
Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis

Abstract
1. Introduction
2. Incidence and Characteristics of CAR T-Associated Cytopenias
3. Factors Associated with Post-CAR T Cytopenias
4. Pre-Treatment Cytopenias and Reduced CAR T Efficacy
5. Implications of Age-Associated Inflammation on Bone Marrow and CAR T Outcomes
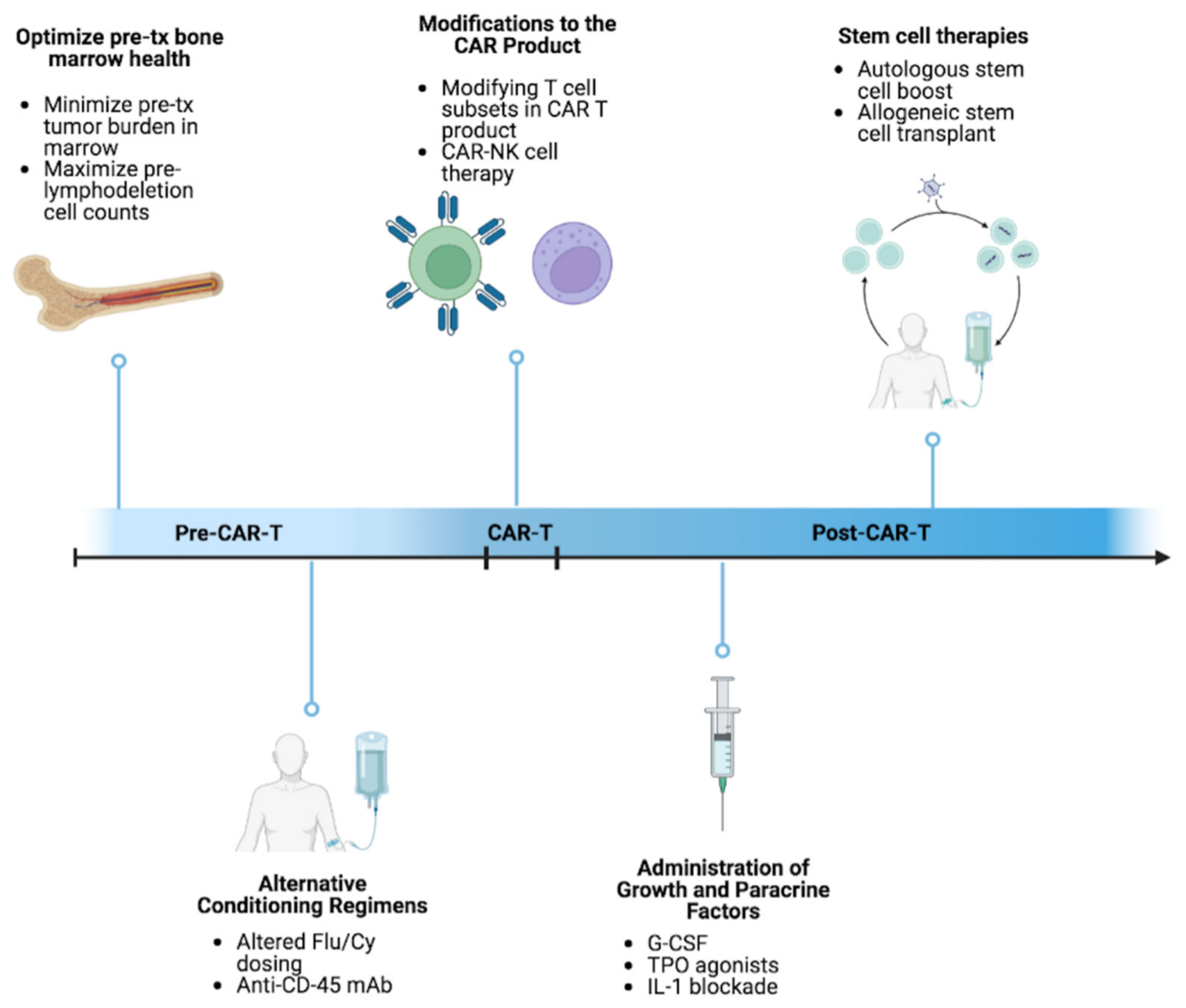
6. Approaches to Mitigating and Managing Cytopenias
6.1. Paracrine Factor Modulation
6.2. Allogeneic Hematopoietic Cell Transplant following CAR T-Cell Therapy
6.3. Autologous Hematopoietic Stem Cell Boost Post CAR T
6.4. Alternative Conditioning Regimens
7. Alternative Approaches to Optimizing CAR T-Cell Therapy Regimens
7.1. Repeat Dosing
7.2. Optimizing the CAR T-Cell Infusion Product Composition
7.3. Optimizing the CAR Construct
8. CAR T and Immune Cell Interaction
9. CAR T and Clonal Hematopoiesis
10. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martino, M.; Alati, C.; Canale, F.A.; Musuraca, G.; Martinelli, G.; Cerchione, C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2150. [Google Scholar] [CrossRef]
- Haradhvala, N.J.; Leick, M.B.; Maurer, K.; Gohil, S.H.; Larson, R.C.; Yao, N.; Gallagher, K.M.E.; Katsis, K.; Frigault, M.J.; Southard, J.; et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 2022, 28, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Reagan, P.M.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; et al. Comparison of 2-year outcomes with CAR T cells (ZUMA-1) vs salvage chemotherapy in refractory large B-cell lymphoma. Blood Adv. 2021, 5, 4149–4155. [Google Scholar] [CrossRef]
- Szász, R.; Telek, B.; Illés, Á. Fludarabine-Cyclophosphamide-Rituximab Treatment in Chronic Lymphocytic Leukemia, Focusing on Long Term Cytopenias Before and After the Era of Targeted Therapies. Pathol. Oncol. Res. 2021, 27, 1609742. [Google Scholar] [CrossRef]
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019, 54, 1643–1650. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Sharma, N.; Reagan, P.M.; Liesveld, J.L. Cytopenia after CAR-T Cell Therapy—A Brief Review of a Complex Problem. Cancers 2022, 14, 1501. [Google Scholar] [CrossRef]
- Schaefer, A.; Huang, Y.; Kittai, A.; Maakaron, J.E.; Saygin, C.; Brammer, J.; Penza, S.; Saad, A.; Jaglowski, S.M.; William, B.M. Cytopenias After CD19 Chimeric Antigen Receptor T-Cells (CAR-T) Therapy for Diffuse Large B-Cell Lymphomas or Transformed Follicular Lymphoma: A Single Institution Experience. Cancer Manag. Res. 2021, 13, 8901–8906. [Google Scholar] [CrossRef]
- Thibaud, S.; Mia, B.; Van Oekelen, O.; Mouhieddine, T.H.; Schaniel, C.; Ghodke-Puranik, Y.; Aleman, A.; Upadhyaya, B.; Lancman, G.; Lagana, A.; et al. Comprehensive Characterization of Prolonged Unexplained Cytopenias in Relapsed/Refractory Multiple Myeloma Patients Following BCMA-Directed CAR-T Cell Therapy. Blood 2022, 140, 614–616. [Google Scholar] [CrossRef]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef]
- Gill, S.; Carney, D.; Ritchie, D.; Wolf, M.; Westerman, D.; Prince, H.M.; Januszewicz, H.; Seymour, J.F. The frequency, manifestations, and duration of prolonged cytopenias after first-line fludarabine combination chemotherapy. Ann. Oncol. 2010, 21, 331–334. [Google Scholar] [CrossRef]
- Rejeski, K.; Perez, A.P.; Sesques, P.; Hoster, E.; Berger, C.S.; Jentzsch, L.; Mougiakakos, D.; Frölich, L.; Ackermann, J.; Buecklein, V.; et al. CAR-HEMATOTOX: A model for CAR T-cell–related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood 2021, 138, 2499–2513. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Shan, M.; Zong, X.; Geng, H.; Li, J.; Chen, G.; Yu, L.; Xu, Y.; Li, C.; et al. Cytopenia after chimeric antigen receptor T cell immunotherapy in relapsed or refractory lymphoma. Front. Immunol. 2022, 13, 997589. [Google Scholar] [CrossRef]
- Juluri, K.R.; Wu, Q.V.; Voutsinas, M.J.M.; Hou, J.; Hirayama, A.V.; Mullane, E.; Miles, N.; Maloney, D.G.; Turtle, C.J.; Bar, M.; et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 2055–2068. [Google Scholar] [CrossRef]
- Jain, T.; Knezevic, A.; Pennisi, M.; Chen, Y.; Ruiz, J.D.; Purdon, T.J.; Devlin, S.M.; Smith, M.; Shah, G.L.; Halton, E.; et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020, 4, 3776–3787. [Google Scholar] [CrossRef] [PubMed]
- Taneja, A.; Jain, T. CAR-T-OPENIA: Chimeric antigen receptor T-cell therapy-associated cytopenias. EJHaem 2022, 3, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.; Bezerra, E.D.; Hirayama, A.V.; Hill, J.A.; Wu, Q.V.; Voutsinas, J.; Sorror, M.L.; Turtle, C.J.; Maloney, D.G.; Bar, M. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol. Blood Marrow Transplant. 2020, 26, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef]
- Hill, J.; Li, D.; Hay, K.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Logue, J.M.; Zucchetti, E.; Bachmeier, C.A.; Krivenko, G.S.; Larson, V.; Ninh, D.; Grillo, G.; Cao, B.; Kim, J.; Chavez, J.C.; et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica 2021, 106, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Dayagi, T.W.; Sherman, G.; Bielorai, B.; Adam, E.; Besser, M.J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E.; Avigdor, A. Characteristics and risk factors of infections following CD28-based CD19 CAR-T cells. Leuk. Lymphoma 2021, 62, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Wudhikarn, K.; Perales, M.-A. Infectious complications, immune reconstitution, and infection prophylaxis after CD19 chimeric antigen receptor T-cell therapy. Bone Marrow Transplant. 2022, 57, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Faramand, R.G.; Davila, M.L. CAR T-cell hematotoxicity: Is inflammation the key? Blood 2021, 138, 2447–2448. [Google Scholar] [CrossRef]
- Nagle, S.J.; Murphree, C.; Raess, P.W.; Schachter, L.; Chen, A.; Hayes-Lattin, B.; Nemecek, E.; Maziarz, R.T. Prolonged hematologic toxicity following treatment with chimeric antigen receptor T cells in patients with hematologic malignancies. Am. J. Hematol. 2021, 96, 455–461. [Google Scholar] [CrossRef]
- Li, X.; Deng, Q.; Henderson, J.; Watson, G.; Deaton, L.; Cain, T.; Fayad, L.; Iyer, S.P.; Hagemeister, F.B.; Nastoupil, L.J.; et al. Targetable Cellular Etiology of Prolonged Cytopenia Following CD19 CAR T-Cell Therapy. Blood 2022, 140, 4502–4503. [Google Scholar] [CrossRef]
- Rejeski, K.; Wu, Z.; Blumenberg, V.; Kunz, W.G.; Mueller, S.; Kajigaya, S.; Gao, S.; Bücklein, V.L.; Frölich, L.; Schmidt, C.; et al. Oligoclonal T-cell expansion in a patient with bone marrow failure after CD19 CAR-T therapy for Richter-transformed DLBCL. Blood 2022, 140, 2175–2179. [Google Scholar] [CrossRef]
- Stewart, A.G.; Henden, A.S. Infectious complications of CAR T-cell therapy: A clinical update. Ther. Adv. Infect. Dis. 2021, 8, 20499361211036773. [Google Scholar] [CrossRef]
- Tabbara, N.; Sharp, J.; Gaut, D.; Pham, T.T.D.; Tang, K.; Oliai, C.; Sim, M.S.; Schiller, G.; Young, P.; Sasine, J.P. Diminished durability of chimeric antigen receptor T-cell efficacy with severe or prolonged postinfusion cytopenias. Am. J. Hematol. 2022, 97, E249–E255. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef]
- Hao, Z.; Li, R.; Meng, L.; Han, Z.; Hong, Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp. Hematol. Oncol. 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Kim, M.Y.; Cooper, M.L.; Ritchey, J.K.; Hollaway, J.; DiPersio, J.F. Normal Myeloid Cells Are Required for Sustained CAR T Cell Activity Against Myeloid Tumor in a Humanized Mouse Model. Blood 2021, 138, 734. [Google Scholar] [CrossRef]
- Bansal, R.; Novo, M.; Al Saleh, A.S.; Guerrico, A.G.; Zhang, H.; Shao, Z.; Babadi, E.; Martinez, K.E.; McCoy, G.A.; Hathcock, M.A.; et al. Peak absolute lymphocyte count after CAR-T infusion predicts clinical response in aggressive lymphoma. Am. J. Hematol. 2022, 97, E241–E244. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Verovskaya, E.; Calero-Nieto, F.; Reynaud, D.; Zhang, S.Y.; Herault, A.; Bakker, S.; Pietras, E.; Svendsen, A.F.; Wang, X.; Kinston, S.; et al. Inflammatory Changes in the Bone Marrow Microenvironment Drive Both Niche and Blood System Remodeling during Aging. Exp. Hematol. 2018, 64, S43–S44. [Google Scholar] [CrossRef]
- Fischer, J.W.; Bhattarai, N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front. Immunol. 2021, 12, 693016. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; O’Brien, S.; Wierda, W.; Kantarjian, H.; Wen, S.; Do, K.-A.; Thomas, D.A.; Cortes, J.; Lerner, S.; Keating, M.J. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008, 112, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Strati, P.; Wierda, W.; Burger, J.; Ferrajoli, A.; Tam, C.; Lerner, S.; Keating, M.J.; O’Brien, S. Myelosuppression after frontline fludarabine, cyclophosphamide, and rituximab in patients with chronic lymphocytic leukemia. Cancer 2013, 119, 3805–3811. [Google Scholar] [CrossRef]
- Bishop, M.R. The benefit of CAR T cells in older patients. Blood 2020, 135, 2020–2021. [Google Scholar] [CrossRef] [PubMed]
- CAR T May Be Beneficial and Safe in Older Patients with Relapsed/Refractory LBCL and Geriatric Vulnerabilities. Available online: https://www.mskcc.org/clinical-updates/car-may-be-beneficial-and-safe-older-patients-relapsed-refractory-lbcl-and-geriatric-vulnerabilities (accessed on 6 December 2022).
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Sidaway, P. CAR T cell therapy efficacious against B-ALL across age groups. Nat. Rev. Clin. Oncol. 2018, 15, 199. [Google Scholar] [CrossRef]
- Liévin, R.; Di Blasi, R.; Morin, F.; Galli, E.; Allain, V.; De Jorna, R.; Vercellino, L.; Parquet, N.; Mebarki, M.; Larghero, J.; et al. Effect of early granulocyte-colony-stimulating factor administration in the prevention of febrile neutropenia and impact on toxicity and efficacy of anti-CD19 CAR-T in patients with relapsed/refractory B-cell lymphoma. Bone Marrow Transplant. 2022, 57, 431–439. [Google Scholar] [CrossRef]
- Mehta, H.M.; Malandra, M.; Corey, S.J. G-CSF and GM-CSF in Neutropenia. J. Immunol. 2015, 195, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Barreto, J.N.; Bansal, R.; Hathcock, M.A.; Doleski, C.J.; Hayne, J.R.; Truong, T.A.; Nedved, A.N.; Ansell, S.M.; Bennani, N.N.; Paludo, J.; et al. The impact of granulocyte colony stimulating factor on patients receiving chimeric antigen receptor T -cell therapy. Am. J. Hematol. 2021, 96, E399–E402. [Google Scholar] [CrossRef] [PubMed]
- Galli, E.; Allain, V.; Di Blasi, R.; Bernard, S.; Vercellino, L.; Morin, F.; Moatti, H.; Caillat-Zucman, S.; Chevret, S.; Thieblemont, C. G-CSF does not worsen toxicities and efficacy of CAR-T cells in refractory/relapsed B-cell lymphoma. Bone Marrow Transplant. 2020, 55, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Bindal, P.; Elavalakanar, P.; Trottier, C.A.; Dodge, L.E.; Logan, E.K.; Sermer, D.J.; Leukam, M.; Weinstock, M.J.; Joyce, R.M.; Liegel, J.; et al. G-CSF Administration Is Associated with Worse Treatment Response and Survival after CAR T-Cell Therapy. Blood 2022, 140, 5238–5240. [Google Scholar] [CrossRef]
- Baur, R.; Jitschin, R.; Kharboutli, S.; Stoll, A.; Völkl, S.; Büttner-Herold, M.; Schmidt, D.; Rösler, W.; Mackensen, A.; Mougiakakos, D. Thrombopoietin receptor agonists for acquired thrombocytopenia following anti-CD19 CAR-T-cell therapy: A case report. J. Immunother. Cancer 2021, 9, e002721. [Google Scholar] [CrossRef]
- Gaut, D.; Tang, K.; Sim, M.S.; Duong, T.; Young, P.; Sasine, J. Filgrastim associations with CAR T-cell therapy. Int. J. Cancer 2021, 148, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Becker, P.S.; Griffiths, E.A.; Alwan, L.M.; Bachiashvili, K.; Brown, A.; Cool, R.; Curtin, P.; Dinner, S.; Gojo, I.; Hicks, A.; et al. NCCN Guidelines Insights: Hematopoietic Growth Factors, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 12–22. [Google Scholar] [CrossRef]
- Wehrli, M.; Gallagher, K.; Chen, Y.-B.; Leick, M.B.; McAfee, S.L.; El-Jawahri, A.R.; DeFilipp, Z.; Horick, N.; O’Donnell, P.; Spitzer, T.; et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J. Immunother. Cancer 2022, 10, e003847. [Google Scholar] [CrossRef]
- Beyar-Katz, O.; Perry, C.; Bar On, Y.; Amit, O.; Gutwein, O.; Wolach, O.; Kedar, R.; Pikovsky, O.; Avivi, I.; Gold, R.; et al. Thrombopoietin receptor agonist for treating bone marrow aplasia following anti-CD19 CAR-T cells—Single-center experience. Ann. Hematol. 2022, 101, 1769–1776. [Google Scholar] [CrossRef]
- Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Di Emidio, P.; Conti, P. CAR-T cell therapy causes inflammation by IL-1 which activates inflammatory cytokine mast cells: Anti-inflammatory role of IL-37. J. Biol. Regul. Homeost. Agents 2019, 33, 1981–1985. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef]
- Jacoby, E. The role of allogeneic HSCT after CAR T cells for acute lymphoblastic leukemia. Bone Marrow Transplant. 2019, 54, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.; Baer, M.R.; Donnellan, W.B.; Carr-O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. End of phase I results of ZUMA-3, a phase 1/2 study of KTE-X19, anti-CD19 chimeric antigen receptor (CAR) T cell therapy, in adult patients (pts) with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2019, 37, 7006. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Annesley, C.; Bleakley, M.; Dahlberg, A.; Jensen, M.C.; Gardner, R. Long Term Follow-up after SCRI-CAR19v1 Reveals Late Recurrences As Well As a Survival Advantage to Consolidation with HCT after CAR T Cell Induced Remission. Blood 2018, 132, 967. [Google Scholar] [CrossRef]
- Gagelmann, N.; Wulf, G.G.; Duell, J.; Glass, B.; van Heteren, P.; von Tresckow, B.; Fischer, M.; Penack, O.; Ayuk, F.A.; Einsele, H.; et al. Hematopoietic stem cell boost for persistent neutropenia after CAR-T cell therapy: A GLA/DRST study. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Rejeski, K.; Burchert, A.; Iacoboni, G.; Sesques, P.; Fransecky, L.; Buecklein, V.; Trenker, C.; Hernani, R.; Naumann, R.; Schäfer, J.A.; et al. Safety and feasibility of stem cell boost as a salvage therapy for severe hematotoxicity after CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 4719–4725. [Google Scholar] [CrossRef]
- Fabrizio, V.A.; Boelens, J.J.; Mauguen, A.; Baggott, C.; Prabhu, S.; Egeler, E.; Mavroukakis, S.; Pacenta, H.L.; Phillips, C.L.; Rossoff, J.; et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR T-cell therapy. Blood Adv. 2022, 6, 1961–1968. [Google Scholar] [CrossRef]
- Bradford, K.L.; Liu, S.; Krajinovic, M.; Ansari, M.; Garabedian, E.; Tse, J.; Wang, X.; Shaw, K.L.; Gaspar, H.B.; Candotti, F.; et al. Busulfan Pharmacokinetics in Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Gene Therapy. Biol. Blood Marrow Transplant. 2020, 26, 1819–1827. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Farber, S.H.; Choi, B.D.; Swartz, A.M.; Shen, S.H.; Gedeon, P.C.; Snyder, D.J.; Herndon, J.E.; Healy, P.; et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance but Requires 4-1BB Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin. Cancer Res. 2019, 25, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef] [PubMed]
- Strati, P.; Jallouk, A.P.; Sun, R.; Choi, J.; Das, K.; Cherng, H.-J.; Ahmed, S.; Lee, H.J.; Iyer, S.P.; Nair, R.; et al. Impact of conditioning chemotherapy on lymphocyte kinetics and outcomes in LBCL patients treated with CAR T-cell therapy. Leukemia 2022, 36, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Turtle, C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs 2017, 77, 237–245. [Google Scholar] [CrossRef]
- Dekker, L.; Calkoen, F.G.; Jiang, Y.; Blok, H.; Veldkamp, S.R.; De Koning, C.C.H.; Spoon, M.; Admiraal, R.; Hoogerbrugge, P.; Vormoor, B.J.; et al. Fludarabine exposure predicts outcome after CD19 CAR T-cell therapy in children and young adults with acute leukemia. Blood Adv. 2022, 6, 1969–1976. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Stokes, J.; Molina, M.S.; Hoffman, E.A.; Simpson, R.J.; Katsanis, E. Immunomodulatory Effects of Bendamustine in Hemato-poietic Cell Transplantation. Cancers 2021, 13, 1702. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kato, T.; Hasegawa, F.; Sunahara, M.; Tsurumaki, Y. The Past, Present, and Future of Clinically Applied Chimeric Antigen Receptor-T-Cell Therapy. Pharmaceuticals 2022, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, G.; Chong, E.A.; Svoboda, J.; Wohlfarth, P.; Nasta, S.D.; Williamson, S.; Landsburg, D.J.; Gerson, J.N.; Barta, S.K.; Pajarillo, R.; et al. Bendamustine Is a Safe and Effective Regimen for Lymphodepletion before Tisagenlecleucel in Patients with Large B-Cell Lymphomas. Blood 2021, 138, 1438. [Google Scholar] [CrossRef]
- Terry, L.A.; Brown, M.H.; Beverley, P.C. The monoclonal antibody, UCHL1, recognizes a 180,000 MW component of the human leucocyte-common antigen, CD45. Immunology 1988, 64, 331–336. [Google Scholar]
- Palchaudhuri, R.; Saez, B.; Hoggatt, J.; Schajnovitz, A.; Sykes, D.B.; Tate, T.A.; Czechowicz, A.; Kfoury, Y.; Ruchika, F.; Rossi, D.J.; et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016, 34, 738–745. [Google Scholar] [CrossRef]
- Courtney, A.H.; Shvets, A.A.; Lu, W.; Griffante, G.; Mollenauer, M.; Horkova, V.; Lo, W.-L.; Yu, S.; Stepanek, O.; Chakraborty, A.K.; et al. CD45 functions as a signaling gatekeeper in T cells. Sci. Signal. 2019, 12, eaaw8151. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, X.; Tu, L.; Cao, J.; Hinrichs, C.S.; Su, X. Size-dependent activation of CAR-T cells. Sci. Immunol. 2022, 7, eabl3995. [Google Scholar] [CrossRef]
- Wellhausen, N.; Rennels, A.K.; Lesch, S.; Agarwal, S.; Charria, B.; Choi, G.; Young, R.M.; Garcia, C.; June, C.H.; Gill, S. Epitope Editing in Hematopoietic Cells Enables CD45-Directed Immune Therapy. Blood 2022, 140, 862–864. [Google Scholar] [CrossRef]
- Gauthier, J.; Bezerra, E.D.; Hirayama, A.V.; Fiorenza, S.; Sheih, A.; Chou, C.K.; Kimble, E.L.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood 2021, 137, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.M.; Yates, B.; Ling, A.; Yuan, C.M.; Wang, H.-W.; Stetler-Stevenson, M.; LaLoggia, M.; Molina, J.C.; Lichtenstein, D.A.; Lee, D.W.; et al. Characterization of extramedullary disease in B-ALL and response to CAR T-cell therapy. Blood Adv. 2022, 6, 2167–2182. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, H.; Shao, M.; Cui, Q.; Wu, Z.; Xiao, L.; Huang, H.; Hu, Y. Lymphodepletion chemotherapy revitalizes chimeric antigen receptor T cells contributing to regression of relapsed B-cell lymphoma: A case report. Medicine 2020, 99, e22510. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Woodhouse, S.; Zhao, Z.; Arya, R.; Govek, K.; Kim, D.; Lundh, S.; Baysoy, A.; Sun, H.; Deng, Y.; et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci. Adv. 2022, 8, eabj2820. [Google Scholar] [CrossRef] [PubMed]
- Good, Z.; Spiegel, J.Y.; Sahaf, B.; Malipatlolla, M.B.; Ehlinger, Z.J.; Kurra, S.; Desai, M.H.; Reynolds, W.D.; Lin, A.W.; Vandris, P.; et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat. Med. 2022, 28, 1860–1871. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.E.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; June, C.H. CAR T-cell Therapy Meets Clonal Hematopoiesis. Blood Cancer Discov. 2022, 3, 382–384. [Google Scholar] [CrossRef]
- Von Bonin, M.; Jambor, H.K.; Teipel, R.; Stölzel, F.; Thiede, C.; Damm, F.; Kroschinsky, F.; Schetelig, J.; Chavakis, T.; Bornhäuser, M. Clonal hematopoiesis and its emerging effects on cellular therapies. Leukemia 2021, 35, 2752–2758. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.Y.; Swoboda, D.M.; Greenbaum, U.; Ma, J.; Patel, R.D.; Devashish, K.; Das, K.; Tanner, M.R.; Strati, P.; Nair, R.; et al. Clonal Hematopoiesis Is Associated with Increased Risk of Severe Neurotoxicity in Axicabtagene Ciloleucel Therapy of Large B-Cell Lymphoma. Blood Cancer Discov. 2022, 3, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Sample Size | Disease | Study | CAR Construct | Incidence of Prolonged Persistent Cytopenia |
|---|---|---|---|---|---|
| Locke et al. [4] | 119 | DLBCL, PMBCL, t FL | ZUMA-1 (phase I/II) | Anti-CD19, CD28 co-stimulatory domain (retroviral) | 7%, 11%, (>3 mo) |
| Schuster SJ et al. [8] | 167 | DLBCL | JULIET (phase II) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | 32%, (>1 mo) |
| Wang et al. [40] | 74 | MCL | KTE-X19 (phase II) | Anti-CD19, CD28 co-stimulatory domain (retroviral) | 16%, 16%, (>3 mo) |
| Maude et al. [48] | 75 | B-ALL (children and young adults) | ELIANA (phase I/II) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | 12%, 53%, (>1 mo) |
| Lee et al. [65] | 53 | B-ALL (children) | NCT01044069 (phase I) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | 33%, (>14 days) |
| Reference | Study | CAR Construct | Anemia | Thrombocytopenia | Neutropenia | CRS | ICANS | Infections |
|---|---|---|---|---|---|---|---|---|
| Locke et al. [4] | ZUMA-1 (phase I/II) | Anti-CD19, CD28 co-stimulatory domain (retroviral) | 43% | 38% | 78% | 13% | 28% | 8% |
| Schuster SJ et al. [8] | JULIET (phase II) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | 39% | 28% | 33% | 22% | 12% | 20% |
| Wang et al. [40] | KTE-X19 (phase II) | Anti-CD19, CD28 co-stimulatory domain (retroviral) | 50% | 51% | 85% | 15% | 31% | 32% |
| Maude et al. [48] | ELIANA (phase I/II) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | Not reported | 41% | 35% | 46% | 13% | 24% |
| Lee et al. [65] | NCT01044069 (phase I) | Anti-CD19, 4-1BB co-stimulatory domain (lentiviral) | 68% | 53% | >50% | 40% | 0% | Not reported |
| Shah B.D. et al. [67] | ZUMA-3 (phase II) | Anti-CD19, CD28 co-stimulatory domain (retroviral) | 49% | 30% | 27% | 24% | 25% | 25% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinhardt, B.; Lee, P.; Sasine, J.P. Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis. Cells 2023, 12, 531. https://doi.org/10.3390/cells12040531
Reinhardt B, Lee P, Sasine JP. Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis. Cells. 2023; 12(4):531. https://doi.org/10.3390/cells12040531
Chicago/Turabian StyleReinhardt, Bryanna, Patrick Lee, and Joshua P. Sasine. 2023. "Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis" Cells 12, no. 4: 531. https://doi.org/10.3390/cells12040531
APA StyleReinhardt, B., Lee, P., & Sasine, J. P. (2023). Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis. Cells, 12(4), 531. https://doi.org/10.3390/cells12040531