


Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas

 ,
,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Methods
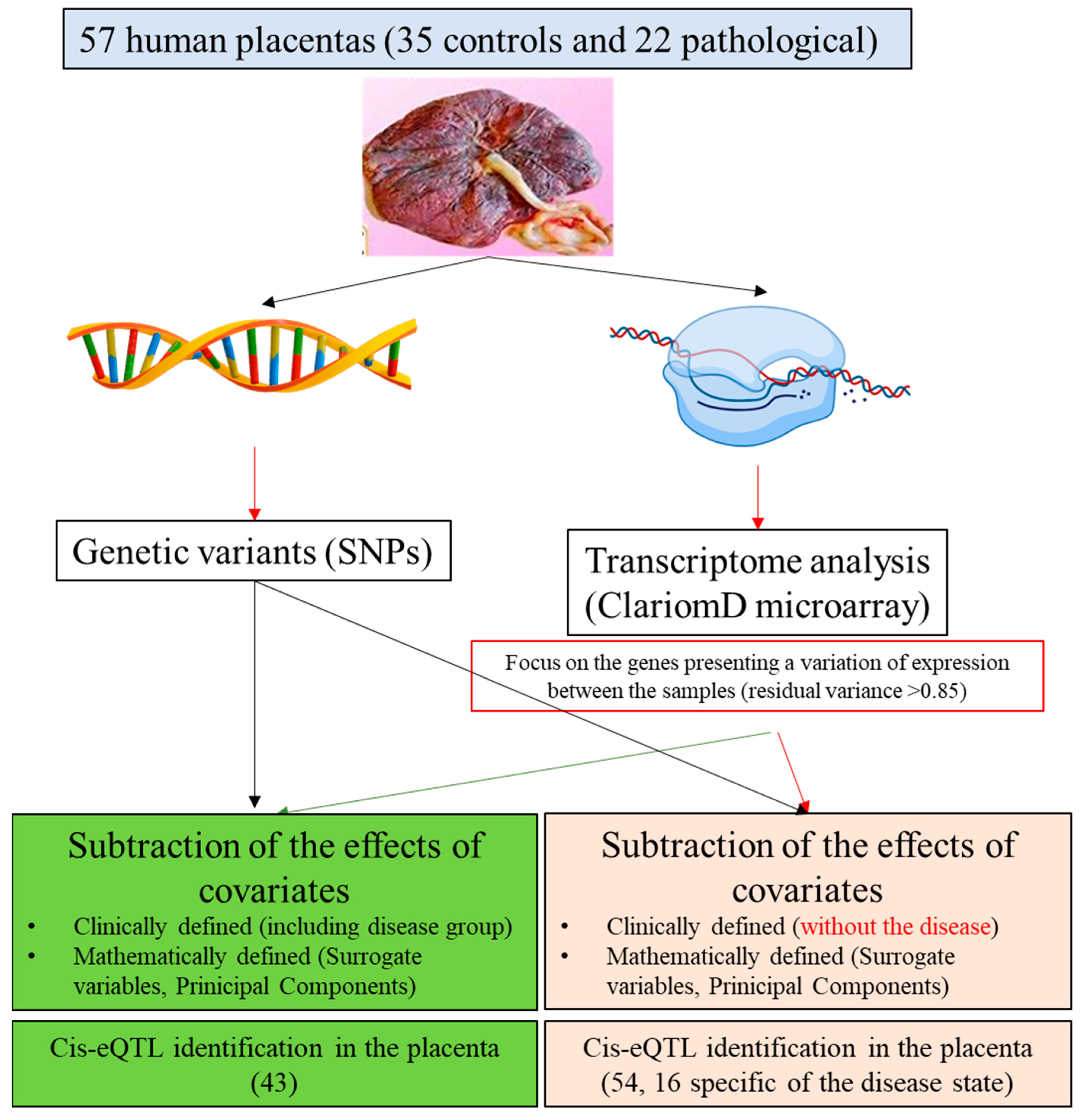
2.1. Summary of the Principle
2.2. Human Placental Samples
2.3. RNA and DNA Extraction
2.4. Transcriptomic Dataset
2.5. Genotype Dataset
2.6. eQTL Analysis Workflow
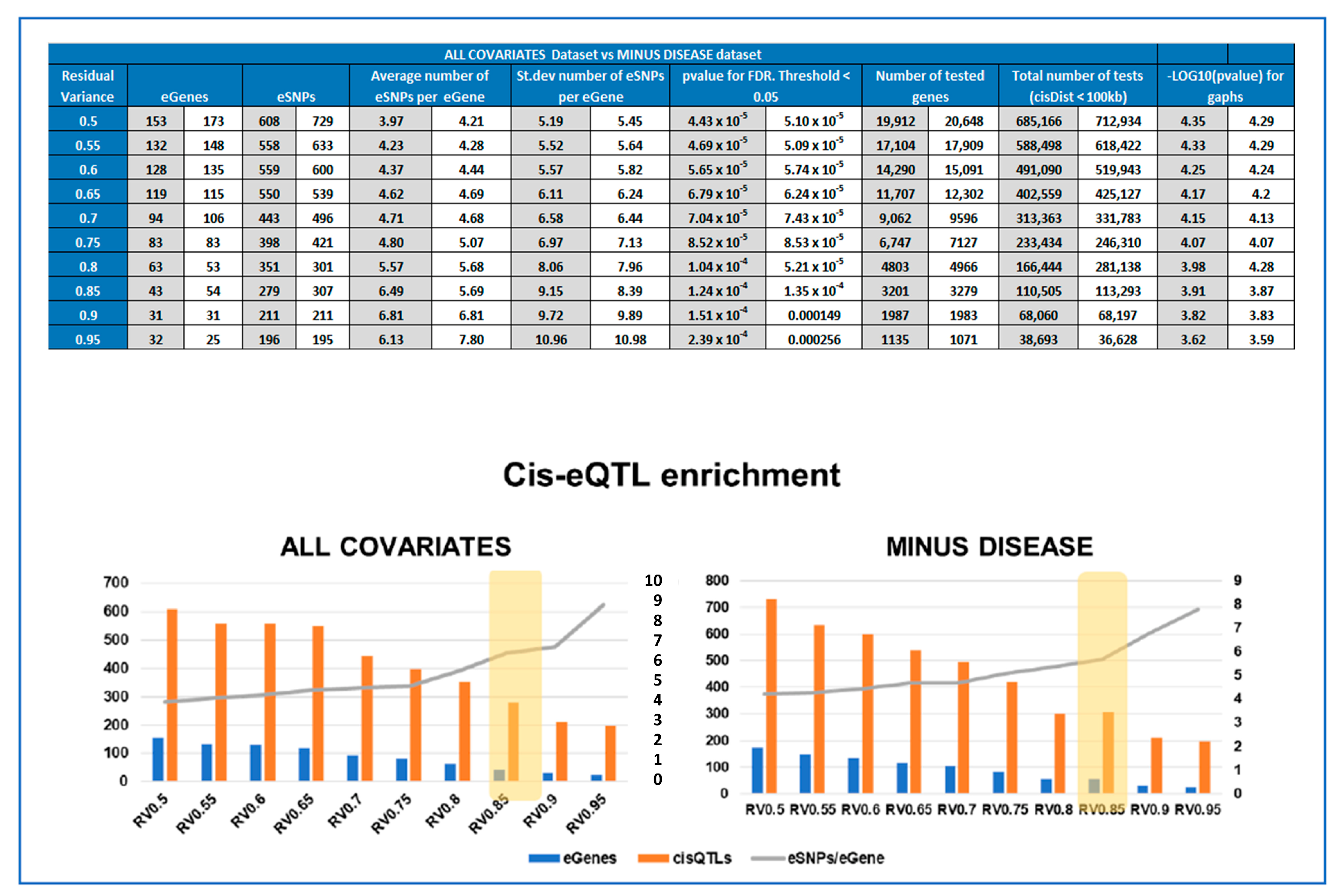
2.7. Cis-QTL Analyses of Gene Expression Subsets
2.8. Calculating Enrichment of Significant cis-QTLs for Each Subset
2.9. Calculating Overlap with Previous Studies
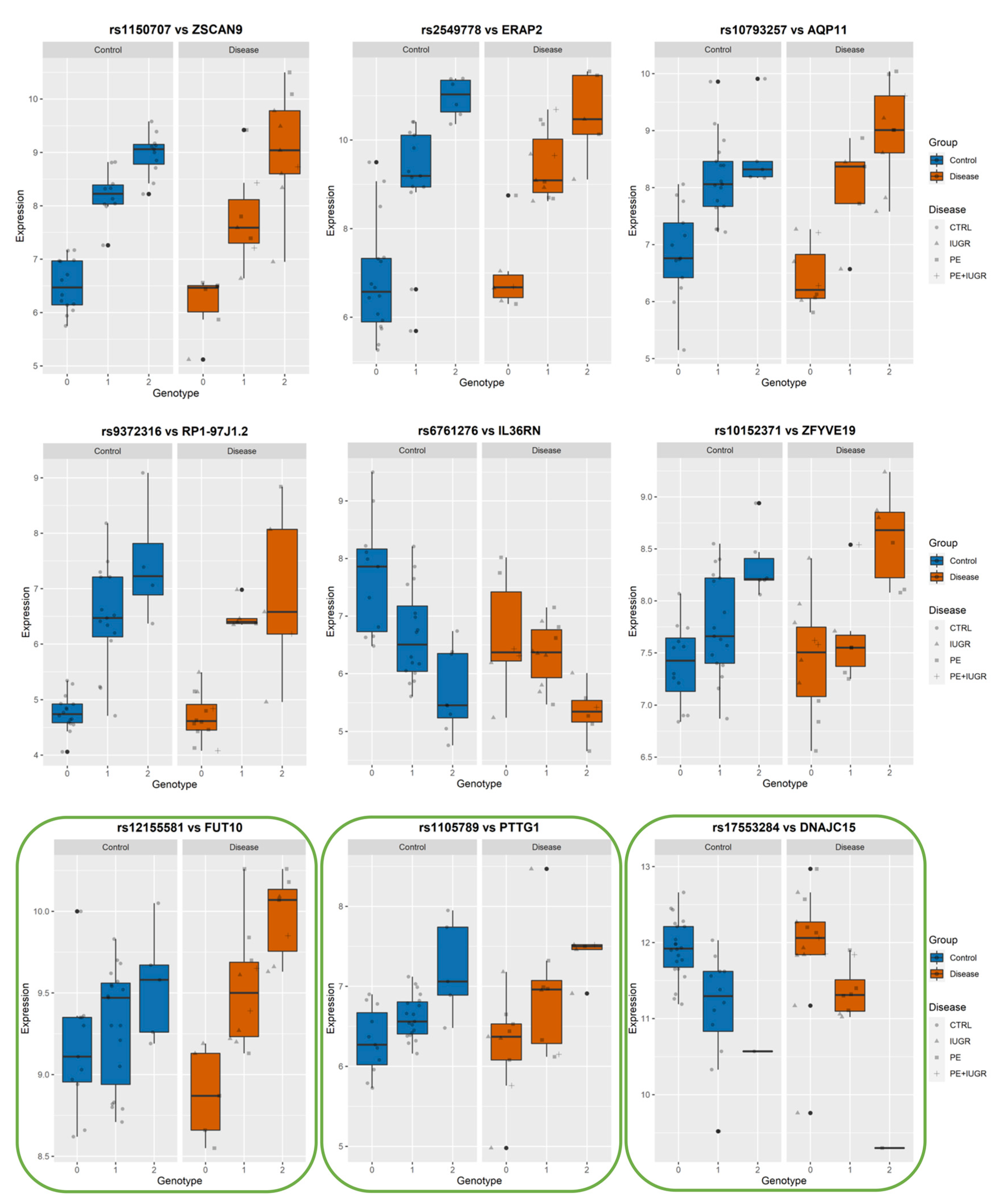
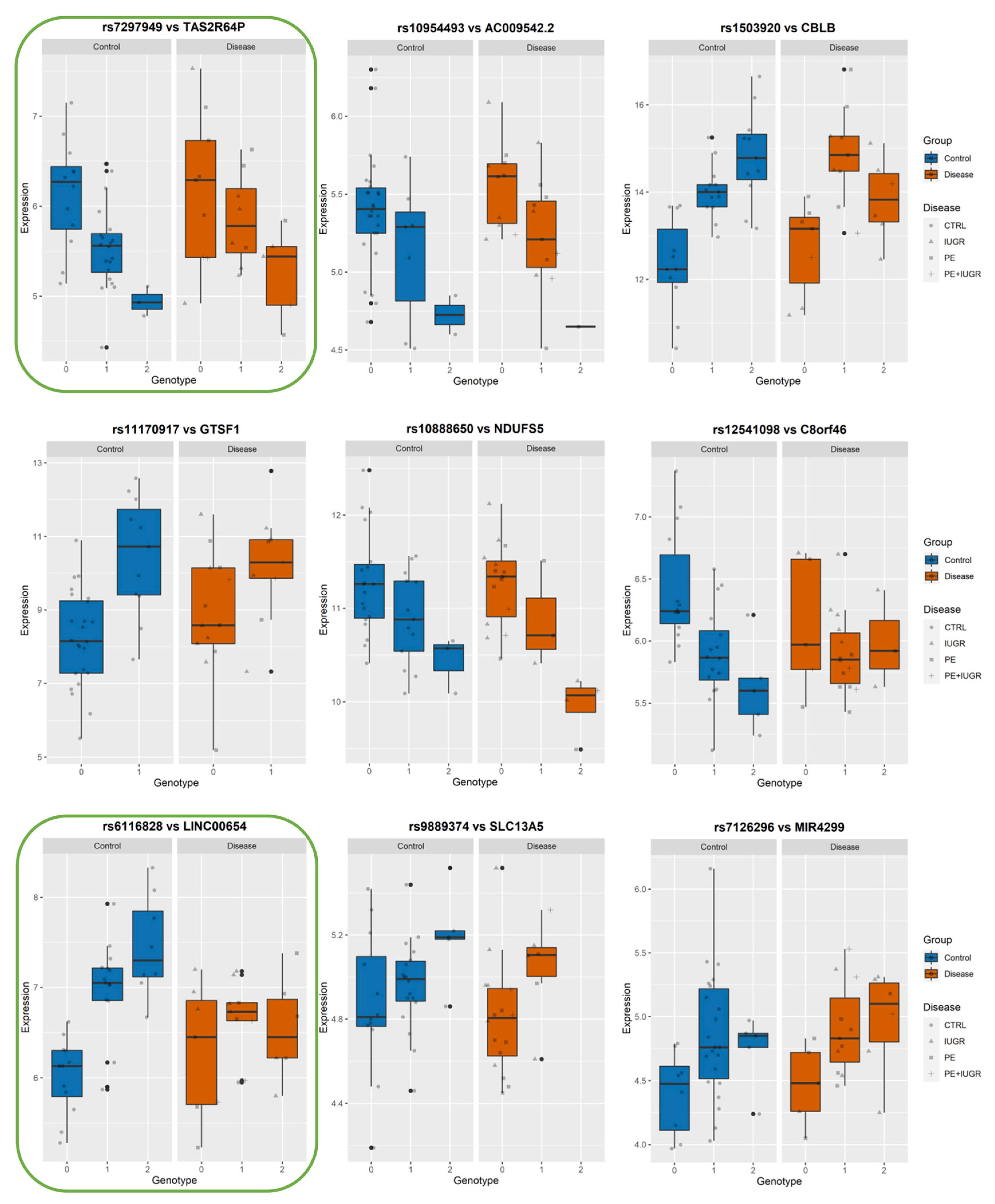
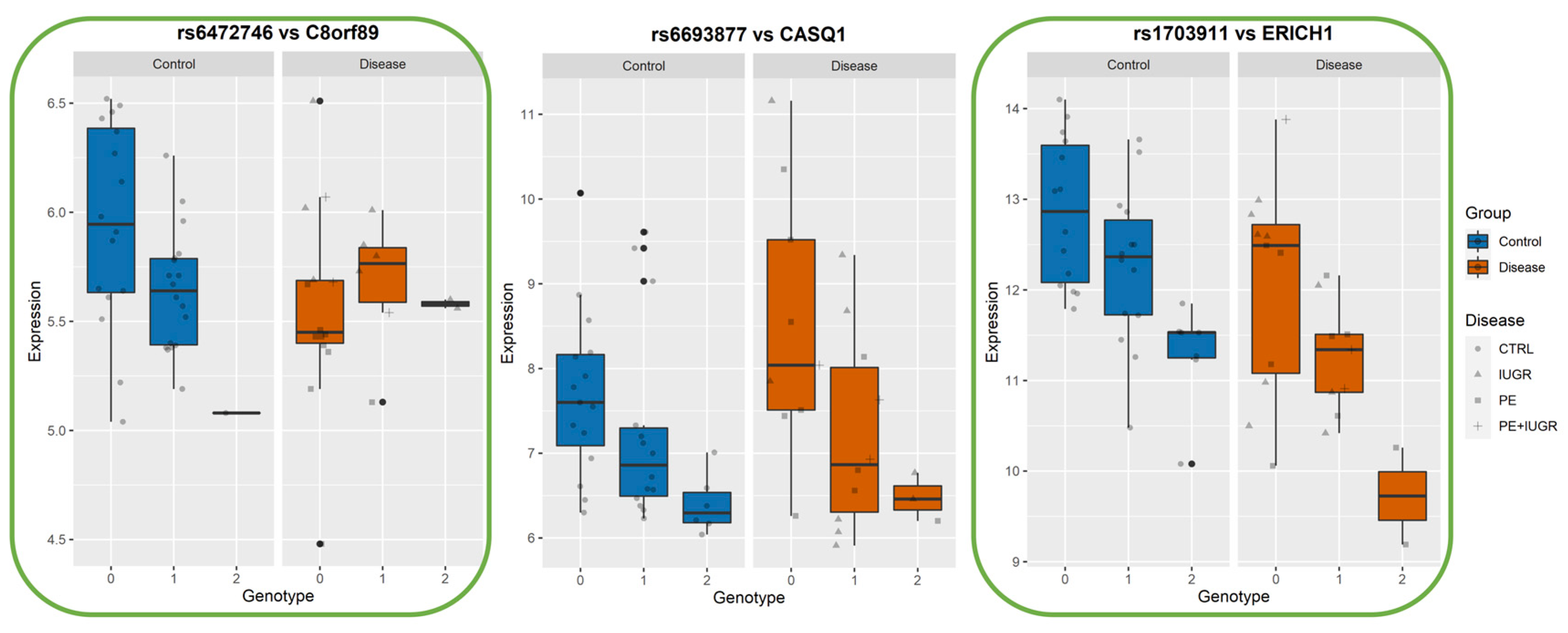
2.10. Calculating Statistical Significance of Interaction between Best-eSNP and Disease on eGene Gene Expression
3. Results
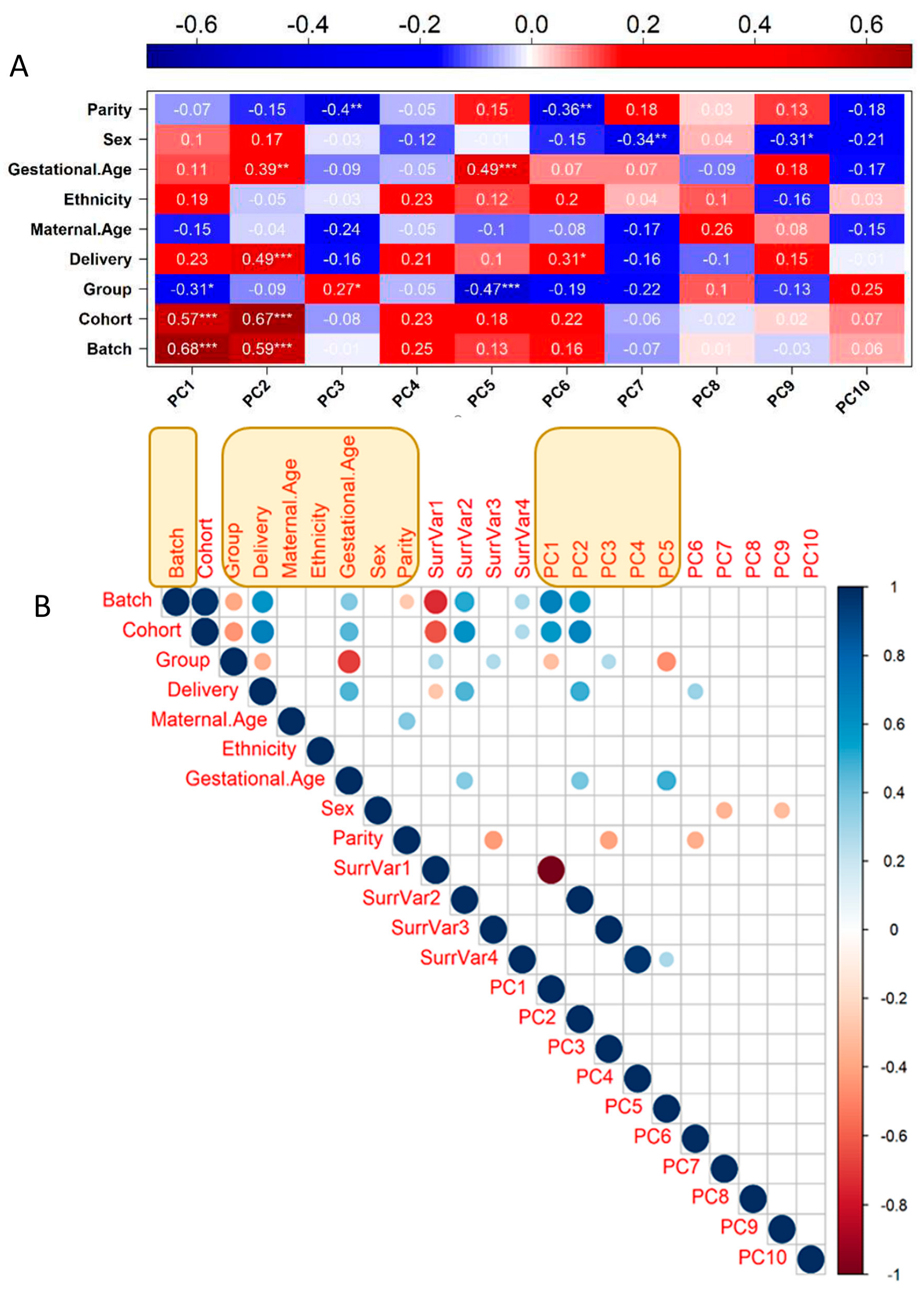
3.1. Transcriptome Identification of Confounding Variables
3.2. Genotyping and Population Stratification
3.3. Optimal Feature Selection for the eQTL Analysis
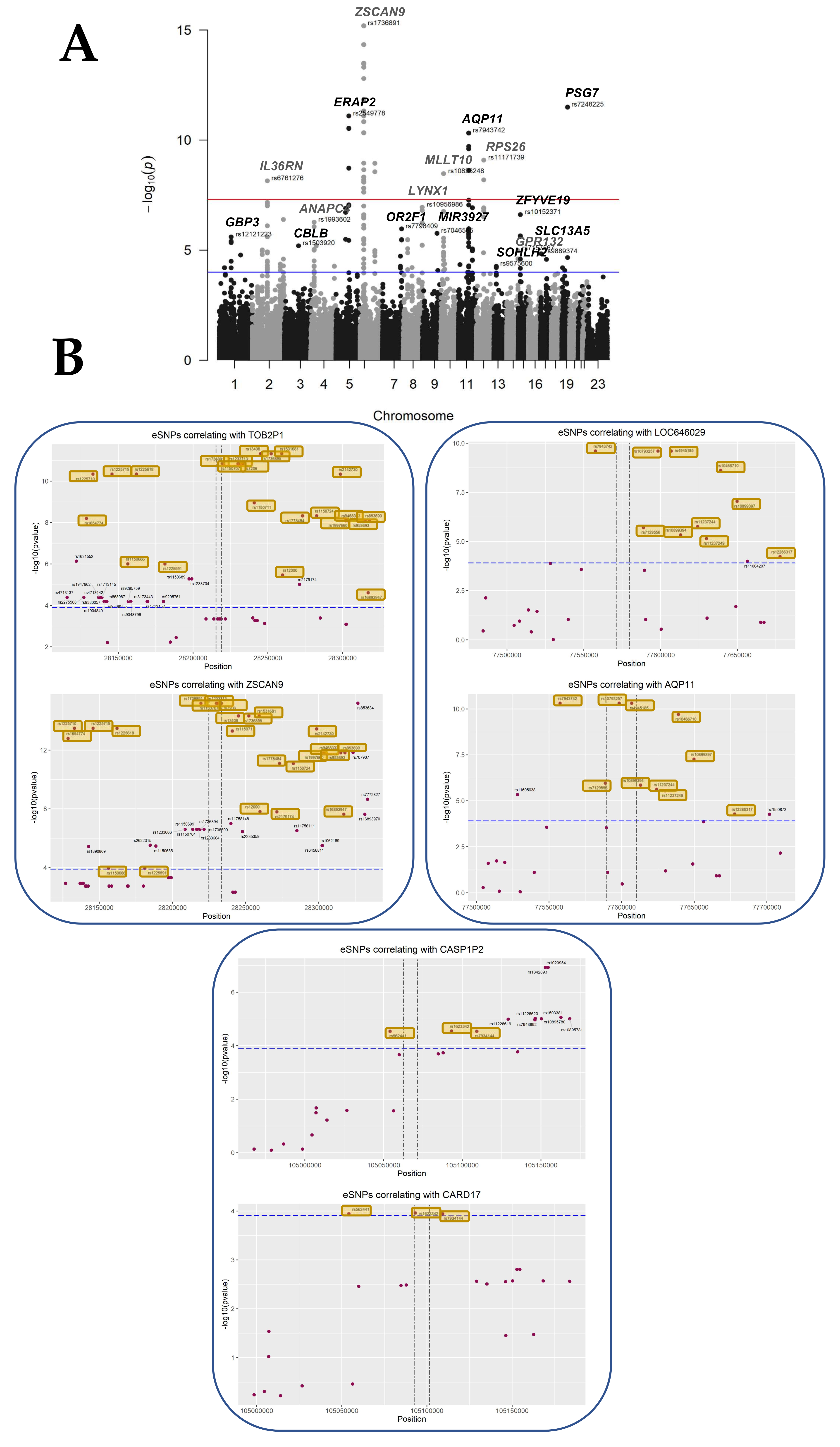
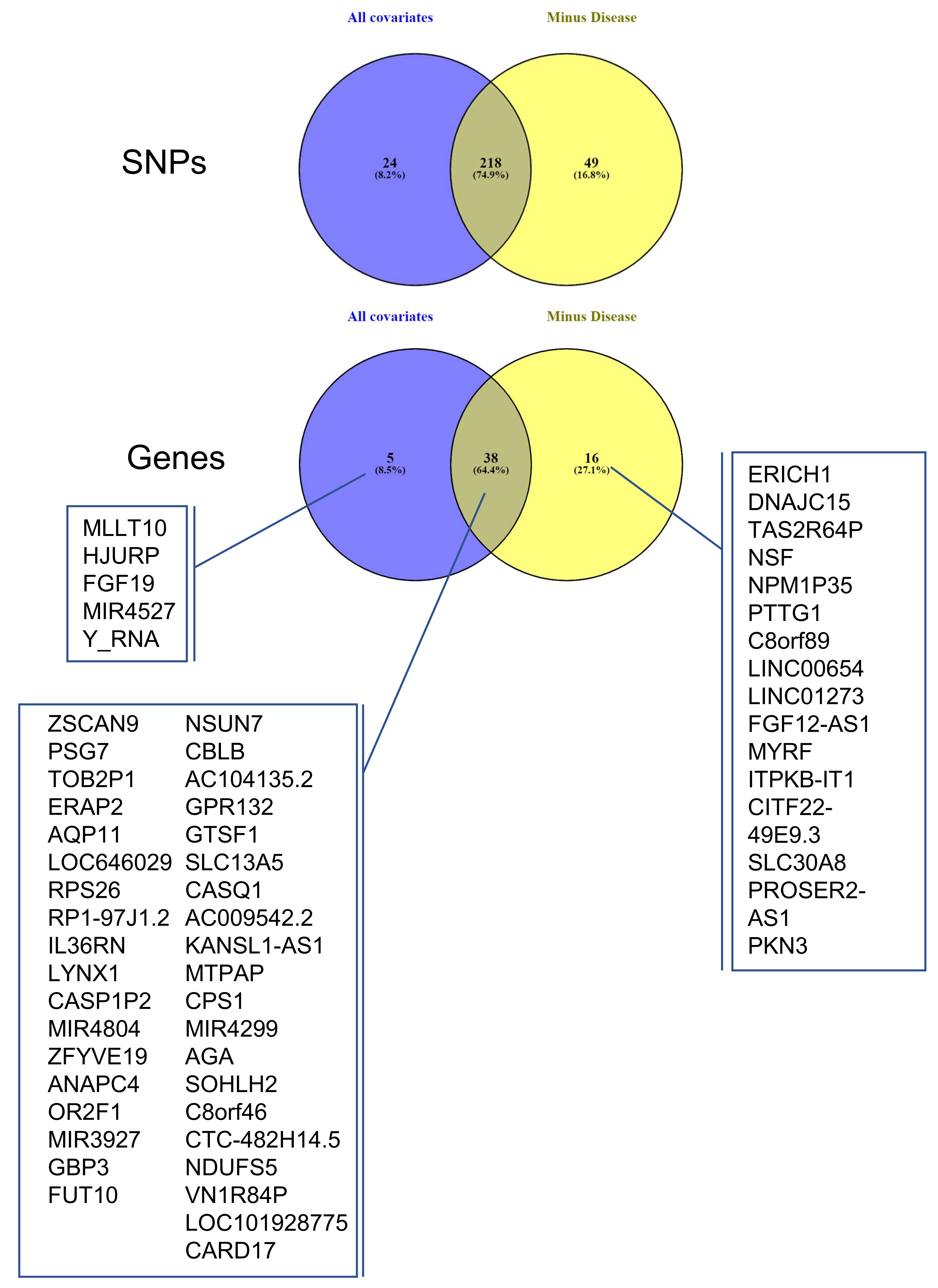
3.4. Identifying eQTLs Involved in the Disease by a Subtraction Strategy
4. Discussion
5. Study Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffith, O.W.; Chavan, A.R.; Protopapas, S.; Maziarz, J.; Romero, R.; Wagner, G.P. Embryo implantation evolved from an ancestral inflammatory attachment reaction. Proc. Natl. Acad. Sci. USA 2017, 114, E6566–E6575. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- Desoye, G.; Hauguel-de Mouzon, S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care 2007, 30 (Suppl. S2), S120–S126. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef]
- Madeleneau, D.; Buffat, C.; Mondon, F.; Grimault, H.; Rigourd, V.; Tsatsaris, V.; Letourneur, F.; Vaiman, D.; Barbaux, S.; Gascoin, G. Transcriptomic analysis of human placenta in intrauterine growth restriction. Pediatr. Res. 2015, 77, 799–807. [Google Scholar] [CrossRef]
- Delahaye, F.; Do, C.; Kong, Y.; Ashkar, R.; Salas, M.; Tycko, B.; Wapner, R.; Hughes, F. Genetic variants influence on the placenta regulatory landscape. PLoS Genet. 2018, 14, e1007785. [Google Scholar] [CrossRef]
- Kikas, T.; Rull, K.; Beaumont, R.N.; Freathy, R.M.; Laan, M. The Effect of Genetic Variation on the Placental Transcriptome in Humans. Front. Genet. 2019, 10, 550. [Google Scholar] [CrossRef]
- Peng, S.; Deyssenroth, M.A.; Di Narzo, A.F.; Lambertini, L.; Marsit, C.J.; Chen, J.; Hao, K. Expression quantitative trait loci (eQTLs) in human placentas suggest developmental origins of complex diseases. Hum. Mol. Genet. 2017, 26, 3432–3441. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Bergstedt, J.; Azzou, S.A.K.; Tsuo, K.; Jaquaniello, A.; Urrutia, A.; Rotival, M.; Lin, D.T.S.; MacIsaac, J.L.; Kobor, M.S.; Albert, M.L.; et al. The immune factors driving DNA methylation variation in human blood. Nat. Commun. 2022, 13, 5895. [Google Scholar] [CrossRef]
- Ruano, C.S.M.; Apicella, C.; Jacques, S.; Gascoin, G.; Gaspar, C.; Miralles, F.; Méhats, C.; Vaiman, D. Alternative splicing in normal and pathological human placentas is correlated to genetic variants. Hum. Genet. 2021, 140, 827–848. [Google Scholar] [CrossRef]
- Chabrun, F.; Huetz, N.; Dieu, X.; Rousseau, G.; Bouzille, G.; Chao de la Barca, J.M.; Procaccio, V.; Lenaers, G.; Blanchet, O.; Legendre, G.; et al. Data-Mining Approach on Transcriptomics and Methylomics Placental Analysis Highlights Genes in Fetal Growth Restriction. Front. Genet. 2019, 10, 1292. [Google Scholar] [CrossRef]
- Gascoin-Lachambre, G.; Buffat, C.; Rebourcet, R.; Chelbi, S.T.; Rigourd, V.; Mondon, F.; Mignot, T.M.; Legras, E.; Simeoni, U.; Vaiman, D.; et al. Cullins in human intra-uterine growth restriction: Expressional and epigenetic alterations. Placenta 2010, 31, 151–157. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2018, 13, 291–310. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Lupattelli, A.; Wood, M.E.; Nordeng, H. Analyzing Missing Data in Perinatal Pharmacoepidemiology Research: Methodological Considerations to Limit the Risk of Bias. Clin. Ther. 2019, 41, 2477–2487. [Google Scholar] [CrossRef]
- Blighe, K.; Lun, A. PCAtools: PCAtools: Everything Principal Components Analysis [WWW Document]. R Packag. Version 2.4.0. 2021. Available online: https://github.com/kevinblighe/PCAtools (accessed on 9 January 2021).
- Tabachnick, B.G.; Fidell, L.S. Using Multivariate Statistics, 7th ed.; Pearson: Boston, MA, USA, 2019. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Listgarten, J.; Kadie, C.; Schadt, E.E.; Heckerman, D. Correction for hidden confounders in the genetic analysis of gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 16465–16470. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.; Kumasaka, N.; De Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA landscape of the human placenta in health and disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef]
- Willyard, C. New human gene tally reignites debate. Nature 2018, 558, 354–355. [Google Scholar] [CrossRef]
- Kikas, T.; Inno, R.; Ratnik, K.; Rull, K.; Laan, M. C-allele of rs4769613 Near FLT1 Represents a High-Confidence Placental Risk Factor for Preeclampsia. Hypertension 2020, 76, 884–891. [Google Scholar] [CrossRef]
- Yan, Y.H.; Yi, P.; Zheng, Y.R.; Yu, L.L.; Han, J.; Han, X.M.; Li, L. Screening for preeclampsia pathogenesis related genes. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 3083–3094. [Google Scholar]
- May, A.P.; Whiteheart, S.W.; Weis, W.I. Unraveling the mechanism of the vesicle transport ATPase NSF, the N-ethylmaleimide-sensitive factor. J. Biol. Chem. 2001, 276, 21991–21994. [Google Scholar] [CrossRef]
- Lim, S.M.; Jang, H.Y.; Lee, J.E.; Shin, J.S.; Park, S.H.; Yoon, B.H.; Kim, G.J. Alteration of Pituitary Tumor Transforming Gene-1 Regulates Trophoblast Invasion via the Integrin/Rho-Family Signaling Pathway. PLoS ONE 2016, 11, e0149371. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Shen, Y.; He, A. Long non-coding RNA-based glycolysis-targeted cancer therapy: Feasibility, progression and limitations. Mol. Biol. Rep. 2021, 48, 2713–2727. [Google Scholar] [CrossRef]
- Steinman, G. The putative etiology and prevention of autism. Prog. Mol. Biol. Transl. Sci. 2020, 173, 1–34. [Google Scholar] [CrossRef]
- Stuebe, A.M.; Lyon, H.; Herring, A.H.; Ghosh, J.; Wise, A.; North, K.E.; Siega-Riz, A.M. Obesity and diabetes genetic variants associated with gestational weight gain. Am. J. Obstet. Gynecol. 2010, 203, 283.e1–283.e17. [Google Scholar] [CrossRef]
- Hamada, H.; Okae, H.; Toh, H.; Chiba, H.; Hiura, H.; Shirane, K.; Sato, T.; Suyama, M.; Yaegashi, N.; Sasaki, H.; et al. Allele-Specific Methylome and Transcriptome Analysis Reveals Widespread Imprinting in the Human Placenta. Am. J. Hum. Genet. 2016, 99, 1045–1058. [Google Scholar] [CrossRef]
- Möpert, K.; Löffler, K.; Röder, N.; Kaufmann, J.; Santel, A. Depletion of protein kinase N3 (PKN3) impairs actin and adherens junctions dynamics and attenuates endothelial cell activation. Eur. J. Cell Biol. 2012, 91, 694–705. [Google Scholar] [CrossRef]
- Kikas, T.; Laan, M.; Kasak, L. Current knowledge on genetic variants shaping placental transcriptome and their link to gestational and postnatal health. Placenta 2021, 116, 2–11. [Google Scholar] [CrossRef]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef]
- Ishigaki, K. Beyond GWAS: From simple associations to functional insights. Semin. Immunopathol. 2022, 44, 3–14. [Google Scholar] [CrossRef]
- Laivuori, H. Pitfalls in setting up genetic studies on preeclampsia. Pregnancy Hypertens. 2013, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medrano-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S. A new water channel aquaporin-11: Extension to renal transplantation. Kidney Res. Clin. Pract. 2015, 34, 188–189. [Google Scholar] [CrossRef]
- Zhang, J.; Jing, S.; Zhang, H.; Zhang, J.; Xie, H.; Feng, L. Low-dose aspirin prevents LPS-induced preeclampsia-like phenotype via AQP-1 and the MAPK/ERK 1/2 pathway. Placenta 2022, 121, 61–69. [Google Scholar] [CrossRef]
- Lv, Z.; Fan, J.; Zhang, X.; Huang, Q.; Han, J.; Wu, F.; Hu, G.; Guo, M.; Jin, Y. Integrative genomic analysis of interleukin-36RN and its prognostic value in cancer. Mol. Med. Rep. 2016, 13, 1404–1412. [Google Scholar] [CrossRef]
- Yu, H.; Li, M.; Wen, X.; Yang, J.; Liang, X.; Li, X.; Bao, X.; Shu, J.; Ren, X.; Chen, W.; et al. Elevation of alpha-1,3 fucosylation promotes the binding ability of TNFR1 to TNF-alpha and contributes to osteoarthritic cartilage destruction and apoptosis. Arthritis Res. Ther. 2022, 24, 93. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef]
- Chatre, L.; Ducat, A.; Spradley, F.T.; Palei, A.C.; Chereau, C.; Couderc, B.; Thomas, K.C.; Wilson, A.R.; Amaral, L.M.; Gaillard, I.; et al. Increased NOS coupling by the metabolite tetrahydrobiopterin (BH4) reduces preeclampsia/IUGR consequences. Redox Biol. 2022, 55, 102406. [Google Scholar] [CrossRef]
- Guo, X.; Mahlakoiv, T.; Ye, Q.; Somanchi, S.; He, S.; Rana, H.; DiFiglia, A.; Gleason, J.; Van der Touw, W.; Hariri, R.; et al. CBLB ablation with CRISPR/Cas9 enhances cytotoxicity of human placental stem cell-derived NK cells for cancer immunotherapy. J. Immunother. Cancer 2021, 9, e001975. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Group | CONTROLS | PE | PE + IUGR | IUGR |
|---|---|---|---|---|
| Delivery mode (Caesarian/Natural) | 23/12 (61.4%) | 9/0 (15.8%) | 3/0 (5.3%) | 10/0 (17.5%) |
| Maternal age (years) | 34.0 ± 3.9 | 34.2 ± 6.0 | 35.3 ± 2.4 | 32.1 ± 6.6 |
| Ethnicity (Afr/Eur) | 8/27 | 5/4 | 1/2 | 1/9 |
| Gestational age (years) | 39.2 ± 1.2 | 34.9 ± 2.6 | 30.0 ± 2.5 | 31.0 ± 2.8 |
| Sex (M/F) | 17/18 | 4/5 | 2/1 | 3/7 |
| Parity | 1.9 ± 1.5 | 1.2 ± 1.1 | 2.0 ± 1.4 | 1.2 ± 0.4 |
| Gene Symbol | Description | eSNPs | Best-eSNP | p-Value | q-Value | Beta | Chr | Pos. | Previous Study |
|---|---|---|---|---|---|---|---|---|---|
| ZSCAN9 | zinc finger and SCAN domain containing 9 | 42 | rs1150707 | 6.46 × 10−16 | 1.43 × 10−11 | 18.80 | 6 | 28229827 | [8] |
| PSG7 | pregnancy specific beta-1-glycoprotein 7 (gene/pseudogene) | 3 | rs7248225 | 3.20 × 10−12 | 1.68 × 10−8 | 27.40 | 19 | 42918847 | [7,8,9] |
| TOB2P1 | transducer of ERBB2, 2 pseudogene 1 | 41 | rs13408 | 4.79 × 10−12 | 2.21 × 10−8 | −17.74 | 6 | 28244970 | [9] |
| ERAP2 | endoplasmic reticulum aminopeptidase 2 | 13 | rs2549778 | 8.01 × 10−12 | 3.28 × 10−8 | 19.34 | 5 | 96868551 | [7,8,9] |
| AQP11 | aquaporin 11 | 12 | rs10793257 | 4.80 × 10−11 | 1.29 × 10−7 | 20.84 | 11 | 77598488 | [7,8,9] |
| LOC646029 | uncharacterized LOC646029 | 11 | rs10793257 | 2.48 × 10−10 | 6.09 × 10−7 | 20.61 | 11 | 77598488 | |
| RPS26 | ribosomal protein S26 | 9 | rs11171739 | 8.12 × 10−10 | 1.95 × 10−6 | 18.98 | 12 | 56076841 | [7] |
| RP1-97J1.2 | putative novel transcript | 7 | rs9372316 | 1.14 × 10−9 | 2.62 × 10−6 | 21.02 | 6 | 112000000 | |
| MLLT10 | myeloid/lymphoid or mixed-lineage leukemia | 15 | rs10828248 | 3.35 × 10−9 | 6.73 × 10−6 | −16.45 | 10 | 21535690 | |
| IL36RN | interleukin 36 receptor antagonist | 25 | rs6761276 | 7.09 × 10−9 | 1.31 × 10−5 | −17.30 | 2 | 113000000 | [8,9] |
| LYNX1 | Ly6/neurotoxin 1 | 9 | rs10956986 | 1.13 × 10−7 | 1.47 × 10−4 | 16.92 | 8 | 143000000 | [9] |
| CASP1P2 | caspase 1 pseudogene 2 | 11 | rs1023954 | 1.18 × 10−7 | 1.50 × 10−4 | 19.23 | 11 | 105000000 | |
| MIR4804 | microRNA 4804 | 5 | rs2253215 | 1.26 × 10−7 | 1.56 × 10−4 | 23.89 | 5 | 72952041 | |
| ZFYVE19 | zinc finger, FYVE domain containing 19 | 6 | rs10152371 | 2.43 × 10−7 | 2.63 × 10−4 | 16.13 | 15 | 40811095 | [7,9] |
| HJURP | Holliday junction recognition protein | 4 | rs2361506 | 4.05 × 10−7 | 4.11 × 10−4 | 18.92 | 2 | 234000000 | |
| ANAPC4 | anaphase promoting complex subunit 4 | 10 | rs1993602 | 5.38 × 10−7 | 5.41 × 10−7 | −15.90 | 4 | 25413484 | [9] |
| OR2F1 | olfactory receptor, family 2, subfamily F, member 1 (gene/pseudogene) | 5 | rs7798409 | 1.08 × 10−6 | 1.00 × 10−3 | −20.39 | 7 | 144000000 | |
| MIR3927 | microRNA 3927 | 1 | rs7046565 | 1.72 × 10−6 | 1.56 × 10−3 | 16.92 | 9 | 110000000 | |
| GBP3 | guanylate binding protein 3 | 5 | rs12121223 | 2.50 × 10−6 | 2.14 × 10−3 | −18.83 | 1 | 89015900 | [9] |
| FUT10 | fucosyltransferase 10 (alpha (1,3) fucosyltransferase) | 5 | rs7018447 | 3.31 × 10−6 | 2.67 × 10−3 | 16.61 | 8 | 33467537 | [9] |
| NSUN7 | NOP2/Sun domain family, member 7 | 3 | rs2437317 | 6.06 × 10−6 | 4.29 × 10−3 | 25.15 | 4 | 40789990 | |
| CBLB | Cbl proto-oncogene B, E3 ubiquitin protein ligase | 1 | rs1503920 | 6.32 × 10−6 | 4.45 × 10−3 | 15.58 | 3 | 106000000 | [8,9] |
| AC104135.2 | novel transcript | 3 | rs7573356 | 9.72 × 10−6 | 6.43 × 10−3 | −20.13 | 2 | 74941537 | |
| GPR132 | G protein-coupled receptor 132 | 2 | rs7157567 | 1.05 × 10−5 | 6.70 × 10−3 | 18.27 | 14 | 105000000 | |
| GTSF1 | gametocyte specific factor 1 | 1 | rs11170917 | 1.31 × 10−5 | 7.98 × 10−3 | 24.42 | 12 | 54472204 | [9] |
| SLC13A5 | solute carrier family 13 (sodium-dependent citrate transporter), member 5 | 1 | rs9889374 | 1.46 × 10−5 | 8.69 × 10−3 | 17.28 | 17 | 6662428 | |
| CASQ1 | calsequestrin 1 (fast-twitch, skeletal muscle) | 1 | rs6693877 | 1.67 × 10−5 | 9.80 × 10−3 | −14.62 | 1 | 160000000 | [9] |
| AC009542.2 | novel transcript, antisense to WDR91 | 3 | rs10954493 | 1.87 × 10−5 | 1.08 × 10−2 | −18.52 | 7 | 135000000 | |
| KANSL1-AS1 | KANSL1 antisense RNA 1 | 1 | rs17585974 | 2.62 × 10−5 | 1.46 × 10−2 | 24.81 | 17 | 46171833 | |
| MTPAP | mitochondrial poly(A) polymerase | 2 | rs1762598 | 4.17× 10−5 | 2.15 × 10−2 | −15.63 | 10 | 30352501 | |
| CPS1 | carbamoyl-phosphate synthase 1 | 3 | rs918233 | 4.27 × 10−5 | 2.20 × 10−2 | −16.64 | 2 | 211000000 | [9] |
| MIR4299 | microRNA 4299 | 1 | rs7126296 | 4.53 × 10−5 | 2.32 × 10−2 | 17.44 | 11 | 11556715 | |
| AGA | aspartylglucosaminidase | 2 | rs4690523 | 5.31 × 10−5 | 2.63 × 10−2 | −15.80 | 4 | 177000000 | [9] |
| SOHLH2 | spermatogenesis and oogenesis specific basic helix-loop-helix 2 | 4 | rs9575600 | 5.41 × 10−5 | 2.64 × 10−2 | 16.53 | 13 | 36223006 | [7] |
| C8orf46 | chromosome 8 open reading frame 46 | 1 | rs12541098 | 6.17 × 10−5 | 2.90 × 10−2 | −15.38 | 8 | 66424147 | [7,8,9] |
| CTC-482H14.5 | novel transcript, antisense to PTPRS | 1 | rs2251127 | 6.29 × 10−5 | 2.90 × 10−2 | 16.34 | 19 | 5138218 | |
| NDUFS5 | NADH dehydrogenase (ubiquinone) Fe-S protein 5, 15kDa (NADH-coenzyme Q reductase) | 2 | rs10888650 | 6.30 × 10−5 | 2.90 × 10−2 | −13.27 | 1 | 39041489 | [8,9] |
| VN1R84P | vomeronasal 1 receptor 84 pseudogene | 1 | rs2015481 | 8.31 × 10−5 | 3.61 × 10−2 | 13.59 | 19 | 21676192 | |
| LOC101928775 | uncharacterized LOC101928775 | 1 | rs10982832 | 8.33 × 10−5 | 3.61 × 10−2 | 16.77 | 9 | 116000000 | |
| FGF19 | fibroblast growth factor 19 | 1 | rs7105655 | 1.01 × 10−4 | 4.24 × 10−2 | 13.69 | 11 | 69738836 | |
| CARD17 | caspase recruitment domain family, member 17 | 3 | rs1623342 | 1.09 × 10−4 | 4.42 × 10−2 | 15.14 | 11 | 105000000 | [7,9] |
| MIR4527 | microRNA 4527 | 1 | rs982265 | 1.17 × 10−4 | 4.67 × 10−2 | −16.51 | 18 | 47444531 | |
| Y_RNA | Y RNA | 1 | rs2248978 | 1.18 × 10−4 | 4.69 × 10−2 | 13.64 | 12 | 105000000 |
| Gene Symbol | Description | Chr | Strand | Group | Keyword | References |
|---|---|---|---|---|---|---|
| ERICH1 | glutamate rich 1 | chr8 | - | Multiple_Complex | PE, IUGR | [32] Identified as eGene by [9] |
| DNAJC15 | DnaJ (Hsp40) homolog, subfamily C, member 15 | chr13 | + | Multiple_Complex | PE | Identified as eGene by [8,9] |
| TAS2R64P | taste receptor, type 2, member 64, pseudogene | chr12 | - | Multiple_Complex | ||
| NSF | N-ethylmaleimide-sensitive factor | chr17 | + | Multiple_Complex | Membrane fusion | [33] Identified as eGene by [7,9] |
| NPM1P35 | nucleophosmin 1 (nucleolar phosphoprotein B23, numatrin) pseudogene 35 | chr11 | + | Pseudogene | NPM1P35 | |
| PTTG1 | pituitary tumor-transforming 1 | chr5 | + | Multiple_Complex | Trophoblast invasion | [34] Identified as eGene by [9] |
| C8orf89 | chromosome 8 open reading frame 89 | chr8 | - | Multiple_Complex | C8orf89 | |
| LINC00654 | long intergenic non-protein coding RNA 654 | chr20 | - | NonCoding | LINC00654 | |
| LINC01273 | long intergenic non-protein coding RNA 1273 | chr20 | + | NonCoding | LINC01273 | |
| FGF12-AS1 | FGF12 antisense RNA 1 | chr3 | + | NonCoding | tumor suppressor | [35] |
| MYRF | myelin regulatory factor | chr11 | + | Multiple_Complex | autism | [36] |
| ITPKB-IT1 | ITPKB intronic transcript 1 | chr1 | - | NonCoding | ||
| CITF22-49E9.3 | novel transcript | chr22 | - | NonCoding | ||
| SLC30A8 | solute carrier family 30 (zinc transporter), member 8 | chr8 | + | Multiple_Complex | gestational weight gain, diabetes | [37] |
| PROSER2-AS1 | PROSER2 antisense RNA 1 | chr10 | - | NonCoding | Placental imprinted, risk for pediatric fracture, | [38] |
| PKN3 | protein kinase N3 | chr9 | + | Multiple_Complex | endothelial cell activation, angiogenesis | [39] |
| From MatrixEQTL Analysis MINUS DISEASE | From Linear Regression to Test Genotype-Disease Interaction | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| eGene | Best-eSNP | p-Value | FDR | beta | Intercept p-Val | eSNP p-Val | Group p-Val | eSNP*Group p-Val | Model p-Val |
| ZSCAN9 | rs1150707 | 1.50 × 10−16 | 3.39 × 10−12 | 18.807 | 0.102647651 | 0.022415409 | 0.00787265 | 0.007064547 | 2.70 × 10−11 |
| ERAP2 | rs2549778 | 1.27 × 10−12 | 6.63 × 10−9 | 18.894 | 0.237759766 | 0.000807833 | 0.029435982 | 0.310364841 | 2.02 × 10−9 |
| TOB2P1 | rs13408 | 1.38 × 10−12 | 6.63 × 10−9 | −17.608 | 0.004647281 | 0.001326778 | 0.886480707 | 0.50710562 | 1.53 × 10−8 |
| PSG7 | rs7248225 | 2.47 × 10−12 | 1.03 × 10−8 | 26.757 | 0.01610927 | 0.000161224 | 0.644681149 | 0.935717136 | 1.80 × 10−8 |
| AQP11 | rs10793257 | 1.10 × 10−11 | 3.37 × 10−8 | 20.693 | 0.936757201 | 0.244954435 | 0.002232625 | 0.009747518 | 3.07 × 10−8 |
| RP1-97J1.2 | rs9372316 | 1.30 × 10−10 | 3.42 × 10−7 | 21.469 | 0.04677023 | 0.02282663 | 0.98324046 | 0.5861815 | 1.34 × 10−5 |
| LOC646029 | rs10793257 | 1.59 × 10−10 | 3.91 × 10−7 | 20.487 | 0.62483721 | 0.449090131 | 0.000702104 | 0.003472929 | 2.96 × 10−8 |
| RPS26 | rs11171739 | 3.39 × 10−10 | 8.00 × 10−7 | 18.460 | 0.05049322 | 0.04455226 | 0.32794464 | 0.08493583 | 3.95 × 10−7 |
| IL36RN | rs6761276 | 2.54 × 10−9 | 5.23 × 10−6 | −17.273 | 0.04374326 | 0.00261548 | 0.37625035 | 0.56160599 | 3.05 × 10−7 |
| CASP1P2 | rs1023954 | 3.22 × 10−8 | 4.76 × 10−5 | 19.003 | 0.08623347 | 0.01572294 | 0.91051179 | 0.45255771 | 2.25 × 10−5 |
| LYNX1 | rs10956986 | 3.60 × 10−8 | 4.86 × 10−5 | 17.143 | 0.077423009 | 0.005369633 | 0.261583699 | 0.381727504 | 6.46 × 10−7 |
| ANAPC4 | rs1993602 | 1.00 × 10−7 | 1.26 × 10−4 | −16.357 | 0.62788117 | 0.22099995 | 0.06658666 | 0.10987541 | 4.00 × 10−6 |
| MIR4804 | rs2253215 | 1.90 × 10−7 | 2.15 × 10−4 | 22.803 | 0.73283414 | 0.05996773 | 0.08001675 | 0.27408907 | 6.05 × 10−6 |
| ZFYVE19 | rs10152371 | 2.32 × 10−7 | 2.54 × 10−4 | 16.012 | 0.29373204 | 0.05488449 | 0.18253459 | 0.21546712 | 4.07 × 10−6 |
| AC104135.2 | rs7573356 | 5.00 × 10−7 | 4.97 × 10−4 | −20.261 | 0.1301216 | 0.146468 | 0.7380213 | 0.2503687 | 3.31 × 10−4 |
| GBP3 | rs12121223 | 8.00 × 10−7 | 7.81 × 10−4 | −19.408 | 0.025397469 | 0.004030585 | 0.591077883 | 0.775339911 | 8.26 × 10−6 |
| OR2F1 | rs7798409 | 9.03 × 10−7 | 8.59 × 10−4 | −20.163 | 0.2916771 | 0.1675941 | 0.5381669 | 0.4819719 | 1.89 × 10−3 |
| FUT10 | rs12155581 | 2.26 × 10−6 | 1.93 × 10−3 | 17.033 | 0.931411621 | 0.806002691 | 0.013310468 | 0.004690609 | 5.07 × 10−6 |
| MIR3927 | rs7046565 | 2.41 × 10−6 | 2.04 × 10−3 | 16.732 | 0.020431665 | 0.002434153 | 0.405350891 | 0.294832332 | 5.07 × 10−4 |
| GPR132 | rs7157567 | 3.86 × 10−6 | 2.96 × 10−3 | 18.710 | 0.06895758 | 0.03121023 | 0.83496731 | 0.76965232 | 2.64 × 10−3 |
| ERICH1 | rs1703911 | 5.35 × 10−6 | 3.90 × 10−3 | −13.888 | 0.048146347 | 0.003527824 | 0.9634337 | 0.940283908 | 3.92 × 10−5 |
| DNAJC15 | rs17553284 | 6.26 × 10−6 | 4.43 × 10−3 | −19.484 | 0.469141 | 0.2646227 | 0.0112012 | 0.0594694 | 2.67 × 10−5 |
| AGA | rs4690523 | 9.97 × 10−6 | 6.57 × 10−3 | −16.364 | 0.50371342 | 0.14900309 | 0.06567993 | 0.18234469 | 7.27 × 10−5 |
| TAS2R64P | rs7297949 | 1.05 × 10−5 | 6.85 × 10−3 | −16.077 | 0.0824359 | 0.02563944 | 0.86790276 | 0.878125 | 2.53 × 10−4 |
| AC009542.2 | rs10954493 | 1.07 × 10−5 | 6.95 × 10−3 | −18.232 | 0.97990037 | 0.03550566 | 0.05021882 | 0.21802336 | 7.21 × 10−6 |
| GTSF1 | rs11170917 | 1.46 × 10−5 | 8.84 × 10−3 | 22.336 | 0.34080682 | 0.02176511 | 0.67946143 | 0.83764288 | 2.02 × 10−3 |
| CBLB | rs1503920 | 1.50 × 10−5 | 9.02 × 10−3 | 14.714 | 0.02302123 | 0.04722184 | 0.47428316 | 0.98231439 | 1.98 × 10−3 |
| SOHLH2 | rs9575600 | 1.65 × 10−5 | 9.76 × 10−3 | 16.140 | 0.10582542 | 0.01653465 | 0.87063264 | 0.92481755 | 3.25 × 10−4 |
| KANSL1-AS1 | rs17585974 | 1.67 × 10−5 | 9.81 × 10−3 | 23.860 | 0.11809498 | 0.06863156 | 0.62204626 | 0.77192668 | 4.91 × 10−3 |
| NSUN7 | rs2437317 | 1.73 × 10−5 | 9.94 × 10−3 | 23.457 | 0.01246184 | 0.01516865 | 0.08784461 | 0.56754666 | 3.00 × 10−3 |
| NSF | rs17698176 | 1.74 × 10−5 | 9.97 × 10−3 | 20.367 | 0.22639048 | 0.00414694 | 0.7413922 | 0.79027276 | 4.09 × 10−5 |
| NDUFS5 | rs10888650 | 1.82 × 10−5 | 1.01 × 10−2 | −13.620 | 0.52728811 | 0.42320815 | 0.24332657 | 0.03161536 | 8.82 × 10−6 |
| CASQ1 | rs6693877 | 1.98 × 10−5 | 1.10 × 10−2 | −14.309 | 0.1020241 | 0.0981154 | 0.9077578 | 0.3968772 | 6.41 × 10−4 |
| NPM1P35 | rs4488202 | 2.38 × 10−5 | 1.29 × 10−2 | −21.404 | 0.6814926 | 0.2694253 | 0.1968538 | 0.1925592 | 1.02 × 10−3 |
| PTTG1 | rs1105789 | 2.43 × 10−5 | 1.32 × 10−2 | 15.471 | 0.0267482 | 0.01723734 | 0.54238551 | 0.79645133 | 7.37 × 10−4 |
| C8orf46 | rs12541098 | 4.07 × 10−5 | 1.98 × 10−2 | −15.621 | 0.01999623 | 0.01458403 | 0.4383046 | 0.67636289 | 2.10 × 10−3 |
| C8orf89 | rs6472746 | 4.65 × 10−5 | 2.19 × 10−2 | −17.620 | 0.004634949 | 0.000225028 | 0.080493823 | 0.022123942 | 3.17 × 10−4 |
| LINC00654 | rs6116828 | 4.93 × 10−5 | 2.29 × 10−2 | 12.740 | 0.16132241 | 0.02684875 | 0.56544041 | 0.76958675 | 4.19 × 10−4 |
| MTPAP | rs1762598 | 5.02 × 10−5 | 2.31 × 10−2 | −15.032 | 0.05145106 | 0.01354065 | 0.49610048 | 0.4799675 | 4.82 × 10−3 |
| CARD17 | rs1623342 | 5.17 × 10−5 | 2.36 × 10−2 | 15.497 | 0.30416008 | 0.03915456 | 0.60188393 | 0.84825756 | 1.63 × 10−3 |
| SLC13A5 | rs9889374 | 5.49 × 10−5 | 2.48 × 10−2 | 15.823 | 0.10733714 | 0.53938538 | 0.84592422 | 0.05661093 | 4.53 × 10−4 |
| LINC01273 | rs6020255 | 5.91 × 10−5 | 2.66 × 10−2 | −14.134 | 0.4831266 | 0.461141 | 0.2441706 | 0.1435129 | 1.01 × 10−3 |
| LOC101928775 | rs10982832 | 5.95 × 10−5 | 2.66 × 10−2 | 16.227 | 0.19619467 | 0.01188732 | 0.8784099 | 0.98711196 | 1.54 × 10−4 |
| VN1R84P | rs2015481 | 6.03 × 10−5 | 2.68 × 10−2 | 13.299 | 0.01188751 | 0.10670774 | 0.3118514 | 0.42971681 | 2.99 × 10−4 |
| CPS1 | rs2250976 | 7.35 × 10−5 | 3.09 × 10−2 | 14.858 | 0.19187507 | 0.09947661 | 0.68926108 | 0.80062325 | 7.05 × 10−3 |
| FGF12-AS1 | rs10937543 | 8.44 × 10−5 | 3.44 × 10−2 | −17.045 | 0.53243677 | 0.07383697 | 0.22123134 | 0.52782373 | 4.57 × 10−4 |
| MYRF | rs7925523 | 1.02 × 10−4 | 4.12 × 10−2 | 18.305 | 0.049161643 | 0.000625781 | 0.393915774 | 0.080254068 | 5.05 × 10−4 |
| ITPKB-IT1 | rs697845 | 1.03 × 10−4 | 4.15 × 10−2 | 19.480 | 0.006181248 | 0.000344546 | 0.057691247 | 0.056088085 | 5.11 × 10−4 |
| MIR4299 | rs7126296 | 1.04 × 10−4 | 4.15 × 10−2 | 15.096 | 0.03207948 | 0.01109053 | 0.62800608 | 0.46298706 | 3.04 × 10−3 |
| CITF22-49E9.3 | rs137878 | 1.12 × 10−4 | 4.38 × 10−2 | −17.668 | 0.20391064 | 0.02392672 | 0.9198819 | 0.67454661 | 4.21 × 10−3 |
| SLC30A8 | rs10505312 | 1.18 × 10−4 | 4.57 × 10−2 | 20.222 | 0.7540111 | 0.457554 | 0.3942176 | 0.4400821 | 3.34 × 10−2 |
| CTC-482H14.5 | rs2620833 | 1.25 × 10−4 | 4.74 × 10−2 | 16.078 | 0.084645173 | 0.008758073 | 0.852309458 | 0.531835913 | 1.04 × 10−3 |
| PROSER2-AS1 | rs7900122 | 1.26 × 10−4 | 4.75 × 10−2 | −15.424 | 0.1660911 | 0.03140426 | 0.69555964 | 0.72078802 | 1.49 × 10−2 |
| PKN3 | rs10819449 | 1.32 × 10−4 | 4.91 × 10−2 | 19.043 | 0.3171833 | 0.416917 | 0.9639612 | 0.2775942 | 9.58 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apicella, C.; Ruano, C.S.M.; Thilaganathan, B.; Khalil, A.; Giorgione, V.; Gascoin, G.; Marcellin, L.; Gaspar, C.; Jacques, S.; Murdoch, C.E.; et al. Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas. Cells 2023, 12, 578. https://doi.org/10.3390/cells12040578
Apicella C, Ruano CSM, Thilaganathan B, Khalil A, Giorgione V, Gascoin G, Marcellin L, Gaspar C, Jacques S, Murdoch CE, et al. Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas. Cells. 2023; 12(4):578. https://doi.org/10.3390/cells12040578
Chicago/Turabian StyleApicella, Clara, Camino S. M. Ruano, Basky Thilaganathan, Asma Khalil, Veronica Giorgione, Géraldine Gascoin, Louis Marcellin, Cassandra Gaspar, Sébastien Jacques, Colin E. Murdoch, and et al. 2023. "Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas" Cells 12, no. 4: 578. https://doi.org/10.3390/cells12040578
APA StyleApicella, C., Ruano, C. S. M., Thilaganathan, B., Khalil, A., Giorgione, V., Gascoin, G., Marcellin, L., Gaspar, C., Jacques, S., Murdoch, C. E., Miralles, F., Méhats, C., & Vaiman, D. (2023). Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas. Cells, 12(4), 578. https://doi.org/10.3390/cells12040578