
- Du, Z.; Liu, X.; Chen, T.; Gao, W.; Wu, Z.; Hu, Z.; Wei, D.; Gao, C.; Li, Q. Targeting a Sirt5-Positive Subpopulation Overcomes Multidrug Resistance in Wild-Type Kras Colorectal Carcinomas. Cell Rep. 2018, 22, 2677–2689. [Google Scholar] [CrossRef]
- Wang, H.L.; Chen, Y.; Wang, Y.Q.; Tao, E.W.; Tan, J.; Liu, Q.Q.; Li, C.M.; Tong, X.M.; Gao, Q.Y.; Hong, J.; et al. Sirtuin5 protects colorectal cancer from DNA damage by keeping nucleotide availability. Nat. Commun. 2022, 13, 6121. [Google Scholar] [CrossRef]
- Wang, K.; Hu, Z.; Zhang, C.; Yang, L.; Feng, L.; Yang, P.; Yu, H. SIRT5 Contributes to Colorectal Cancer Growth by Regulating T Cell Activity. J. Immunol. Res. 2020, 2020, 3792409. [Google Scholar] [CrossRef]
- Guan, J.; Jiang, X.; Gai, J.; Sun, X.; Zhao, J.; Li, J.; Li, Y.; Cheng, M.; Du, T.; Fu, L.; et al. Sirtuin 5 regulates the proliferation, invasion and migration of prostate cancer cells through acetyl-CoA acetyltransferase 1. J. Cell. Mol. Med. 2020, 24, 14039–14049. [Google Scholar] [CrossRef]
- Yan, D.; Franzini, A.; Pomicter, A.D.; Halverson, B.J.; Antelope, O.; Mason, C.C.; Ahmann, J.M.; Senina, A.V.; Vellore, N.A.; Jones, C.L. SIRT5 is a druggable metabolic vulnerability in acute myeloid leukemia. Blood Cancer Discov. 2021, 2, 266–287. [Google Scholar] [CrossRef]
- Rajabi, N.; Hansen, T.N.; Nielsen, A.L.; Nguyen, H.T.; Bæk, M.; Bolding, J.E.; Bahlke, O.Ø.; Petersen, S.E.G.; Bartling, C.R.O.; Strømgaard, K.; et al. Investigation of Carboxylic Acid Isosteres and Prodrugs for Inhibition of the Human SIRT5 Lysine Deacylase Enzyme. Angew. Chem. Int. Ed. Engl. 2022, 61, e202115805. [Google Scholar] [CrossRef]
- Liu, X.; Rong, F.; Tang, J.; Zhu, C.; Chen, X.; Jia, S.; Wang, Z.; Sun, X.; Deng, H.; Zha, H.; et al. Repression of p53 function by SIRT5-mediated desuccinylation at Lysine 120 in response to DNA damage. Cell Death Diff. 2022, 29, 722–736. [Google Scholar] [CrossRef]
- Hu, T.; Shukla, S.K.; Vernucci, E.; He, C.; Wang, D.; King, R.J.; Jha, K.; Siddhanta, K.; Mullen, N.J.; Attri, K.S.; et al. Metabolic Rewiring by Loss of Sirt5 Promotes Kras-Induced Pancreatic Cancer Progression. Gastroenterology 2021, 161, 1584–1600. [Google Scholar] [CrossRef]
- Lu, X.; Yang, P.; Zhao, X.; Jiang, M.; Hu, S.; Ouyang, Y.; Zeng, L.; Wu, J. OGDH mediates the inhibition of SIRT5 on cell proliferation and migration of gastric cancer. Exp. Cell Res. 2019, 382, 111483. [Google Scholar] [CrossRef]
- Tang, Z.; Li, L.; Tang, Y.; Xie, D.; Wu, K.; Wei, W.; Xiao, Q. CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Sci. 2018, 109, 2590–2598. [Google Scholar] [CrossRef]
- Clark, O.; Yen, K.; Mellinghoff, I.K. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin. Cancer Res. 2016, 22, 1837–1842. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Kwon, O.K.; Bang, I.H.; Choi, S.Y.; Jeon, J.M.; Na, A.Y.; Gao, Y. SIRT5 Is the desuccinylase of LDHA as novel cancer metastatic stimulator in aggressive prostate cancer. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Walter, M.; Chen, I.P.; Vallejo-Gracia, A.; Kim, I.J.; Bielska, O.; Lam, V.L.; Hayashi, J.M.; Cruz, A.; Shah, S.; Soveg, F.W.; et al. SIRT5 is a proviral factor that interacts with SARS-CoV-2 Nsp14 protein. PLoS Pathog. 2022, 18, e1010811. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, H.; Zhang, H.; Sui, L.; Li, L.; Xu, W.; Du, S.; Hao, P.; Jiang, Y.; Chen, J.; et al. The global succinylation of SARS-CoV-2-infected host cells reveals drug targets. Proc. Natl. Acad. Sci. USA 2022, 119, e2123065119. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus NSP14-NSP10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, C.; Zha, H.; Tang, J.; Rong, F.; Chen, X.; Fan, S.; Xu, C.; Du, J.; Zhu, J.; et al. SIRT5 impairs aggregation and activation of the signaling adaptor MAVS through catalyzing lysine desuccinylation. EMBO J. 2020, 39, e103285. [Google Scholar] [CrossRef]
- Buler, M.; Aatsinki, S.M.; Izzi, V.; Uusimaa, J.; Hakkola, J. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014, 28, 3225–3237. [Google Scholar] [CrossRef]
- Cowell, R.M.; Talati, P.; Blake, K.R.; Meador-Woodruff, J.H.; Russell, J.W. Identification of novel targets for PGC-1alpha and histone deacetylase inhibitors in neuroblastoma cells. Biochem. Biophys. Res. Commun. 2009, 379, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Sutton, N.R.; Wang, Z.; Goonewardena, S.N.; Holinstat, M. Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID-19. Eur. J. Pharmacol. 2021, 898, 173988. [Google Scholar] [CrossRef] [PubMed]
- Suenkel, B.; Valente, S.; Zwergel, C.; Weiss, S.; Di Bello, E.; Fioravanti, R.; Aventaggiato, M.; Amorim, J.A.; Garg, N.; Kumar, S.; et al. Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5. J. Med. Chem. 2022, 65, 14015–14031. [Google Scholar] [CrossRef]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; De Fabiani, E.; Altucci, L.; et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar] [CrossRef]
- Schuetz, A.; Min, J.; Antoshenko, T.; Wang, C.L.; Allali-Hassani, A.; Dong, A.; Loppnau, P.; Vedadi, M.; Bochkarev, A.; Sternglanz, R.; et al. Structural Basis of Inhibition of the Human NAD+-Dependent Deacetylase SIRT5 by Suramin. Structure 2007, 15, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Suenkel, B.; Fischer, F.; Steegborn, C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg. Med. Chem. Lett. 2013, 23, 143–146. [Google Scholar] [CrossRef]
- Yao, J.; Yin, Y.; Han, H.; Chen, S.; Zheng, Y.; Liang, B.; Wu, M.; Shu, K.; Debnath, B.; Lombard, D.B.; et al. Pyrazolone derivatives as potent and selective small-molecule SIRT5 inhibitors. Eur. J. Med. Chem. 2023, 247, 115024. [Google Scholar] [CrossRef]
- Maurer, B.; Rumpf, T.; Scharfe, M.; Stolfa, D.A.; Schmitt, M.L.; He, W.; Verdin, E.; Sippl, W.; Jung, M. Inhibitors of the NAD(+)-dependent protein desuccinylase and demalonylase Sirt5. ACS Med. Chem. Lett. 2012, 3, 1050. [Google Scholar] [CrossRef] [PubMed]
- Trapp, J.; Meier, R.; Hongwiset, D.; Kassack, M.U.; Sippl, W.; Jung, M. Structure-activity studies on suramin analogues as inhibitors of NAD +-dependent histone deacetylases (sirtuins). ChemMedChem 2007, 2, 1419–1431. [Google Scholar] [CrossRef]
- Guetschow, E.D.; Kumar, S.; Lombard, D.B.; Kennedy, R.T. Identification of sirtuin 5 inhibitors by ultrafast microchip electrophoresis using nanoliter volume samples. Anal. Bioanal. Chem. 2016, 408, 721–731. [Google Scholar] [CrossRef]
- Glas, C.; Dietschreit, J.C.B.; Wössner, N.; Urban, L.; Ghazy, E.; Sippl, W.; Jung, M.; Ochsenfeld, C.; Bracher, F. Identification of the subtype-selective Sirt5 inhibitor balsalazide through systematic SAR analysis and rationalization via theoretical investigations. Eur. J. Med. Chem. 2020, 206, 112676. [Google Scholar] [CrossRef]
- Glas, C.; Naydenova, E.; Lechner, S.; Wossner, N.; Yang, L.; Dietschreit, J.C.B.; Sun, H.; Jung, M.; Kuster, B.; Ochsenfeld, C.; et al. Development of hetero-triaryls as a new chemotype for subtype-selective and potent Sirt5 inhibition. Eur. J. Med. Chem. 2022, 240, 114594. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Peltier, R.; Zhang, M.; Song, D.; Huang, H.; Chen, G.; Chen, Y.; Zhou, F.; Hao, Q.; Bian, L.; et al. Desuccinylation-Triggered Peptide Self-Assembly: Live Cell Imaging of SIRT5 Activity and Mitochondrial Activity Modulation. J. Am. Chem. Soc. 2020, 142, 18150–18159. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Feraco, A.; Mirabilii, S.; Saladini, S.; Sansone, L.; Vernucci, E.; Tomaselli, G.; Marzolla, V.; Rotili, D.; Russo, M.A.; et al. Sirt5 inhibition induces brown fat-like phenotype in 3t3-l1 preadipocytes. Cells 2021, 10, 1126. [Google Scholar] [CrossRef] [PubMed]
- Stevens, G.J.; Hitchcock, K.; Karen Wang, Y.; Coppola, G.M.; Versace, R.W.; Chin, J.A.; Shapiro, M.; Suwanrumpha, S.; Mangold, B.L.K. In vitro metabolism of N-(5-chloro-2-methylphenyl)-N’-(2-methylpropyl)thiourea: Species comparison and identification of a novel thiocarbamide-glutathione adduct. Chem. Res. Toxicol. 1997, 10, 733–741. [Google Scholar] [CrossRef]
- Neal, R.A.; Halpert, J. Toxicology of thiono-sulfur compounds. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 321–339. [Google Scholar] [CrossRef]
- Poulsen, L.L.; Hyslop, R.M.; Ziegler, D.M. S-oxygenation of N-substituted thioureas catalyzed by the pig liver microsomal FAD-containing monooxygenase. Arch. Biochem. Biophys. 1979, 198, 78–88. [Google Scholar] [CrossRef]
- Ziegler, D.M. Flavin-containing monooxygenases: Catalytic mechanism and substrate specificities. Drug Metab. Rev. 1988, 19, 1–32. [Google Scholar] [CrossRef]
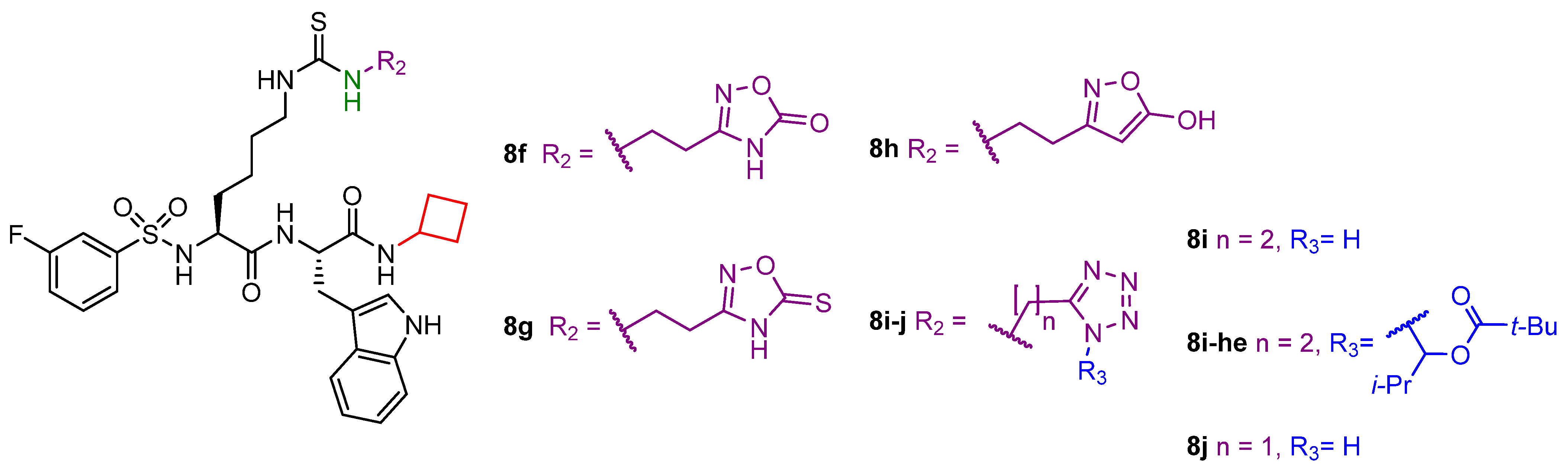
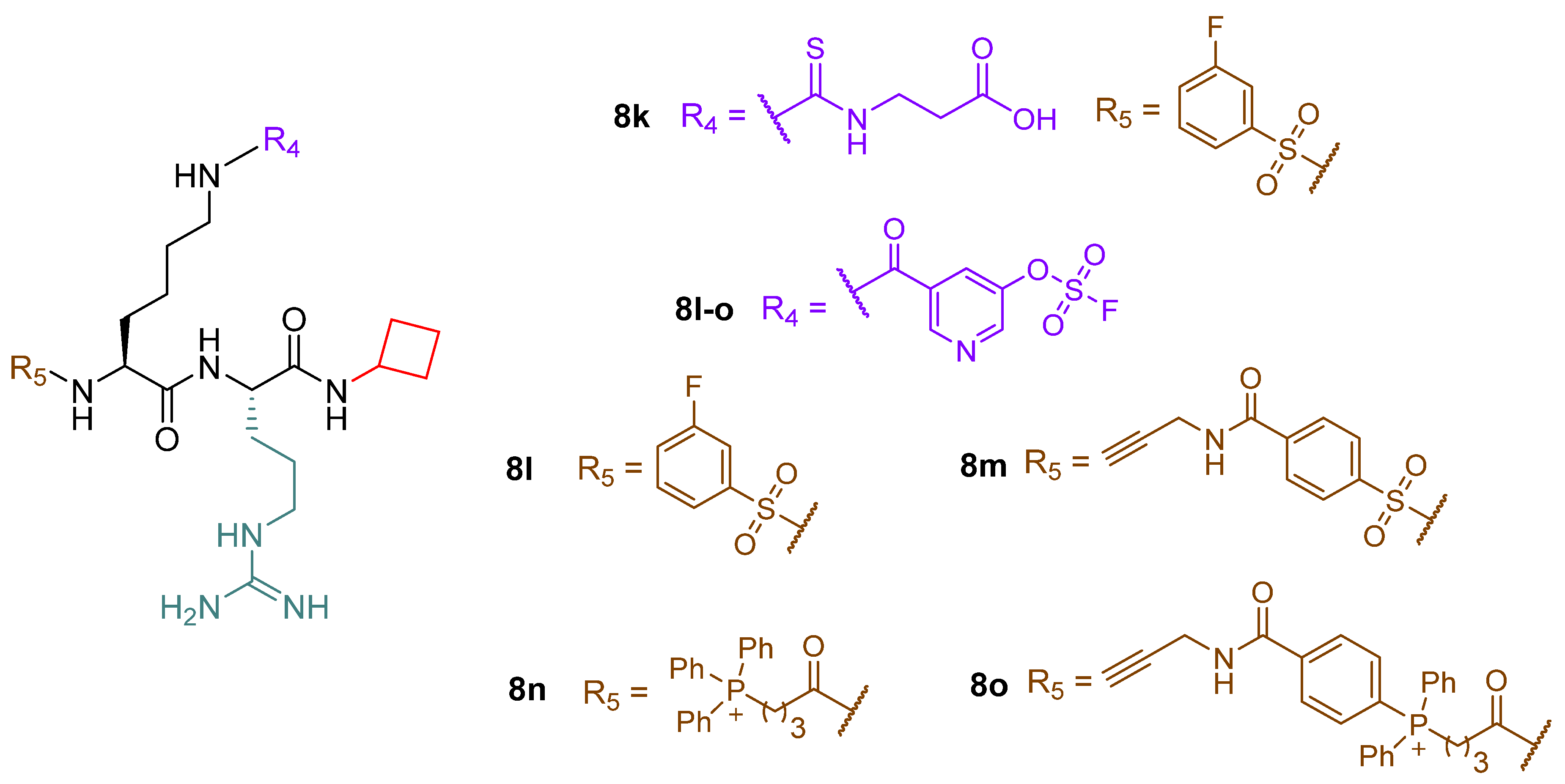
- Rajabi, N.; Auth, M.; Troelsen, K.R.; Pannek, M.; Bhatt, D.P.; Fontenas, M.; Hirschey, M.D.; Steegborn, C.; Madsen, A.S.; Olsen, C.A. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure–Activity Relationship, Biostructural, and Kinetic Insight. Angew. Chem. Int. Ed. Engl. 2017, 56, 14836–14841. [Google Scholar] [CrossRef]
- Bolding, J.E.; Martin-Gago, P.; Rajabi, N.; Gamon, L.F.; Hansen, T.N.; Bartling, C.R.O.; Stromgaard, K.; Davies, M.J.; Olsen, C.A. Aryl Fluorosulfate Based Inhibitors that Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem. Int. Ed. Engl. 2022, 61, e202204565. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbrizi, E.; Fiorentino, F.; Carafa, V.; Altucci, L.; Mai, A.; Rotili, D. Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells 2023, 12, 852. https://doi.org/10.3390/cells12060852
Fabbrizi E, Fiorentino F, Carafa V, Altucci L, Mai A, Rotili D. Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells. 2023; 12(6):852. https://doi.org/10.3390/cells12060852
Chicago/Turabian StyleFabbrizi, Emanuele, Francesco Fiorentino, Vincenzo Carafa, Lucia Altucci, Antonello Mai, and Dante Rotili. 2023. "Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection" Cells 12, no. 6: 852. https://doi.org/10.3390/cells12060852
APA StyleFabbrizi, E., Fiorentino, F., Carafa, V., Altucci, L., Mai, A., & Rotili, D. (2023). Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells, 12(6), 852. https://doi.org/10.3390/cells12060852