
Pathogenic Aspects and Therapeutic Avenues of Autophagy in Parkinson’s Disease

Abstract
1. Introduction
2. Parkinson’s Disease
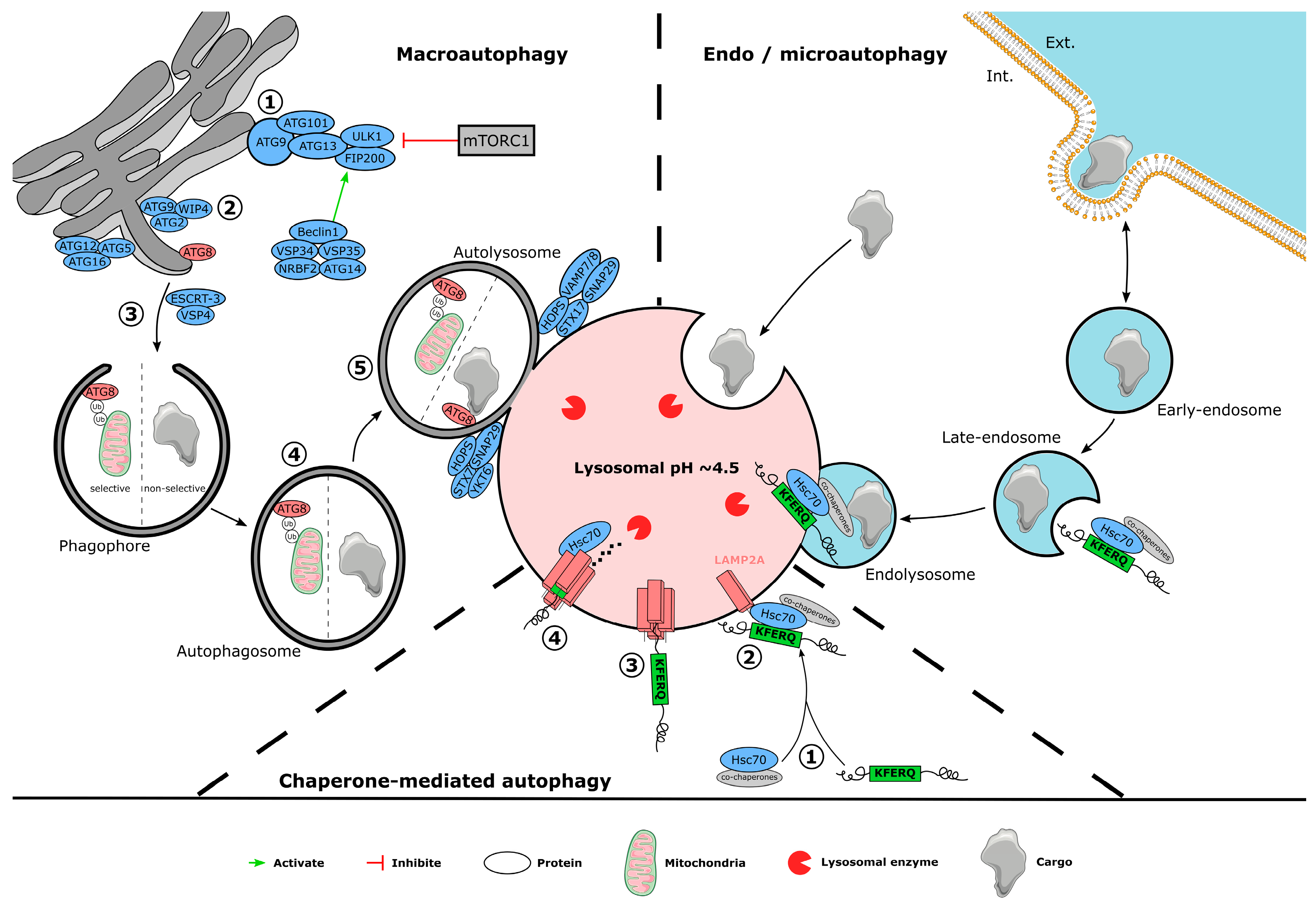
3. Autophagy Mechanism
3.1. Macroautophagy
3.2. Chaperone-Mediated Autophagy
3.3. Endosomal Microautophagy
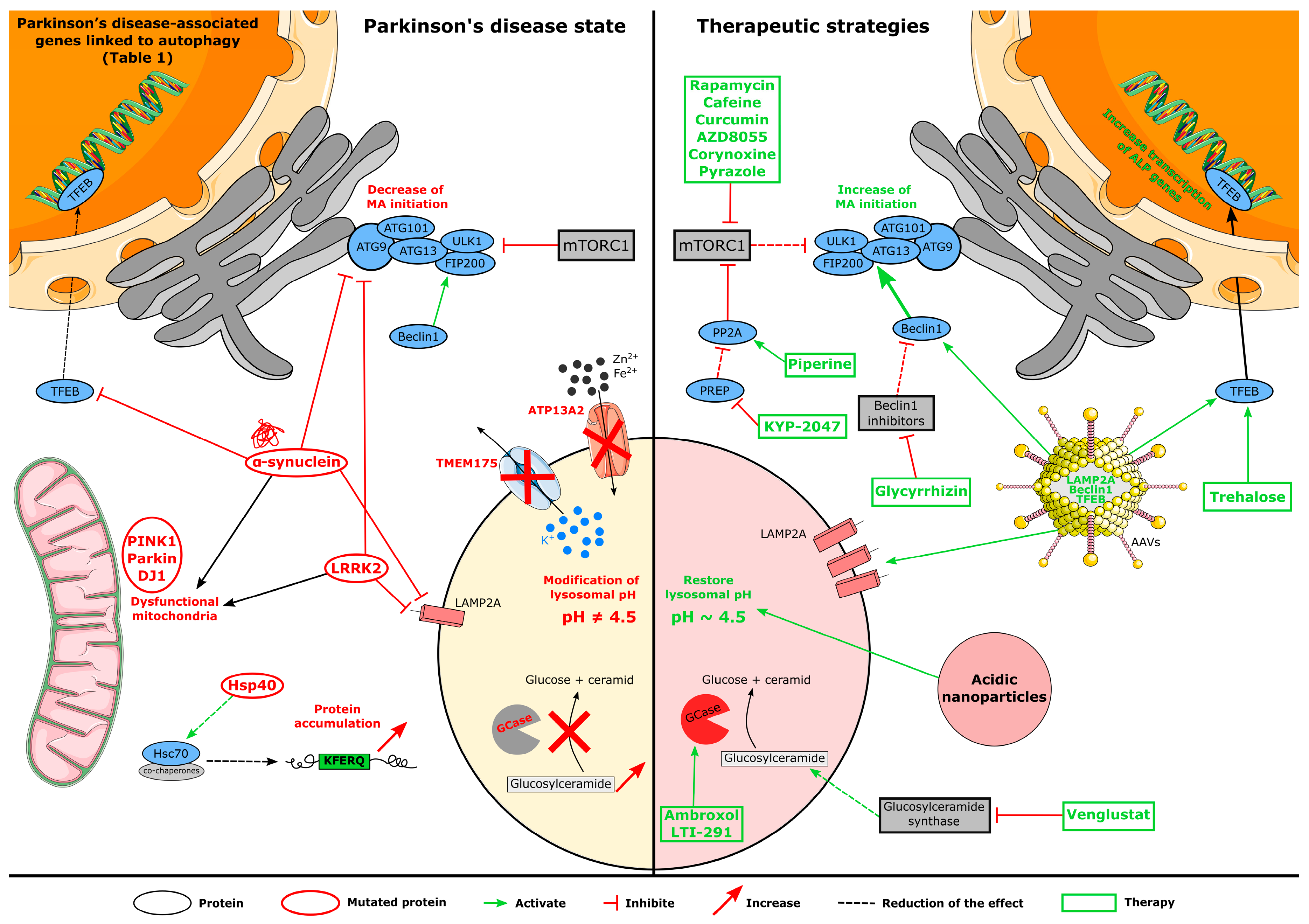
4. Autophagy Disruption in Parkinson’s Disease
4.1. Genetic Implication in Autophagy Dysfunction
4.2. Rodent Models Linked to ALP Dysfunctions
5. Autophagy-Related Therapies for PD
5.1. Pharmacological Treatments
5.1.1. mTOR-Dependent Drugs
5.1.2. mTOR-Independent Drugs
5.2. Gene Therapies
5.3. Biotechnological Strategy
5.4. Drugs Currently in Clinical Trials
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236; discussion 222. [Google Scholar] [CrossRef] [PubMed]
- Ehringer, H.; Hornykiewicz, O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin. Wochenschr. 1960, 38, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.L.; Ng, L.K.; Chase, T.N. Long-lasting dyskinesia induced by levodopa. Lancet 1971, 1, 1016–1017. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Jenner, P.; Chen, S.D. Monoamine Oxidase-B Inhibitors for the Treatment of Parkinson’s Disease: Past, Present, and Future. J. Parkinsons Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef]
- Fabbri, M.; Ferreira, J.J.; Rascol, O. COMT Inhibitors in the Management of Parkinson’s Disease. CNS Drugs 2022, 36, 261–282. [Google Scholar] [CrossRef]
- Limousin, P.; Foltynie, T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat. Rev. Neurol. 2019, 15, 234–242. [Google Scholar] [CrossRef]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinsons Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Aubignat, M.; Tir, M.; Krystkowiak, P. Non-motor symptoms of Parkinson’s disease from pathophysiology to early diagnosis. Rev. Med. Interne 2021, 42, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and non-motor circuit disturbances in early Parkinson disease: Which happens first? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Costa, R.M. Networking brainstem and basal ganglia circuits for movement. Nat. Rev. Neurosci. 2022, 23, 342–360. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Gai, W.P.; Yuan, H.X.; Li, X.Q.; Power, J.T.; Blumbergs, P.C.; Jensen, P.H. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Plowey, E.D.; Cherra, S.J., 3rd; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Takao, M.; Aoyama, M.; Ishikawa, K.; Sakiyama, Y.; Yomono, H.; Saito, Y.; Kurisaki, H.; Mihara, B.; Murayama, S. Spinocerebellar ataxia type 2 is associated with Parkinsonism and Lewy body pathology. BMJ Case Rep. 2011, 2011. [Google Scholar] [CrossRef]
- Marcelo, A.; Afonso, I.T.; Afonso-Reis, R.; Brito, D.V.C.; Costa, R.G.; Rosa, A.; Alves-Cruzeiro, J.; Ferreira, B.; Henriques, C.; Nobre, R.J.; et al. Autophagy in Spinocerebellar ataxia type 2, a dysregulated pathway, and a target for therapy. Cell Death Dis. 2021, 12, 1117. [Google Scholar] [CrossRef]
- Grotzsch, H.; Pizzolato, G.P.; Ghika, J.; Schorderet, D.; Vingerhoets, F.J.; Landis, T.; Burkhard, P.R. Neuropathology of a case of dopa-responsive dystonia associated with a new genetic locus, DYT14. Neurology 2002, 58, 1839–1842. [Google Scholar] [CrossRef]
- Cookson, M.R.; Lockhart, P.J.; McLendon, C.; O’Farrell, C.; Schlossmacher, M.; Farrer, M.J. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum. Mol. Genet. 2003, 12, 2957–2965. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Muqit, M.M.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhaes, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef]
- Chien, H.F.; Rodriguez, R.D.; Bonifati, V.; Nitrini, R.; Pasqualucci, C.A.; Gelpi, E.; Barbosa, E.R. Neuropathologic Findings in a Patient With Juvenile-Onset Levodopa-Responsive Parkinsonism Due to ATP13A2 Mutation. Neurology 2021, 97, 763–766. [Google Scholar] [CrossRef]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Mishima, T.; Fujioka, S. Perry Disease: Concept of a New Disease and Clinical Diagnostic Criteria. J. Mov. Disord. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Ishikawa, K.I.; Saiki, S.; Furuya, N.; Imamichi, Y.; Tsuboi, Y.; Hattori, N. p150(glued) deficiency impairs effective fusion between autophagosomes and lysosomes due to their redistribution to the cell periphery. Neurosci. Lett. 2019, 690, 181–187. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef]
- Ramirez-Valle, F.; Braunstein, S.; Zavadil, J.; Formenti, S.C.; Schneider, R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008, 181, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Muller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue-Gervais, I.G.; Doiron, K.; Champagne, C.; Mayes, L.; Leiva-Torres, G.A.; Vanie, P., Jr.; Douglas, T.; Vidal, S.M.; Alnemri, E.S.; Saleh, M. The mitochondrial protease HtrA2 restricts the NLRP3 and AIM2 inflammasomes. Sci. Rep. 2018, 8, 8446. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Kuru, S.; Yoshida, M.; Tatsumi, S.; Mimuro, M. Immunohistochemical localization of spatacsin in alpha-synucleinopathies. Neuropathology 2014, 34, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Nishioka, K.; Meng, H.; Takanashi, M.; Hasegawa, I.; Inoshita, T.; Shiba-Fukushima, K.; Li, Y.; Yoshino, H.; Mori, A.; et al. Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum. Mol. Genet. 2019, 28, 3895–3911. [Google Scholar] [CrossRef]
- Quadri, M.; Mandemakers, W.; Grochowska, M.M.; Masius, R.; Geut, H.; Fabrizio, E.; Breedveld, G.J.; Kuipers, D.; Minneboo, M.; Vergouw, L.J.M.; et al. LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: A genome-wide linkage and sequencing study. Lancet Neurol. 2018, 17, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.R.; Sim, J.C.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef]
- Niu, M.; Zheng, N.; Wang, Z.; Gao, Y.; Luo, X.; Chen, Z.; Fu, X.; Wang, Y.; Wang, T.; Liu, M.; et al. RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 2020, 8, 598622. [Google Scholar] [CrossRef] [PubMed]
- Arasaratnam, C.J.; Singh-Bains, M.K.; Waldvogel, H.J.; Faull, R.L.M. Neuroimaging and neuropathology studies of X-linked dystonia parkinsonism. Neurobiol. Dis. 2021, 148, 105186. [Google Scholar] [CrossRef]
- Deng, H.X.; Shi, Y.; Yang, Y.; Ahmeti, K.B.; Miller, N.; Huang, C.; Cheng, L.; Zhai, H.; Deng, S.; Nuytemans, K.; et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 2016, 48, 733–739. [Google Scholar] [CrossRef]
- Kim, M.J.; Deng, H.X.; Wong, Y.C.; Siddique, T.; Krainc, D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 2017, 26, 729–741. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal Dysfunction and alpha-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Arotcarena, M.L.; Teil, M.; Dehay, B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef]
- Bourdenx, M.; Dehay, B. What lysosomes actually tell us about Parkinson’s disease? Ageing Res. Rev. 2016, 32, 140–149. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Eskelinen, E.L. The vacuole versus the lysosome: When size matters. Autophagy 2014, 10, 185–187. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagne, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Guissart, C.; Olahova, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A.; et al. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N. Engl. J. Med. 2021, 384, 2406–2417. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef]
- van der Vaart, A.; Griffith, J.; Reggiori, F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2010, 21, 2270–2284. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Gubas, A.; Dikic, I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022, 289, 75–89. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Biol. 2017, 96, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Manjithaya, R.; Nazarko, T.Y.; Farre, J.C.; Subramani, S. Molecular mechanism and physiological role of pexophagy. FEBS Lett. 2010, 584, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef]
- Otto, F.B.; Thumm, M. Mechanistic dissection of macro- and micronucleophagy. Autophagy 2021, 17, 626–639. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Mesquita, A.; Glenn, J.; Jenny, A. Differential activation of eMI by distinct forms of cellular stress. Autophagy 2021, 17, 1828–1840. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; International Parkinson’s Disease Genomics Consortium; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, F.; Mueller, S.H.; Szymczak, S.; Junge, O.; Tittmann, L.; May, S.; Lohmann, K.; Grallert, H.; Lieb, W.; Strauch, K.; et al. Rare Variants in Specific Lysosomal Genes Are Associated With Parkinson’s Disease. Mov. Disord. 2020, 35, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Smolders, S.; Van den Haute, C.; Heeman, B.; van Veen, S.; Crosiers, D.; Beletchi, I.; Verstraeten, A.; Gossye, H.; Gelders, G.; et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 2020, 139, 1001–1024. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Liu, H.; Koros, C.; Strohaker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibanez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Tang, B.; Xiong, H.; Sun, P.; Zhang, Y.; Wang, D.; Hu, Z.; Zhu, Z.; Ma, H.; Pan, Q.; Xia, J.H.; et al. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum. Mol. Genet. 2006, 15, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mamais, A.; Kluss, J.H.; Bonet-Ponce, L.; Landeck, N.; Langston, R.G.; Smith, N.; Beilina, A.; Kaganovich, A.; Ghosh, M.C.; Pellegrini, L.; et al. Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia. PLoS Biol. 2021, 19, e3001480. [Google Scholar] [CrossRef] [PubMed]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B. Altered alpha-synuclein homeostasis causing Parkinson’s disease: The potential roles of dardarin. Trends Neurosci. 2005, 28, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Rios, P.; Romo-Lozano, M.; Madero-Perez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef]
- Wulansari, N.; Darsono, W.H.W.; Woo, H.J.; Chang, M.Y.; Kim, J.; Bae, E.J.; Sun, W.; Lee, J.H.; Cho, I.J.; Shin, H.; et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv. 2021, 7, eabb1540. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Jinn, S.; Blauwendraat, C.; Toolan, D.; Gretzula, C.A.; Drolet, R.E.; Smith, S.; Nalls, M.A.; Marcus, J.; Singleton, A.B.; Stone, D.J. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet. 2019, 28, 3244–3254. [Google Scholar] [CrossRef]
- Wie, J.; Liu, Z.; Song, H.; Tropea, T.F.; Yang, L.; Wang, H.; Liang, Y.; Cang, C.; Aranda, K.; Lohmann, J.; et al. A growth-factor-activated lysosomal K(+) channel regulates Parkinson’s pathology. Nature 2021, 591, 431–437. [Google Scholar] [CrossRef]
- Hu, M.; Li, P.; Wang, C.; Feng, X.; Geng, Q.; Chen, W.; Marthi, M.; Zhang, W.; Gao, C.; Reid, W.; et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 2022, 185, 2292–2308.e2220. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Lin, B.; Zeng, W.; Fan, C.; Wu, H.; Ge, Y.; Li, Q.; Li, C.; Wei, Y.; Xin, J.; et al. Lysosomal K(+) channel TMEM175 promotes apoptosis and aggravates symptoms of Parkinson’s disease. EMBO Rep. 2022, 23, e53234. [Google Scholar] [CrossRef] [PubMed]
- Krohn, L.; Ozturk, T.N.; Vanderperre, B.; Ouled Amar Bencheikh, B.; Ruskey, J.A.; Laurent, S.B.; Spiegelman, D.; Postuma, R.B.; Arnulf, I.; Hu, M.T.M.; et al. Genetic, Structural, and Functional Evidence Link TMEM175 to Synucleinopathies. Ann. Neurol. 2020, 87, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Palomba, N.P.; Fortunato, G.; Pepe, G.; Modugno, N.; Pietracupa, S.; Damiano, I.; Mascio, G.; Carrillo, F.; Di Giovannantonio, L.G.; Ianiro, L.; et al. Common and Rare Variants in TMEM175 Gene Concur to the Pathogenesis of Parkinson’s Disease in Italian Patients. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Pan, P.Y.; Sheehan, P.; Wang, Q.; Zhu, X.; Zhang, Y.; Choi, I.; Li, X.; Saenz, J.; Zhu, J.; Wang, J.; et al. Synj1 haploinsufficiency causes dopamine neuron vulnerability and alpha-synuclein accumulation in mice. Hum. Mol. Genet. 2020, 29, 2300–2312. [Google Scholar] [CrossRef]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar] [CrossRef]
- Tu, H.Y.; Yuan, B.S.; Hou, X.O.; Zhang, X.J.; Pei, C.S.; Ma, Y.T.; Yang, Y.P.; Fan, Y.; Qin, Z.H.; Liu, C.F.; et al. alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef]
- Cataldi, S.; Follett, J.; Fox, J.D.; Tatarnikov, I.; Kadgien, C.; Gustavsson, E.K.; Khinda, J.; Milnerwood, A.J.; Farrer, M.J. Altered dopamine release and monoamine transporters in Vps35 p.D620N knock-in mice. NPJ Parkinsons Dis. 2018, 4, 27. [Google Scholar] [CrossRef]
- Niu, M.; Zhao, F.; Bondelid, K.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Liu, J.; Zhu, X. VPS35 D620N knockin mice recapitulate cardinal features of Parkinson’s disease. Aging Cell 2021, 20, e13347. [Google Scholar] [CrossRef]
- Ramonet, D.; Daher, J.P.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 2011, 6, e18568. [Google Scholar] [CrossRef]
- Arbez, N.; He, X.; Huang, Y.; Ren, M.; Liang, Y.; Nucifora, F.C.; Wang, X.; Pei, Z.; Tessarolo, L.; Smith, W.W.; et al. G2019S-LRRK2 mutation enhances MPTP-linked Parkinsonism in mice. Hum. Mol. Genet. 2020, 29, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Gimenez, J.; Cuadros, T.; Bove, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Penuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Wray, S. Modelling neurodegenerative disease using brain organoids. Semin. Cell Dev. Biol. 2021, 111, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wey, M.C.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.F.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 2015, 5, 28743. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Masini, D.; Bonito-Oliva, A.; Bertho, M.; Fisone, G. Inhibition of mTORC1 Signaling Reverts Cognitive and Affective Deficits in a Mouse Model of Parkinson’s Disease. Front. Neurol. 2018, 9, 208. [Google Scholar] [CrossRef]
- Saiki, S.; Sasazawa, Y.; Imamichi, Y.; Kawajiri, S.; Fujimaki, T.; Tanida, I.; Kobayashi, H.; Sato, F.; Sato, S.; Ishikawa, K.; et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176–187. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef]
- Zia, A.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Samarghandian, S. The role of curcumin in aging and senescence: Molecular mechanisms. Biomed. Pharmacother. 2021, 134, 111119. [Google Scholar] [CrossRef]
- Lee, J.H.; Rao, M.V.; Yang, D.S.; Stavrides, P.; Im, E.; Pensalfini, A.; Huo, C.; Sarkar, P.; Yoshimori, T.; Nixon, R.A. Transgenic expression of a ratiometric autophagy probe specifically in neurons enables the interrogation of brain autophagy in vivo. Autophagy 2019, 15, 543–557. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Y.; Yu, X.; Lu, J.; Jia, W.; Song, J.; Liu, L.; Wang, Y.; Huang, Y.; Xie, J.; et al. Corynoxine Protects Dopaminergic Neurons Through Inducing Autophagy and Diminishing Neuroinflammation in Rotenone-Induced Animal Models of Parkinson’s Disease. Front. Pharmacol. 2021, 12, 642900. [Google Scholar] [CrossRef] [PubMed]
- Oleksak, P.; Nepovimova, E.; Chrienova, Z.; Musilek, K.; Patocka, J.; Kuca, K. Contemporary mTOR inhibitor scaffolds to diseases breakdown: A patent review (2015–2021). Eur. J. Med. Chem. 2022, 238, 114498. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lu, Y.; Zhang, Q.; Liu, W.; Yang, R.; Jiao, J.; Liu, J.; Gao, G.; Yang, H. Piperine promotes autophagy flux by P2RX4 activation in SNCA/alpha-synuclein-induced Parkinson disease model. Autophagy 2022, 18, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Cui, H.; Kilpelainen, T.; Zouzoula, L.; Auno, S.; Trontti, K.; Kurvonen, S.; Norrbacka, S.; Hovatta, I.; Jensen, P.H.; Myohanen, T.T. Prolyl oligopeptidase inhibition reduces alpha-synuclein aggregation in a cellular model of multiple system atrophy. J. Cell Mol. Med. 2021, 25, 9634–9646. [Google Scholar] [CrossRef]
- Rostami, J.; Jantti, M.; Cui, H.; Rinne, M.K.; Kukkonen, J.P.; Falk, A.; Erlandsson, A.; Myohanen, T. Prolyl oligopeptidase inhibition by KYP-2407 increases alpha-synuclein fibril degradation in neuron-like cells. Biomed. Pharmacother. 2020, 131, 110788. [Google Scholar] [CrossRef]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Mannisto, P.T.; Maguire-Zeiss, K.A.; Myohanen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Yang, G.; Li, J.; Cai, Y.; Yang, Z.; Li, R.; Fu, W. Glycyrrhizic Acid Alleviates 6-Hydroxydopamine and Corticosterone-Induced Neurotoxicity in SH-SY5Y Cells Through Modulating Autophagy. Neurochem. Res. 2018, 43, 1914–1926. [Google Scholar] [CrossRef]
- Santoro, M.; Maetzler, W.; Stathakos, P.; Martin, H.L.; Hobert, M.A.; Rattay, T.W.; Gasser, T.; Forrester, J.V.; Berg, D.; Tracey, K.J.; et al. In-vivo evidence that high mobility group box 1 exerts deleterious effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model and Parkinson’s disease which can be attenuated by glycyrrhizin. Neurobiol. Dis. 2016, 91, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Kwon, Y.; Huh, Y.E.; Choi, H.J. Trehalose ameliorates prodromal non-motor deficits and aberrant protein accumulation in a rotenone-induced mouse model of Parkinson’s disease. Arch. Pharm. Res. 2022, 45, 417–432. [Google Scholar] [CrossRef]
- Howson, P.A.; Johnston, T.H.; Ravenscroft, P.; Hill, M.P.; Su, J.; Brotchie, J.M.; Koprich, J.B. Beneficial Effects of Trehalose on Striatal Dopaminergic Deficits in Rodent and Primate Models of Synucleinopathy in Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2019, 369, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Kakoty, V.; K, C.S.; Dubey, S.K.; Yang, C.H.; Taliyan, R. Neuroprotective Effects of Trehalose and Sodium Butyrate on Preformed Fibrillar Form of alpha-Synuclein-Induced Rat Model of Parkinson’s Disease. ACS Chem. Neurosci. 2021, 12, 2643–2660. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.; Gaubert, A.; Verget, J.; Thiolat, M.L.; Barthelemy, P.; Latxague, L.; Dehay, B. Trehalose-Based Nucleolipids as Nanocarriers for Autophagy Modulation: An In Vitro Study. Pharmaceutics 2022, 14, 857. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef]
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Cherif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol. Cell 2017, 65, 917–931.e916. [Google Scholar] [CrossRef]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Arotcarena, M.L.; Bourdenx, M.; Dutheil, N.; Thiolat, M.L.; Doudnikoff, E.; Dovero, S.; Ballabio, A.; Fernagut, P.O.; Meissner, W.G.; Bezard, E.; et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight 2019, 4, e129719. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef]
- Cunha, A.; Gaubert, A.; Latxague, L.; Dehay, B. PLGA-Based Nanoparticles for Neuroprotective Drug Delivery in Neurodegenerative Diseases. Pharmaceutics 2021, 13, 1042. [Google Scholar] [CrossRef]
- Brouillard, M.; Barthelemy, P.; Dehay, B.; Crauste-Manciet, S.; Desvergnes, V. Nucleolipid Acid-Based Nanocarriers Restore Neuronal Lysosomal Acidification Defects. Front. Chem. 2021, 9, 736554. [Google Scholar] [CrossRef]
- Baltazar, G.C.; Guha, S.; Lu, W.; Lim, J.; Boesze-Battaglia, K.; Laties, A.M.; Tyagi, P.; Kompella, U.B.; Mitchell, C.H. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS ONE 2012, 7, e49635. [Google Scholar] [CrossRef]
- Lee, H.J.; Han, J.; Jang, Y.; Kim, S.J.; Park, J.H.; Seo, K.S.; Jeong, S.; Shin, S.; Lim, K.; Heo, J.Y.; et al. Docosahexaenoic acid prevents paraquat-induced reactive oxygen species production in dopaminergic neurons via enhancement of glutathione homeostasis. Biochem. Biophys. Res. Commun. 2015, 457, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Prevot, G.; Soria, F.N.; Thiolat, M.L.; Daniel, J.; Verlhac, J.B.; Blanchard-Desce, M.; Bezard, E.; Barthelemy, P.; Crauste-Manciet, S.; Dehay, B. Harnessing Lysosomal pH through PLGA Nanoemulsion as a Treatment of Lysosomal-Related Neurodegenerative Diseases. Bioconjug. Chem. 2018, 29, 4083–4089. [Google Scholar] [CrossRef] [PubMed]
- Senturk, M.; Cakir, M.; Tekin, A.; Kucukkartallar, T.; Yildirim, M.A.; Alkan, S.; Findik, S. Comparison of primary repair and repair with polyglycolic acid coated tube in recurrent laryngeal nerve cuts (an experimental study). Am. J. Surg. 2020, 219, 632–636. [Google Scholar] [CrossRef] [PubMed]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Guha, S.; Liu, J.; Baltazar, G.; Laties, A.M.; Mitchell, C.H. Rescue of compromised lysosomes enhances degradation of photoreceptor outer segments and reduces lipofuscin-like autofluorescence in retinal pigmented epithelial cells. Adv. Exp. Med. Biol. 2014, 801, 105–111. [Google Scholar] [CrossRef]
- Xue, X.; Wang, L.R.; Sato, Y.; Jiang, Y.; Berg, M.; Yang, D.S.; Nixon, R.A.; Liang, X.J. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano Lett. 2014, 14, 5110–5117. [Google Scholar] [CrossRef]
- Zhang, X.; Misra, S.K.; Moitra, P.; Zhang, X.; Jeong, S.J.; Stitham, J.; Rodriguez-Velez, A.; Park, A.; Yeh, Y.S.; Gillanders, W.E.; et al. Use of acidic nanoparticles to rescue macrophage lysosomal dysfunction in atherosclerosis. Autophagy 2022, 1–18. [Google Scholar] [CrossRef]
- Sanchez-Mirasierra, I.; Ghimire, S.; Hernandez-Diaz, S.; Soukup, S.F. Targeting Macroautophagy as a Therapeutic Opportunity to Treat Parkinson’s Disease. Front. Cell Dev. Biol. 2022, 10, 1–18. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hallqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Saiki, H.; Hatano, T.; Gasser, T.; Isaacson, S.H.; Gaemers, S.J.M.; Minini, P.; Saubadu, S.; Sharma, J.; Walbillic, S.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial. J. Parkinsons Dis. 2022, 12, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.A.; Martinez, J.; Millar Vernetti, P.; Ma, T.; Perez, M.A.; Zhong, J.; Qian, Y.; Dutta, S.; Maina, K.N.; Siddique, I.; et al. mTOR Inhibition with Sirolimus in Multiple System Atrophy: A Randomized, Double-Blind, Placebo-Controlled Futility Trial and 1-Year Biomarker Longitudinal Analysis. Mov. Disord. 2022, 37, 778–789. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.; Zhu, L.; Chen, Y.; Liu, B. Unraveling the Roles of Protein Kinases in Autophagy: An Update on Small-Molecule Compounds for Targeted Therapy. J. Med. Chem. 2022, 65, 5870–5885. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Olopade, P.; Selby, S.T.; Pagan, F.; Moussa, C.E. Tyrosine Kinase Inhibition Regulates Early Systemic Immune Changes and Modulates the Neuroimmune Response in alpha-Synucleinopathy. J. Clin. Cell Immunol. 2014, 5, 259. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Arellano, J.; Howard, H.H.; et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol. Res. Perspect. 2019, 7, e00470. [Google Scholar] [CrossRef]
- Pagan, F.L.; Wilmarth, B.; Torres-Yaghi, Y.; Hebron, M.L.; Mulki, S.; Ferrante, D.; Matar, S.; Ahn, J.; Moussa, C. Long-Term Safety and Clinical Effects of Nilotinib in Parkinson’s Disease. Mov. Disord. 2021, 36, 740–749. [Google Scholar] [CrossRef]
- Xie, X.; Yuan, P.; Kou, L.; Chen, X.; Li, J.; Li, Y. Nilotinib in Parkinson’s disease: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 996217. [Google Scholar] [CrossRef]
- Xu, Y.; Du, S.; Marsh, J.A.; Horie, K.; Sato, C.; Ballabio, A.; Karch, C.M.; Holtzman, D.M.; Zheng, H. TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol. Psychiatry 2021, 26, 5925–5939. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Kim, M.Y.; Lee, S.Y.; Kim, M.J.; Hong, T.J.; Jeon, J.K.; Ganbat, D.; Kim, H.T.; Kim, S.S.; Kam, T.I.; et al. MEK1/2 inhibition rescues neurodegeneration by TFEB-mediated activation of autophagic lysosomal function in a model of Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 4770–4780. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Protein | Inheritance | LB | ALP Involvement |
|---|---|---|---|---|
| SNCA | α-synuclein | AD | Yes [5] | Yes [24] |
| LRRK2 | Leucine-rich repeat kinase 2 | AD/AR | Yes [25] | Yes [26] |
| VPS35 | Vacuolar protein sorting ortholog 35 | AD | Unknown | Yes [27] |
| ATXN2 | Ataxin 2 | AD | Yes [28] | Yes [29] |
| GCH1 | GTP cyckihydrolase 1 | AD | No [30] | Unknown |
| PRKN | Parkin RBR E3 ubiquitin protein ligase | AR | No [31] | Yes [32] |
| PINK1 | PTEN-induced putative kinase 1 | AR | Yes [33] | Yes [34] |
| PARK7 | Parkinsonism-associated deglycase/DJ1 | AR | Yes [35] | Yes [36] |
| ATP13A2 | ATPase cation transporting 13A2 | AR | No [37] | Yes [38] |
| DCTN1 | Dynactin subunit 1 | AD | No/few [39] | Yes [40] |
| DNAJC6 | DnaJ Heat Shock Protein Family (Hsp40) Member C6 | AR | Unknown | Yes [41] |
| DNAJC13 | DnaJ Heat Shock Protein Family (Hsp40) Member C13 | AD | Yes [42] | Yes [42] |
| EIF4G1 | Eukaryotic translation initiation factor 4G1 | AD | Yes [43] | Yes [44] |
| FBXO7 | F-Box Protein 7 | AR | Unknown | Yes [45] |
| HTRA2 | HTRA serine peptidase 2 | AD | Yes [46] | Yes [47] |
| PLA2G6 | Phospholipase A2 group 6 | AR | Yes [48] | Yes [49] |
| SYNJ1 | Synaptojanin 1 | AR | Unknown | Yes [50] |
| SPG11 | Spatacsin | AR | Yes [51] | Yes [52] |
| CHCHD2 | Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 2 | AD | Yes [53] | Yes [53] |
| LRP10 | LDL Receptor-Related Protein 10 | AD | Yes [54] | Unknown |
| RAB39B | Ras-related protein Rab-39B | XL-D | Yes [55] | Yes [56] |
| TAF1 | TATA-box binding protein associated factor 1 | XL-D | No [57] | Unknown |
| TMEM230 | Transmembrane Protein 230 | AD | Yes [58] | Yes [59] |
| UQCRC1 | Ubiquinol-Cytochrome C Reductase Core Protein 1 | AD | Unknown | Unknown |
| VPS13C | Vacuolar Protein Sorting 13 Homolog C | AR | Yes [60] | Yes [60] |
| TMEM175 | Endosomal/lysosomal proton channel TMEM175 | Unknown | Yes [61] | Yes [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinet, R.; Dehay, B. Pathogenic Aspects and Therapeutic Avenues of Autophagy in Parkinson’s Disease. Cells 2023, 12, 621. https://doi.org/10.3390/cells12040621
Kinet R, Dehay B. Pathogenic Aspects and Therapeutic Avenues of Autophagy in Parkinson’s Disease. Cells. 2023; 12(4):621. https://doi.org/10.3390/cells12040621
Chicago/Turabian StyleKinet, Rémi, and Benjamin Dehay. 2023. "Pathogenic Aspects and Therapeutic Avenues of Autophagy in Parkinson’s Disease" Cells 12, no. 4: 621. https://doi.org/10.3390/cells12040621
APA StyleKinet, R., & Dehay, B. (2023). Pathogenic Aspects and Therapeutic Avenues of Autophagy in Parkinson’s Disease. Cells, 12(4), 621. https://doi.org/10.3390/cells12040621