
Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2

 , , , , , ,
, , , , , ,  ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Generation of the Mice and Genotyping
2.3. Ultrasound Echocardiography
2.4. Embryo Collection and Fixation, Theiler Staging
2.5. Proteasomal Activity Assays
2.6. High Resolution Episcopic Microscopy
2.7. Immunofluorescence Staining on Cryosections
2.8. Ex Vivo Studies
2.9. Mass Spectrometry for Identification
2.10. Proteomics
2.11. Wholemount Immunohistochemistry
2.12. Electron Microscopy
2.13. Image Analysis
2.14. Statistics
3. Results
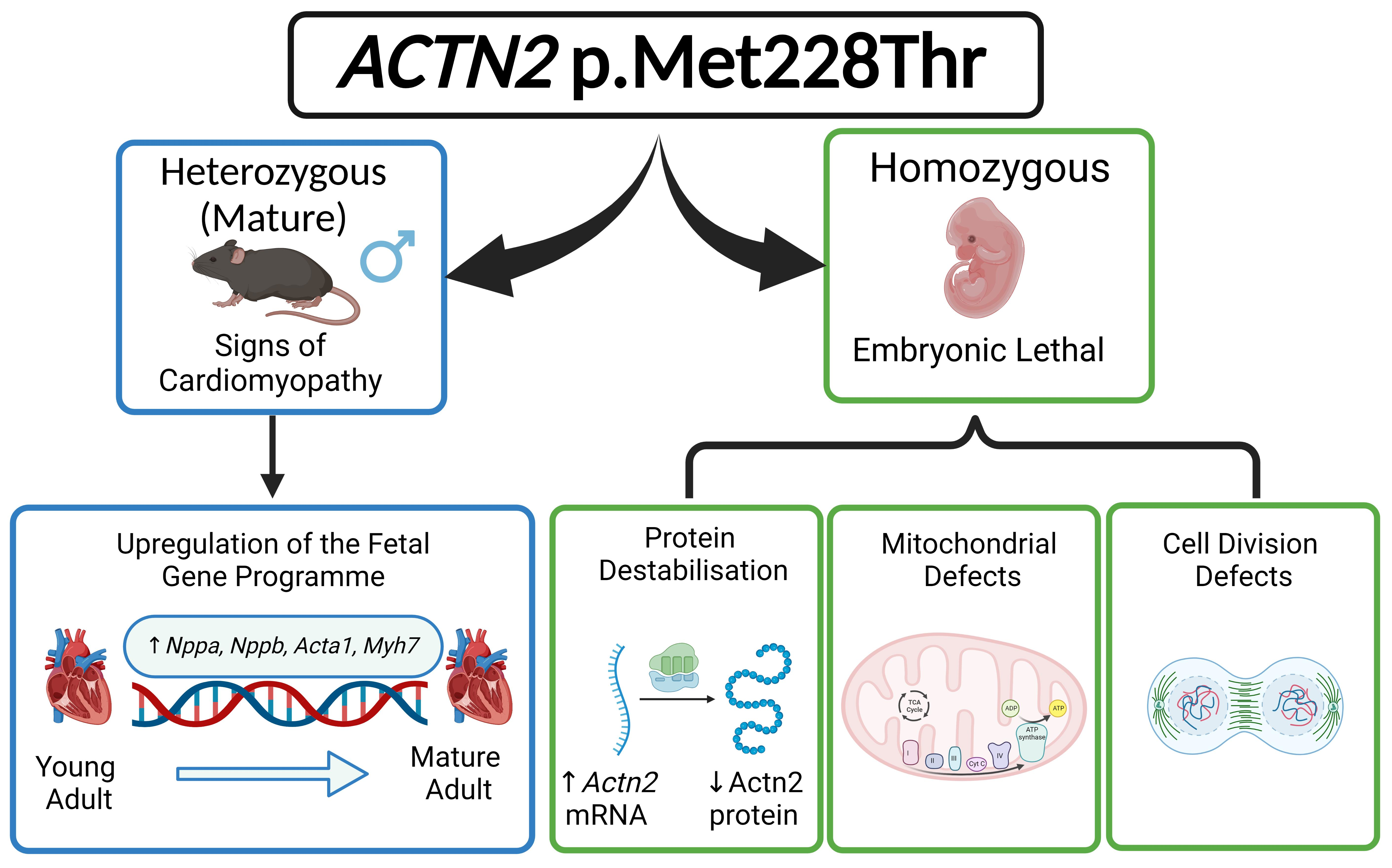
3.1. Generation of a Mouse Model for a Hypertrophic Cardiomyopathy-Causing Genetic Variant
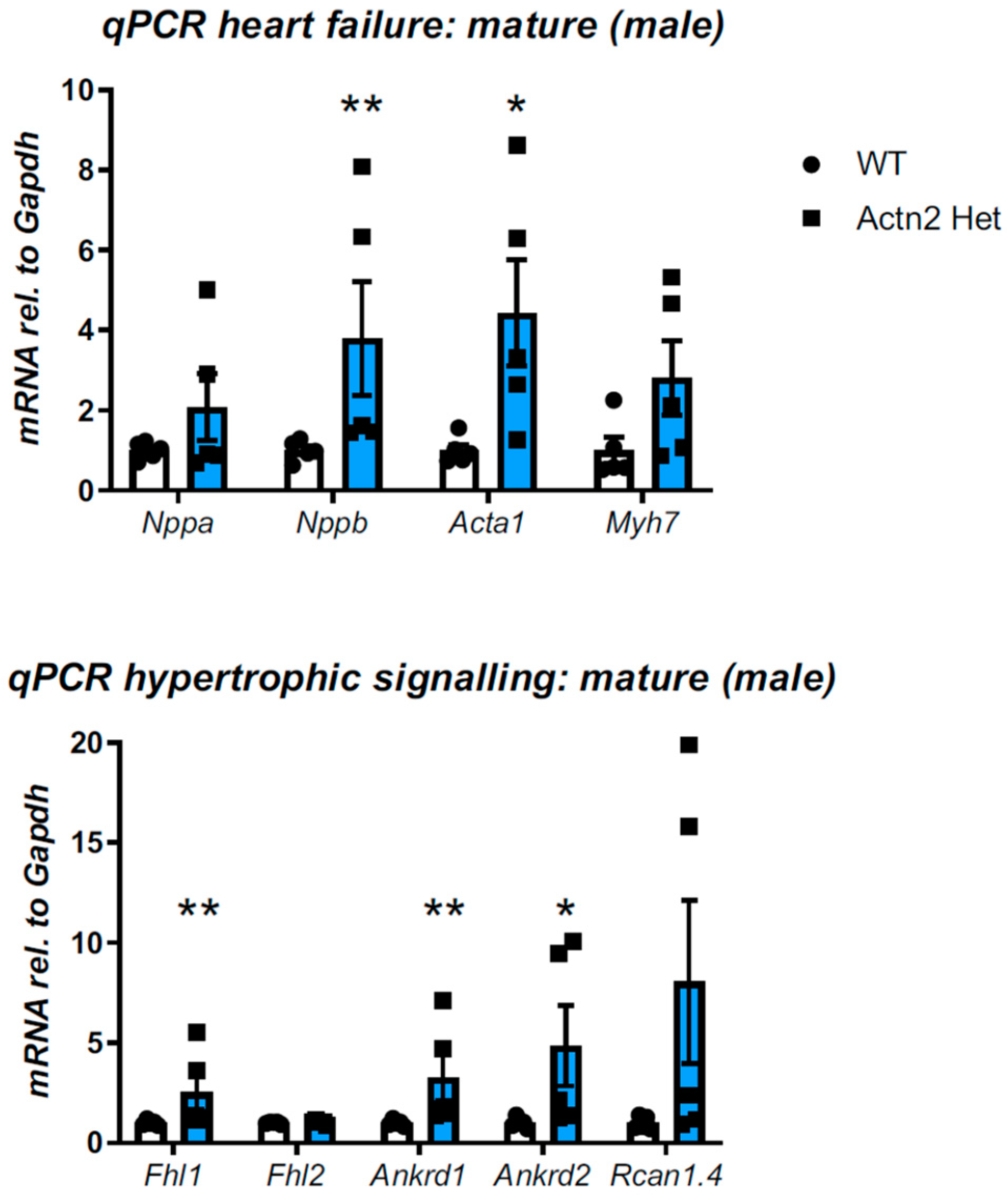
3.2. Cardiac Phenotyping of Adult Mice with the Actn2 p.Met228Thr Variant
3.3. Actn2 p.Met228Thr Is Embryonic Lethal in the Homozygous Setting
3.4. Detailed Morphological Analysis of Hom Actn2 p.Met228Thr E15.5 Embryos
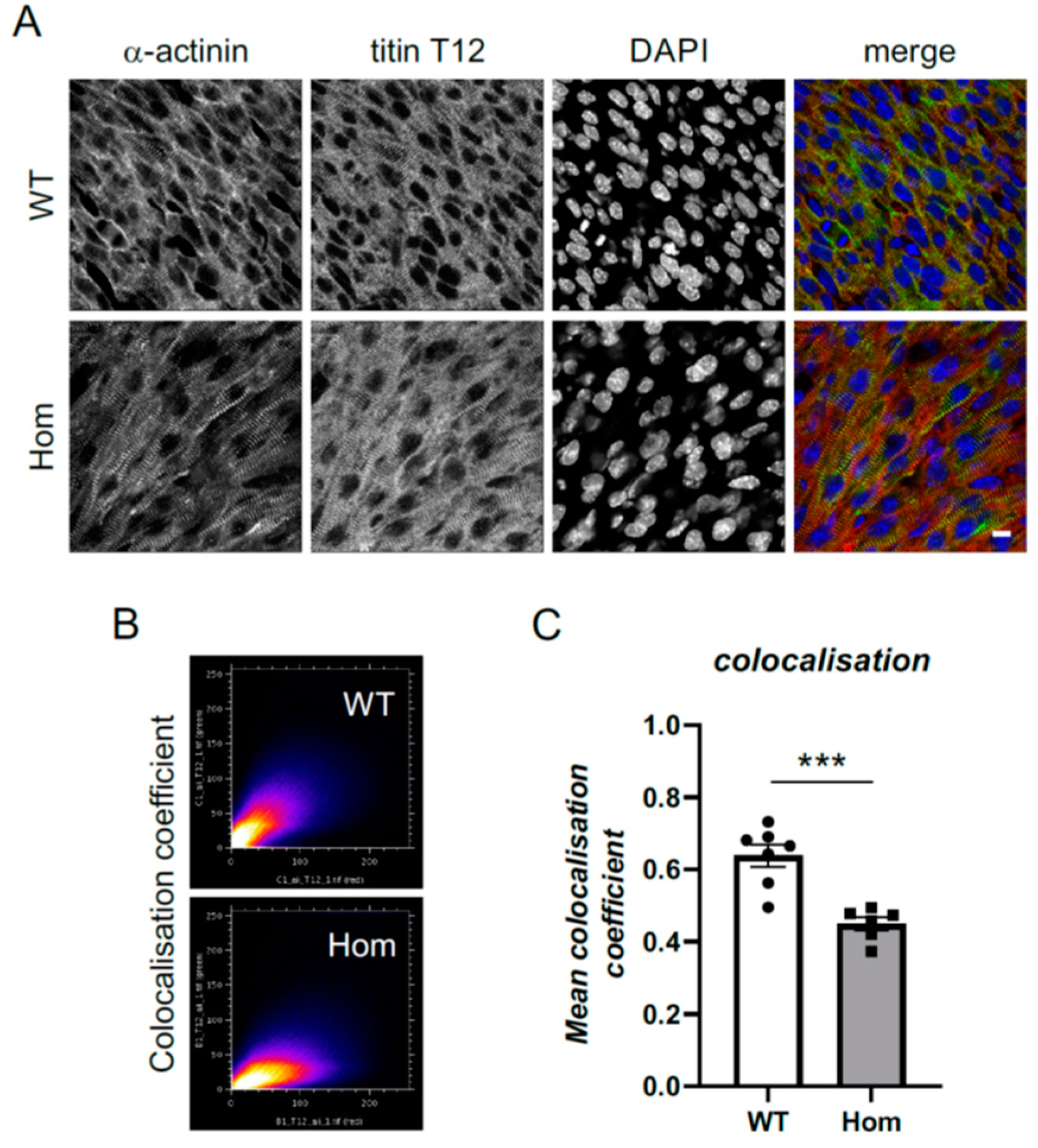
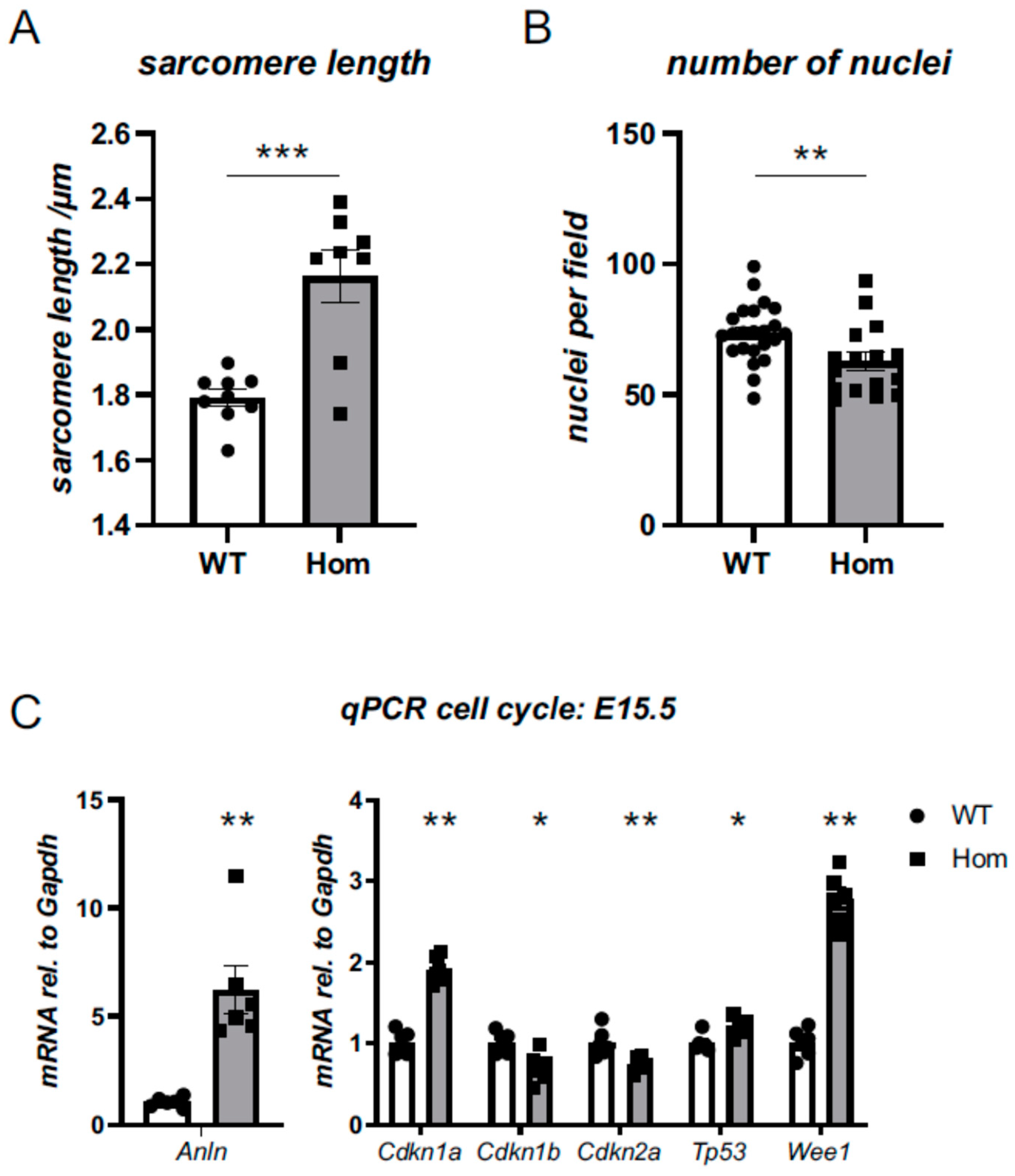
3.5. Sarcomeric and Nuclear Organisation in Hom Hearts
3.6. Cell-Cycle Defects in Hom Hearts
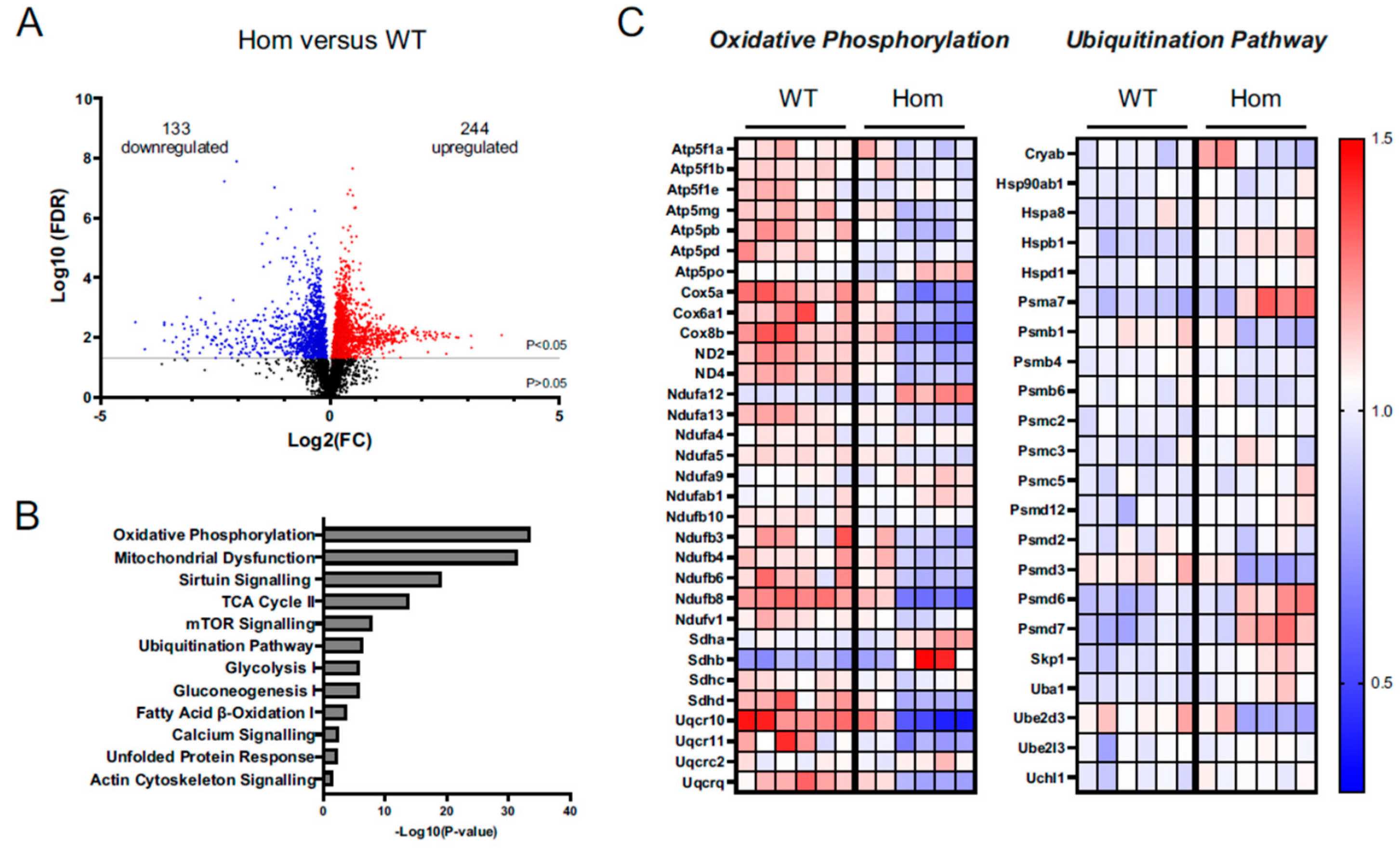
3.7. Proteomics Analysis Gives Insights into Disturbances in the Hom Hearts
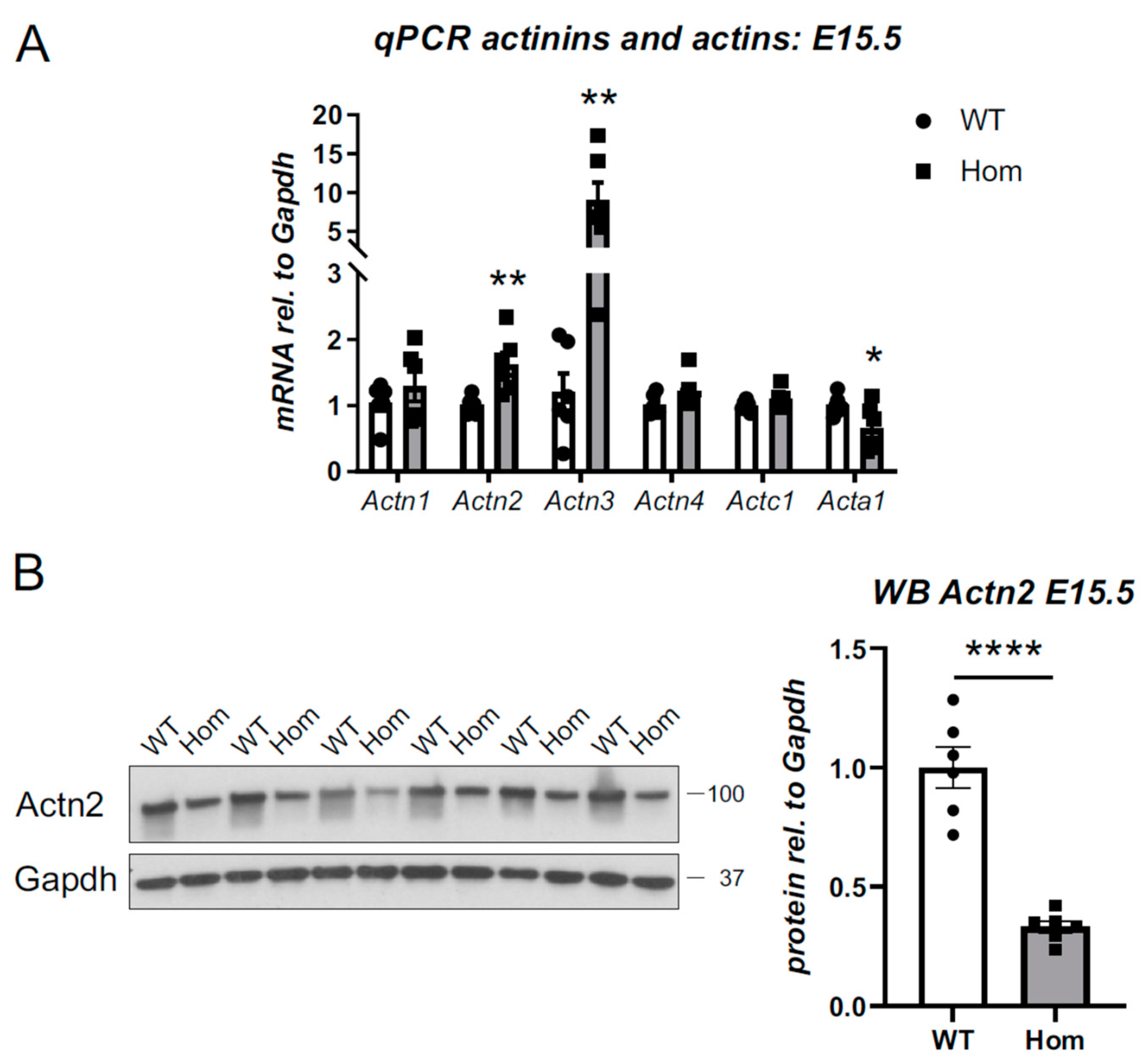
3.8. Destabilisation of Actn2 Protein in the Hom Hearts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lange, S.; Pinotsis, N.; Agarkova, I.; Ehler, E. The M-band: The underestimated part of the sarcomere. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118440. [Google Scholar] [CrossRef] [PubMed]
- Wadmore, K.; Azad, A.J.; Gehmlich, K. The Role of Z-disc Proteins in Myopathy and Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 3058. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Poloni, G.; Sontayananon, N.; Jiang, H.; Gehmlich, K. The giant titin: How to evaluate its role in cardiomyopathies. J. Muscle Res. Cell Motil. 2019, 40, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Frey, N. Cardiac Z-disc signaling network. J. Biol. Chem. 2011, 286, 9897–9904. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Ede, A., Jr.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjoblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle alpha-actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef]
- Avery, A.W.; Fealey, M.E.; Wang, F.; Orlova, A.; Thompson, A.R.; Thomas, D.D.; Hays, T.S.; Egelman, E.H. Structural basis for high-affinity actin binding revealed by a beta-III-spectrin SCA5 missense mutation. Nat. Commun. 2017, 8, 1350. [Google Scholar] [CrossRef] [PubMed]
- Galkin, V.E.; Orlova, A.; Salmazo, A.; Djinovic-Carugo, K.; Egelman, E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct Mol. Biol. 2010, 17, 614–616. [Google Scholar] [CrossRef]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef]
- Girolami, F.; Iascone, M.; Tomberli, B.; Bardi, S.; Benelli, M.; Marseglia, G.; Pescucci, C.; Pezzoli, L.; Sana, M.E.; Basso, C.; et al. Novel alpha-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circ. Cardiovasc. Genet. 2014, 7, 741–750. [Google Scholar] [CrossRef]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Theis, J.L.; Bos, J.M.; Bartleson, V.B.; Will, M.L.; Binder, J.; Vatta, M.; Towbin, J.A.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 351, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Haywood, N.J.; Wolny, M.; Rogers, B.; Trinh, C.H.; Shuping, Y.; Edwards, T.A.; Peckham, M. Hypertrophic cardiomyopathy mutations in the calponin-homology domain of ACTN2 affect actin binding and cardiomyocyte Z-disc incorporation. Biochem. J. 2016, 473, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Zech, A.T.L.; Prondzynski, M.; Singh, S.R.; Pietsch, N.; Orthey, E.; Alizoti, E.; Busch, J.; Madsen, A.; Behrens, C.S.; Meyer-Jens, M.; et al. ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes. Cells 2022, 11, 2745. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.C.; Hugill, A.; Dear, N.T.; Ashcroft, F.M.; Cox, R.D. Deletion of nicotinamide nucleotide transhydrogenase: A new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 2006, 55, 2153–2156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hooper, C.; Kelly, M.; Steeples, V.; Simon, J.N.; Beglov, J.; Azad, A.J.; Leinhos, L.; Bennett, P.; Ehler, E.; et al. Functional analysis of a gene-edited mouse model to gain insights into the disease mechanisms of a titin missense variant. Basic Res. Cardiol. 2021, 116, 14. [Google Scholar] [CrossRef] [PubMed]
- Geyer, S.H.; Reissig, L.; Rose, J.; Wilson, R.; Prin, F.; Szumska, D.; Ramirez-Solis, R.; Tudor, C.; White, J.; Mohun, T.J.; et al. A staging system for correct phenotype interpretation of mouse embryos harvested on embryonic day 14 (E14.5). J. Anat. 2017, 230, 710–719. [Google Scholar] [CrossRef]
- Strucksberg, K.H.; Tangavelou, K.; Schröder, R.; Clemen, C.S. Proteasomal activity in skeletal muscle: A matter of assay design, muscle type, and age. Anal. Biochem. 2010, 399, 225–229. [Google Scholar] [CrossRef]
- Kalisch-Smith, J.I.; Ved, N.; Szumska, D.; Munro, J.; Troup, M.; Harris, S.E.; Rodriguez-Caro, H.; Jacquemot, A.; Miller, J.J.; Stuart, E.M.; et al. Maternal iron deficiency perturbs embryonic cardiovascular development in mice. Nat. Commun. 2021, 12, 3447. [Google Scholar] [CrossRef]
- Gehmlich, K.; Dodd, M.S.; Allwood, J.W.; Kelly, M.; Bellahcene, M.; Lad, H.V.; Stockenhuber, A.; Hooper, C.; Ashrafian, H.; Redwood, C.S.; et al. Changes in the cardiac metabolome caused by perhexiline treatment in a mouse model of hypertrophic cardiomyopathy. Mol. Biosyst. 2015, 11, 564–573. [Google Scholar] [CrossRef]
- Davis, S.; Scott, C.; Ansorge, O.; Fischer, R. Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain. J. Proteome Res. 2019, 18, 1787–1795. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.F.J.; Breusegem, S.Y.; Larrieu, D. Current Methods and Pipelines for Image-Based Quantitation of Nuclear Shape and Nuclear Envelope Abnormalities. Cells 2022, 11, 347. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.A.; Naik, J.; Fibben, K.S.; Kong, X.; Kiyono, T.; Yokomori, K.; Grosberg, A. Striated myocyte structural integrity: Automated analysis of sarcomeric z-discs. PLoS Comput. Biol. 2020, 16, e1007676. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010, 52, 9–25. [Google Scholar] [PubMed]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, M.; Kelly, M.; Hooper, C.; Yavari, A.; Beglov, J.; Bellahcene, M.; Ghataorhe, K.; Poloni, G.; Goel, A.; Kyriakou, T.; et al. Mutant Muscle LIM Protein C58G causes cardiomyopathy through protein depletion. J. Mol. Cell Cardiol. 2018, 121, 287–296. [Google Scholar] [CrossRef]
- Vignier, N.; Schlossarek, S.; Fraysse, B.; Mearini, G.; Kramer, E.; Pointu, H.; Mougenot, N.; Guiard, J.; Reimer, R.; Hohenberg, H.; et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ. Res. 2009, 105, 239–248. [Google Scholar] [CrossRef]
- Singh, S.R.; Zech, A.T.L.; Geertz, B.; Reischmann-Dusener, S.; Osinska, H.; Prondzynski, M.; Kramer, E.; Meng, Q.; Redwood, C.; van der Velden, J.; et al. Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circ. Heart Fail. 2017, 10, e004140. [Google Scholar] [CrossRef]
- Heinonen, I.; Sorop, O.; van Dalen, B.M.; Wust, R.C.I.; van de Wouw, J.; de Beer, V.J.; Octavia, Y.; van Duin, R.W.B.; Hoogstrate, Y.; Blonden, L.; et al. Cellular, mitochondrial and molecular alterations associate with early left ventricular diastolic dysfunction in a porcine model of diabetic metabolic derangement. Sci. Rep. 2020, 10, 13173. [Google Scholar] [CrossRef]
- Olsson, M.C.; Palmer, B.M.; Leinwand, L.A.; Moore, R.L. Gender and aging in a transgenic mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1136–H1144. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Englmann, D.R.; Sultan, K.R.; Sauer, M.; Eschenhagen, T.; Carrier, L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 2012, 107, 235. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.J.; Schroeder, M.E.; Aguado, B.A.; Anseth, K.S.; Leinwand, L.A. Matters of the heart: Cellular sex differences. J. Mol. Cell Cardiol. 2021, 160, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.S., Jr.; Speck, A.E.; Amaral, I.M.; Canas, P.M.; Cunha, R.A. The exercise sex gap and the impact of the estrous cycle on exercise performance in mice. Sci. Rep. 2018, 8, 10742. [Google Scholar] [CrossRef] [PubMed]
- Barsha, G.; Denton, K.M.; Mirabito Colafella, K.M. Sex- and age-related differences in arterial pressure and albuminuria in mice. Biol. Sex. Differ. 2016, 7, 57. [Google Scholar] [CrossRef]
- Campinho, P.; Lamperti, P.; Boselli, F.; Vilfan, A.; Vermot, J. Blood Flow Limits Endothelial Cell Extrusion in the Zebrafish Dorsal Aorta. Cell Rep. 2020, 31, 107505. [Google Scholar] [CrossRef]
- Espinosa, M.G.; Taber, L.A.; Wagenseil, J.E. Reduced embryonic blood flow impacts extracellular matrix deposition in the maturing aorta. Dev. Dyn. 2018, 247, 914–923. [Google Scholar] [CrossRef]
- Digilio, M.; Marino, B. Clinical manifestations of Noonan syndrome. Images Paediatr. Cardiol. 2001, 3, 19–30. [Google Scholar]
- Burch, M.; Sharland, M.; Shinebourne, E.; Smith, G.; Patton, M.; McKenna, W. Cardiologic abnormalities in Noonan syndrome: Phenotypic diagnosis and echocardiographic assessment of 118 patients. J. Am. Coll Cardiol. 1993, 22, 1189–1192. [Google Scholar] [CrossRef]
- Ikenishi, A.; Okayama, H.; Iwamoto, N.; Yoshitome, S.; Tane, S.; Nakamura, K.; Obayashi, T.; Hayashi, T.; Takeuchi, T. Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev. Growth Differ. 2012, 54, 731–738. [Google Scholar] [CrossRef]
- Ahuja, P.; Perriard, E.; Perriard, J.C.; Ehler, E. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J. Cell Sci. 2004, 117, 3295–3306. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Bahrudin, U.; Morisaki, H.; Morisaki, T.; Ninomiya, H.; Higaki, K.; Nanba, E.; Igawa, O.; Takashima, S.; Mizuta, E.; Miake, J.; et al. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J. Mol. Biol. 2008, 384, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Hom, J.R.; Quintanilla, R.A.; Hoffman, D.L.; de Mesy Bentley, K.L.; Molkentin, J.D.; Sheu, S.S.; Porter, G.A., Jr. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 2011, 21, 469–478. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef]
- Guo, Y.; Cao, Y.; Jardin, B.D.; Sethi, I.; Ma, Q.; Moghadaszadeh, B.; Troiano, E.C.; Mazumdar, N.; Trembley, M.A.; Small, E.M.; et al. Sarcomeres regulate murine cardiomyocyte maturation through MRTF-SRF signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2008861118. [Google Scholar] [CrossRef]
- Ladha, F.A.; Thakar, K.; Pettinato, A.M.; Legere, N.; Ghahremani, S.; Cohn, R.; Romano, R.; Meredith, E.; Chen, Y.S.; Hinson, J.T. Actinin BioID reveals sarcomere crosstalk with oxidative metabolism through interactions with IGF2BP2. Cell Rep. 2021, 36, 109512. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gustafsson Å, B. Cell death in the myocardium: My heart won’t go on. IUBMB Life 2013, 65, 651–656. [Google Scholar] [CrossRef]
- van der Velden, J.; Asselbergs, F.W.; Bakkers, J.; Batkai, S.; Bertrand, L.; Bezzina, C.R.; Bot, I.; Brundel, B.; Carrier, L.; Chamuleau, S.; et al. Animal models and animal-free innovations for cardiovascular research: Current status and routes to be explored. Consensus document of the ESC working group on myocardial function and the ESC Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2022, 118, 3016–3051. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broadway-Stringer, S.; Jiang, H.; Wadmore, K.; Hooper, C.; Douglas, G.; Steeples, V.; Azad, A.J.; Singer, E.; Reyat, J.S.; Galatik, F.; et al. Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2. Cells 2023, 12, 721. https://doi.org/10.3390/cells12050721
Broadway-Stringer S, Jiang H, Wadmore K, Hooper C, Douglas G, Steeples V, Azad AJ, Singer E, Reyat JS, Galatik F, et al. Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2. Cells. 2023; 12(5):721. https://doi.org/10.3390/cells12050721
Chicago/Turabian StyleBroadway-Stringer, Sophie, He Jiang, Kirsty Wadmore, Charlotte Hooper, Gillian Douglas, Violetta Steeples, Amar J. Azad, Evie Singer, Jasmeet S. Reyat, Frantisek Galatik, and et al. 2023. "Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2" Cells 12, no. 5: 721. https://doi.org/10.3390/cells12050721
APA StyleBroadway-Stringer, S., Jiang, H., Wadmore, K., Hooper, C., Douglas, G., Steeples, V., Azad, A. J., Singer, E., Reyat, J. S., Galatik, F., Ehler, E., Bennett, P., Kalisch-Smith, J. I., Sparrow, D. B., Davies, B., Djinovic-Carugo, K., Gautel, M., Watkins, H., & Gehmlich, K. (2023). Insights into the Role of a Cardiomyopathy-Causing Genetic Variant in ACTN2. Cells, 12(5), 721. https://doi.org/10.3390/cells12050721