
PD-1/PD-L1 and DNA Damage Response in Cancer

 ,
,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
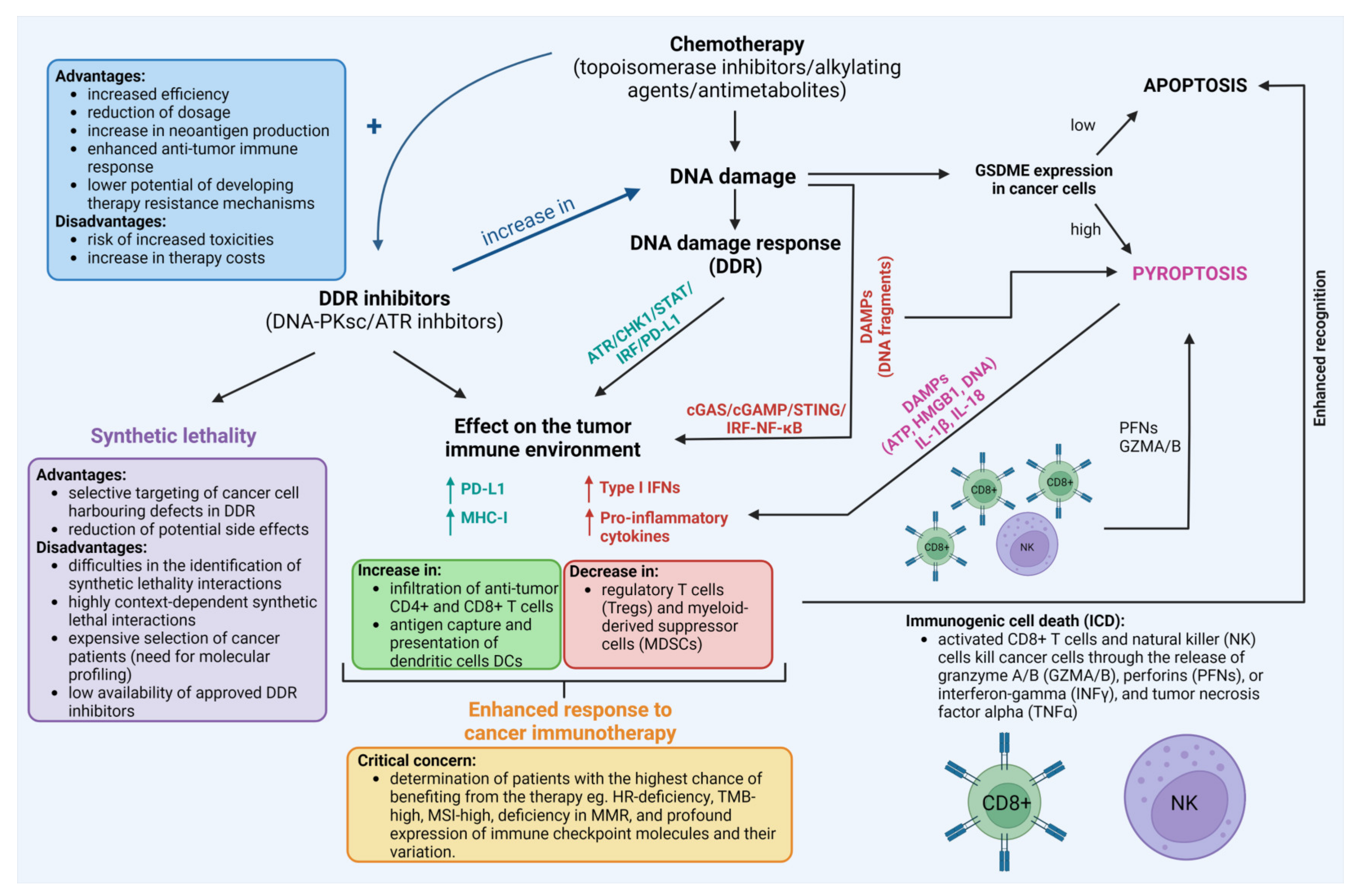{kind=link}
Abstract
1. Introduction

2. PD-1/PD-L1 Pathway
3. Canonical DNA Damage Response
4. DNA Strand Break Repair
5. DNA Damage Response and PD-1/PD-L1
6. Cytotoxic Agents, DDR, and Immunotherapy
6.1. Irinotecan
6.2. Etoposide
6.3. Cisplatin and Other Platinum Compounds
7. Current Challenges and Future Prospects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 53BP1 | TP53-binding protein 1 |
| 5-FU | 5-fluorouracil |
| AKT | RAC-alpha serine/threonine-protein kinase |
| APC | Antigen-presenting cell |
| APAF-1 | Apoptotic protease-activating factor 1 |
| ATM | Serine-protein kinases ataxia telangiectasia mutated |
| ATRIP | ATR-interacting protein |
| BAD | Bcl2-associated agonist of cell death |
| BAP31 | B-cell receptor-associated protein 31 |
| BER | Base excision repair |
| BRAF | Serine/threonine-protein kinase B-raf |
| BRCA1/2 | Breast cancer type 1 susceptibility protein 1/2 |
| CDC25A/C | M-phase inducer phosphatases |
| CDK | Cyclin-dependent kinase |
| cGAS | Cyclic GMP-AMP synthase |
| CHK1/2 | Serine/threonine-protein kinase CHK1/2 |
| CK2 | Casein kinase II |
| COX2 | Cyclooxygenase-2 |
| CRT | Calreticulin |
| CSC | Cancer stem cells |
| CtIP | CtBP-interacting protein |
| DAMPS | Damage-associated molecular pattern molecules |
| DCs | Dendritic cells |
| DDR | DNA damage response |
| DISC | Death-inducing signaling complex |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DSBR | Double-strand break repair |
| DSBs | Double-strand breaks |
| ED | Extracellular domain |
| eIF2α | Eukaryotic translation initiation factor 2A |
| EMT | Epithelial-to-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| ES-SCLC | Extensive-stage small cell lung cancer |
| EXO1 | Exonuclease 1 |
| FA | Fanconi anemia |
| FADD | FAS-associated death domain protein |
| FdUMP | 5-fluoro-2′-deoxyuridine 5′-monophosphate |
| FdUTP | Fluorodeoxyuridine triphosphate |
| FLIP | Flice-like inhibitory protein |
| FOXO1 | Forkhead box protein O1 |
| GADD45 | 45kd-growth arrest and DNA damage |
| GDSM | Gasdermin |
| GZMA/B | Granzyme A/B |
| HIF-1α | Hypoxia-inducible factor alpha |
| HMG1/2 | Nonhistone chromosomal high-mobility group 1 and 2 |
| HMGB1 | High mobility group protein 1 |
| HR | Homologous recombination |
| hUBF | Highly similar to Human upstream binding factor |
| ICD | Immunogenic cell death |
| ICLs | Interstrand crosslinks |
| ICT | Intracellular cytoplasmic tail |
| IFN | Interferon |
| IL | Interleukin |
| ILC2s | Group 2 innate lymphoid cells |
| irAEs | Immune-related adverse effects |
| IRF | Interferon regulatory factor |
| ITIM | Immunoreceptor tyrosine-based inhibitory motif |
| ITSM | Immunoreceptor tyrosine-based switch motif |
| JAK | Tyrosine-protein kinase |
| JNK | Jun amino-terminal kinase |
| KU70/80 | X-ray repair cross-complementing protein 5/6 |
| M2 | Tumor-associated macrophages M2 |
| mAbs | Monoclonal antibodies |
| MCM2/7 | Minichromosome maintenance complex |
| MDC-1 | Mediator of DNA damage checkpoint protein 1 |
| MDM2 | Mouse double minute 2 homolog |
| MDSC | Myeloid-derived suppressor cells |
| MDSCs | Myeloid-derived suppressor cells |
| MEK | Dual specificity mitogen-activated protein kinase kinase |
| MHC | Major histocompatibility complex |
| MLH1 | DNA mismatch repair protein MLH1 |
| MMR | Mismatch repair |
| mPGES1 | Prostaglandin E synthase |
| MRE11 | Double-strand break repair protein MRE11 |
| MSH2/6 | DNA mismatch repair protein MSH2/6 |
| MSI | Microsatellite instability |
| mtDNA | Mitochondrial DNA |
| mTOR | Serine/threonine-protein kinase mTOR |
| Nal-IRI | Liposomal irinotecan |
| NBS1 | Nibrin |
| nDNA | Nuclear DNA |
| NER | Nucleotide excision repair |
| NF-Kβ | Nuclear factor NF-kappa-B p105 subunit |
| NHEJ | Non-homologous end joining |
| NK | Natural killer cells |
| OMI | Serine protease HTRA2 |
| OS | Overall survival |
| P2RX7 | P2X purinoceptor 7 |
| PAR | Polymer ADP-ribose residues |
| PARP-1 | Poly [ADP-ribose] polymerase 1 |
| PARPi | PARP inhibitor |
| PD-1/PD-L1 | Programmed death receptor-1/programmed death ligand-1 |
| PERK | Eukaryotic translation initiation factor 2-alpha kinase 3 |
| PFN | Perforins |
| PFS | Progression-free survival |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PKC | Protein kinase C |
| PLCg-1 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 |
| PLK | Serine/threonine-protein kinase PLK |
| PRR | Pattern recognition receptors |
| PS | Phosphatidylserine |
| PTEN | Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase |
| RAD50 | DNA repair protein RAD50 |
| RAD51 | DNA repair protein RAD51 homolog 1 |
| RAD52 | DNA repair protein RAD52 homolog |
| RAP80 | Receptor-associated protein 80 |
| RNF4, -8, -168 | E3 ubiquitin-protein ligases |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| SCLC | Small-cell lung cancer |
| SHP1/2 | Src homology region 2 domain containing phosphatases |
| SMACK | Diablo IAP-binding mitochondrial protein |
| SSBR | Single-strand break repair |
| ssDNA | Single-stranded DNA |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon genes protein |
| TBP | TATA binding protein |
| TCR | T-cell receptor |
| TGF-β | Transforming growth factor β |
| TICs | Tumor-initiating cells |
| TILs | Tumor infiltrating lymphocytes |
| TLR | Toll-like receptor |
| TLS | Translesion DNA synthesis |
| TM | Transmembrane domain |
| TMB | Tumor mutational burden |
| TNF-α | Tumor necrosis factor α |
| TOPBP1 | Topoisomerase 2-binding protein 1 |
| TP53 | Cellular tumor antigen p53 |
| Tregs | Regulatory T cells |
| WAF1/CIP1 | Cyclin-dependent kinase inhibitor 1 |
| XLF | Non-homologous end-joining factor 1 |
| XRCC4 | X-ray repair cross-complementing protein 4 |
References
- Dillman, R.O. Cancer Immunotherapy. Cancer Biother. Radiopharm. 2011, 26, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D. An Overview of Cancer Immunotherapy. Immunol. Cell. Biol. 2000, 78, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Gamrekelashvili, J.; Krüger, C.; von Wasielewski, R.; Hoffmann, M.; Huster, K.M.; Busch, D.H.; Manns, M.P.; Korangy, F.; Greten, T.F. Necrotic Tumor Cell Death in Vivo Impairs Tumor-Specific Immune Responses. J. Immunol. 2007, 178, 1573–1580. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Nowak, A.K.; Lake, R.A. Contribution of the Immune System to the Chemotherapeutic Response. Semin. Immunopathol. 2011, 33, 353–367. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined Cytotoxic Chemotherapy and Immunotherapy of Cancer: Modern Times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Park, S.-J.; Ye, W.; Xiao, R.; Silvin, C.; Padget, M.; Hodge, J.W.; Van Waes, C.; Schmitt, N.C. Cisplatin and Oxaliplatin Induce Similar Immunogenic Changes in Preclinical Models of Head and Neck Cancer. Oral Oncol. 2019, 95, 127–135. [Google Scholar] [CrossRef]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer Metal Drugs and Immunogenic Cell Death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. Engl. 2018, 57, 4840–4848. [Google Scholar] [CrossRef] [PubMed]
- Inthagard, J.; Edwards, J.; Roseweir, A.K. Immunotherapy: Enhancing the Efficacy of This Promising Therapeutic in Multiple Cancers. Clin. Sci. 2019, 133, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in Cancer Immunotherapy: Clinical Implications and Future Considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 Pathway: Basic Biology and Role in Cancer Immunotherapy. J. Cell Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef] [PubMed]
- LV, B.; Wang, Y.; Ma, D.; Cheng, W.; Liu, J.; Yong, T.; Chen, H.; Wang, C. Immunotherapy: Reshape the Tumor Immune Microenvironment. Front. Immunol. 2022, 13, 844142. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liu, Q.; Xiang, Y.; Gou, X.; Li, W. Role of the Tumor Immune Microenvironment in Tumor Immunotherapy. Oncol. Lett. 2022, 23, 53. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, S.H.; Heo, Y.-S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef]
- Descourt, R.; Greillier, L.; Perol, M.; Ricordel, C.; Auliac, J.-B.; Falchero, L.; Gervais, R.; Veillon, R.; Vieillot, S.; Guisier, F.; et al. First-Line Single-Agent Pembrolizumab for PD-L1-Positive (Tumor Proportion Score ≥ 50%) Advanced Non-Small Cell Lung Cancer in the Real World: Impact in Brain Metastasis: A National French Multicentric Cohort (ESCKEYP GFPC Study). Cancer Immunol. Immunother. 2022, 72, 91–99. [Google Scholar] [CrossRef]
- Duan, H.; Shao, C.; Pan, M.; Liu, H.; Dong, X.; Zhang, Y.; Tong, L.; Feng, Y.; Wang, Y.; Wang, L.; et al. Neoadjuvant Pembrolizumab and Chemotherapy in Resectable Esophageal Cancer: An Open-Label, Single-Arm Study (PEN-ICE). Front. Immunol. 2022, 13, 849984. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Yuan, W.; Pal, A.; Crespo, M.; Ferreira, A.; Gurel, B.; Prout, T.; Ameratunga, M.; Chénard-Poirier, M.; Curcean, A.; et al. Phase 1, Dose-Escalation Study of Guadecitabine (SGI-110) in Combination with Pembrolizumab in Patients with Solid Tumors. J. Immunother. Cancer 2022, 10, e004495. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Qin, S.; Wang, L.; Tan, C.; Peng, Y.; Zeng, X.; Luo, X.; Yi, L.; Wan, X. Cost-Effectiveness of Pembrolizumab Plus Chemotherapy as First-Line Therapy for Advanced Oesophageal Cancer. Front. Pharmacol. 2022, 13, 881787. [Google Scholar] [CrossRef] [PubMed]
- Miron, B.; Geynisman, D.M. Bempegaldesleukin/Nivolumab and Challenges in First-Line Treatment of Metastatic Urothelial Carcinoma. Eur. Urol. 2022, 82, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Bennani, N.N.; Kim, H.J.; Pederson, L.D.; Atherton, P.J.; Micallef, I.N.; Thanarajasingam, G.; Nowakowski, G.S.; Witzig, T.; Feldman, A.L.; Ansell, S.M. Nivolumab in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Modest Activity and Cases of Hyperprogression. J. Immunother. Cancer 2022, 10, e004984. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Yasumatsu, R.; Masuda, M.; Yamauchi, M.; Wakasaki, T.; Hashimoto, K.; Jiromaru, R.; Manako, T.; Nakagawa, T. Five-Year Follow-up of Patients With Head and Neck Cancer Treated With Nivolumab and Long-Term Responders for Over Two Years. In Vivo 2022, 36, 1881–1886. [Google Scholar] [CrossRef]
- Stein, A.; Paschold, L.; Tintelnot, J.; Goekkurt, E.; Henkes, S.-S.; Simnica, D.; Schultheiss, C.; Willscher, E.; Bauer, M.; Wickenhauser, C.; et al. Efficacy of Ipilimumab vs FOLFOX in Combination With Nivolumab and Trastuzumab in Patients With Previously Untreated ERBB2-Positive Esophagogastric Adenocarcinoma: The AIO INTEGA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1150–1158. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, J.; Zhang, Z.; Cen, C.; Chen, W.; Jiang, B.; Meng, Y.; Wang, Y.; Berglund, B.; Zhai, G.; et al. Comprehensive Evaluation of Anti-PD-1, Anti-PD-L1, Anti-CTLA-4 and Their Combined Immunotherapy in Clinical Trials: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 883655. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, C.; Ding, D.; Tian, H.; Yu, C. Comparative Efficacy and Safety of Anti-PD-1/PD-L1 for the Treatment of Non-Small Cell Lung Cancer: A Network Meta-Analysis of 13 Randomized Controlled Studies. Front. Oncol. 2022, 12, 827050. [Google Scholar] [CrossRef]
- Imai, H.; Nagai, Y.; Minemura, H.; Tsuda, T.; Yamada, Y.; Wasamoto, S.; Kishikawa, T.; Shiono, A.; Shiihara, J.; Yamaguchi, O.; et al. Efficacy and Safety of Amrubicin Monotherapy after Atezolizumab plus Carboplatin and Etoposide in Patients with Relapsed Small-Cell Lung Cancer. Investig. New Drugs 2022, 40, 1066–1079. [Google Scholar] [CrossRef]
- Kuwano, A.; Yada, M.; Narutomi, F.; Nagasawa, S.; Tanaka, K.; Kurosaka, K.; Ohishi, Y.; Masumoto, A.; Motomura, K. Therapeutic Efficacy of Atezolizumab plus Bevacizumab for Hepatocellular Carcinoma with WNT/β-Catenin Signal Activation. Oncol. Lett. 2022, 24, 216. [Google Scholar] [CrossRef]
- Gürbüz, M.; Kutlu, Y.; Akkuş, E.; Köksoy, E.B.; Köse, N.; Öven, B.B.; Uluç, B.O.; Demiray, A.G.; Erdem, D.; Demir, B.; et al. Atezolizumab Combined with Chemotherapy in the First-Line Treatment of Extensive-Stage Small Cell Lung Cancer: A Real-Life Data of the Turkish Oncology Group. J. Cancer Res. Clin. Oncol. 2022, 148, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Rupp, A.-K.; Poschke, I.; Bunse, T.; Lindner, K.; Wick, A.; Blobner, J.; Misch, M.; Tabatabai, G.; Glas, M.; et al. AMPLIFY-NEOVAC: A Randomized, 3-Arm Multicenter Phase I Trial to Assess Safety, Tolerability and Immunogenicity of IDH1-Vac Combined with an Immune Checkpoint Inhibitor Targeting Programmed Death-Ligand 1 in Isocitrate Dehydrogenase 1 Mutant Gliomas. Neurol. Res. Pract. 2022, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.; Rodriguez-Freixinos, V.; Doherty, M.; Wasson, K.; Iscoe, N.; Raskin, W.; Hallet, J.; Myrehaug, S.; Law, C.; Thawer, A.; et al. Avelumab in Unresectable/Metastatic, Progressive, Grade 2-3 Neuroendocrine Neoplasms (NENs): Combined Results from NET-001 and NET-002 Trials. Eur. J. Cancer 2022, 169, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Park, S.H.; Kozlov, V.; Dao, T.V.; Castellano, D.; Li, J.-R.; Mukherjee, S.D.; Howells, K.; Dry, H.; Lanasa, M.C.; et al. Durvalumab Plus Olaparib in Previously Untreated, Platinum-Ineligible Patients With Metastatic Urothelial Carcinoma: A Multicenter, Randomized, Phase II Trial (BAYOU). J. Clin. Oncol. 2022, 41, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kawanaka, Y.; Yasuda, Y.; Tanizaki, J.; Iwashima, D.; Nonagase, Y.; Uemasu, K.; Hirayama, Y.; Ogura, M.; Ozaki, T.; Takahashi, K.-I. The Safety and Efficacy of Durvalumab Consolidation Therapy in the Management of Patients with Stage III Non-Small-Cell Lung Cancer and Preexisting Interstitial Lung Disease. Respir. Investig. 2022, 60, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Uchimiak, K.; Badowska-Kozakiewicz, A.M.; Sobiborowicz-Sadowska, A.; Deptała, A. Current State of Knowledge on the Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer Treatment: Approaches, Efficacy, and Challenges. Clin. Med. Insights Oncol. 2022, 16, 11795549221099868. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, L.; Zuo, Y.; Qian, H.; Liu, C. Immune Checkpoint Blockade in Cancer Immunotherapy: Mechanisms, Clinical Outcomes, and Safety Profiles of PD-1/PD-L1 Inhibitors. Arch. Immunol. Ther. Exp. 2020, 68, 36. [Google Scholar] [CrossRef]
- Xin Yu, J.; Hodge, J.P.; Oliva, C.; Neftelinov, S.T.; Hubbard-Lucey, V.M.; Tang, J. Trends in Clinical Development for PD-1/PD-L1 Inhibitors. Nat. Rev. Drug Discov. 2020, 19, 163–164. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Reynoso, E.D.; Elpek, K.G.; Francisco, L.; Bronson, R.; Bellemare-Pelletier, A.; Sharpe, A.H.; Freeman, G.J.; Turley, S.J. Intestinal Tolerance Is Converted to Autoimmune Enteritis upon PD-1 Ligand Blockade. J. Immunol. 2009, 182, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and Its Ligands Are Important Immune Checkpoints in Cancer. Oncotarget 2016, 8, 2171–2186. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical Signaling of PD-1 on T Cells and Its Functional Implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The ’Shp’ing News: SH2 Domain-Containing Tyrosine Phosphatases in Cell Signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3zeta Signalosome and Downstream Signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 Immunoreceptor Inhibits B Cell Receptor-Mediated Signaling by Recruiting Src Homology 2-Domain-Containing Tyrosine Phosphatase 2 to Phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol. Cell Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 Is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical Blockade of PD1 and LAG3--Potential Mechanisms of Action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Philips, E.A.; Garcia-España, A.; Tocheva, A.S.; Ahearn, I.M.; Adam, K.R.; Pan, R.; Mor, A.; Kong, X.-P. The Structural Features That Distinguish PD-L2 from PD-L1 Emerged in Placental Mammals. J. Biol. Chem. 2020, 295, 4372–4380. [Google Scholar] [CrossRef]
- Cheng, X.; Veverka, V.; Radhakrishnan, A.; Waters, L.C.; Muskett, F.W.; Morgan, S.H.; Huo, J.; Yu, C.; Evans, E.J.; Leslie, A.J.; et al. Structure and Interactions of the Human Programmed Cell Death 1 Receptor. J. Biol. Chem. 2013, 288, 11771–11785. [Google Scholar] [CrossRef] [PubMed]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The Intracellular Signalosome of PD-L1 in Cancer Cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/MPGES1/PGE2 Pathway Regulates PD-L1 Expression in Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Van Duijn, A.; Willemsen, K.J.; van Uden, N.O.P.; Hoyng, L.; Erades, S.; Koster, J.; Luiten, R.M.; Bakker, W.J. A Secondary Role for Hypoxia and HIF1 in the Regulation of (IFNγ-Induced) PD-L1 Expression in Melanoma. Cancer Immunol. Immunother. 2022, 71, 529–540. [Google Scholar] [CrossRef]
- Zhou, L.; Cha, G.; Chen, L.; Yang, C.; Xu, D.; Ge, M. HIF1α/PD-L1 Axis Mediates Hypoxia-Induced Cell Apoptosis and Tumor Progression in Follicular Thyroid Carcinoma. OncoTargets Ther. 2019, 12, 6461–6470. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-ΚB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-MTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Taghiloo, S.; Norozi, S.; Asgarian-Omran, H. The Effects of PI3K/Akt/MTOR Signaling Pathway Inhibitors on the Expression of Immune Checkpoint Ligands in Acute Myeloid Leukemia Cell Line. Iran. J. Allergy Asthma Immunol. 2022, 21, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Fan, S.; Li, J.; Zhou, X.; Liao, Q.; Tang, F.; Liu, W. Programmed Death-Ligand 1 Signaling and Expression Are Reversible by Lycopene via PI3K/AKT and Raf/MEK/ERK Pathways in Tongue Squamous Cell Carcinoma. Genes Nutr. 2022, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK Pathway Activity Plays a Key Role in PD-L1 Expression of Lung Adenocarcinoma Cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef]
- Xing, S.; Chen, S.; Yang, X.; Huang, W. Role of MAPK Activity in PD-L1 Expression in Hepatocellular Carcinoma Cells. J. BUON 2020, 25, 1875–1882. [Google Scholar]
- Bu, L.L.; Yu, G.T.; Wu, L.; Mao, L.; Deng, W.W.; Liu, J.F.; Kulkarni, A.B.; Zhang, W.F.; Zhang, L.; Sun, Z.J. STAT3 Induces Immunosuppression by Upregulating PD-1/PD-L1 in HNSCC. J. Dent. Res. 2017, 96, 1027–1034. [Google Scholar] [CrossRef]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Kołat, D.; Kałuzińska, Ż.; Kontek, R. Cancer-Associated Transcription Factors in DNA Damage Response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188757. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair-Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-Dependent Kinases in DNA Damage Response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef] [PubMed]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 Kinase in the DNA Damage Response and Beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Huang, S. The Role of Cdc25A in the Regulation of Cell Proliferation and Apoptosis. Anticancer Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Nussenzweig, A. Causes and Consequences of the DNA Damage Response. Cell Cycle 2007, 6, 2339–2340. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of Non-Homologous End Joining (NHEJ) in Maintaining Genomic Integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and Anti-PD-L1 Treatment Synergistically Promote Antitumor Immunity in Mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Wu, C.-T.; Chen, W.-C.; Chang, Y.-H.; Lin, W.-Y.; Chen, M.-F. The Role of PD-L1 in the Radiation Response and Clinical Outcome for Bladder Cancer. Sci. Rep. 2016, 6, 19740. [Google Scholar] [CrossRef]
- Bedi, D.; Henderson, H.J.; Manne, U.; Samuel, T. Camptothecin Induces PD-L1 and Immunomodulatory Cytokines in Colon Cancer Cells. Medicines 2019, 6, 51. [Google Scholar] [CrossRef]
- Gilad, Y.; Eliaz, Y.; Yu, Y.; Han, S.J.; O’Malley, B.W.; Lonard, D.M. Drug-Induced PD-L1 Expression and Cell Stress Response in Breast Cancer Cells Can Be Balanced by Drug Combination. Sci. Rep. 2019, 9, 15099. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune Induction Strategies in Metastatic Triple-Negative Breast Cancer to Enhance the Sensitivity to PD-1 Blockade: The TONIC Trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Wang, J.; Hu, C.; Wang, J.; Shen, Y.; Bao, Q.; He, F.; Wang, H.; Gong, L.; Liu, Z.; Hu, F.; et al. Checkpoint Blockade in Combination With Doxorubicin Augments Tumor Cell Apoptosis in Osteosarcoma. J. Immunother. 2019, 42, 321–330. [Google Scholar] [CrossRef]
- Iwai, T.; Sugimoto, M.; Wakita, D.; Yorozu, K.; Kurasawa, M.; Yamamoto, K. Topoisomerase I Inhibitor, Irinotecan, Depletes Regulatory T Cells and up-Regulates MHC Class I and PD-L1 Expression, Resulting in a Supra-Additive Antitumor Effect When Combined with Anti-PD-L1 Antibodies. Oncotarget 2018, 9, 31411–31421. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, J.; Cai, H.; Lang, J. Carboplatin and Programmed Death-Ligand 1 Blockade Synergistically Produce a Similar Antitumor Effect to Carboplatin Alone in Murine ID8 Ovarian Cancer Model. J. Obstet. Gynaecol. Res. 2018, 44, 303–311. [Google Scholar] [CrossRef]
- Wahba, J.; Natoli, M.; Whilding, L.M.; Parente-Pereira, A.C.; Jung, Y.; Zona, S.; Lam, E.W.-F.; Smith, J.R.; Maher, J.; Ghaem-Maghami, S. Chemotherapy-Induced Apoptosis, Autophagy and Cell Cycle Arrest Are Key Drivers of Synergy in Chemo-Immunotherapy of Epithelial Ovarian Cancer. Cancer Immunol. Immunother. 2018, 67, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin Increases PD-L1 Expression and Optimizes Immune Check-Point Blockade in Non-Small Cell Lung Cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.-J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.-Y.; Kim, S.; Keam, B.; Kim, S.; Ahn, Y.-O.; Chung, E.-J.; Kim, J.-H.; Kim, T.M.; Kwon, S.K.; Jeon, Y.K.; et al. Changes in Programmed Death-Ligand 1 Expression during Cisplatin Treatment in Patients with Head and Neck Squamous Cell Carcinoma. Oncotarget 2017, 8, 97920–97927. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.-F.; Lin, J.-F.; Lin, Y.-C.; Chou, K.-Y.; Chen, H.-E.; Ho, C.-Y.; Chen, P.-C.; Hwang, T.I.-S. Cisplatin Contributes to Programmed Death-Ligand 1 Expression in Bladder Cancer through ERK1/2-AP-1 Signaling Pathway. Biosci. Rep. 2019, 39, BSR20190362. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin Induces Programmed Death-1-Ligand 1(PD-L1) over-Expression in Hepatoma H22 Cells via Erk /MAPK Signaling Pathway. Cell. Mol. Biol. 2010, 56, 1366–1372. [Google Scholar]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Pham, T.; Mansfield, A.S.; Yan, Y.; et al. Targeting B7-H1 (PD-L1) Sensitizes Cancer Cells to Chemotherapy. Heliyon 2018, 4, e01039. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Wang, H.; Li, A.; Xu, Y.; Tang, L.; Chen, Q.; Zhang, C.; Gao, Y.; Song, J.; Du, Z. Decitibine Improve the Efficiency of Anti-PD-1 Therapy via Activating the Response to IFN/PD-L1 Signal of Lung Cancer Cells. Oncogene 2018, 37, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Bensaid, D.; Blondy, T.; Deshayes, S.; Dehame, V.; Bertrand, P.; Grégoire, M.; Errami, M.; Blanquart, C. Assessment of New HDAC Inhibitors for Immunotherapy of Malignant Pleural Mesothelioma. Clin. Epigenet. 2018, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Lailler, C.; Lamuraglia, M.; Racine, F.; Louandre, C.; Godin, C.; Chauffert, B.; Galmiche, A.; Saidak, Z. DNA Damage Response- and JAK-Dependent Regulation of PD-L1 Expression in Head and Neck Squamous Cell Carcinoma (HNSCC) Cells Exposed to 5-Fluorouracil (5-FU). Transl. Oncol. 2021, 14, 101110. [Google Scholar] [CrossRef]
- Van Der Kraak, L.; Goel, G.; Ramanan, K.; Kaltenmeier, C.; Zhang, L.; Normolle, D.P.; Freeman, G.J.; Tang, D.; Nason, K.S.; Davison, J.M.; et al. 5-Fluorouracil Upregulates Cell Surface B7-H1 (PD-L1) Expression in Gastrointestinal Cancers. J. Immunother. Cancer 2016, 4, 65. [Google Scholar] [CrossRef]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT Pathway Is Involved in the Upregulation of PD-L1 Expression in Pancreatic Cancer Cell Lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 Expression in Colorectal Cancer Is Associated with Microsatellite Instability, BRAF Mutation, Medullary Morphology and Cytotoxic Tumor-Infiltrating Lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Roberts, J.; Salaria, S.N.; Cates, J.; Wang, Y.; Vnencak-Jones, C.; Berlin, J.; Shi, C. PD-L1 Expression Patterns in Microsatellite Instability-High Intestinal Adenocarcinoma Subtypes. Am. J. Clin. Pathol. 2019, 152, 384–391. [Google Scholar] [CrossRef]
- Morihiro, T.; Kuroda, S.; Kanaya, N.; Kakiuchi, Y.; Kubota, T.; Aoyama, K.; Tanaka, T.; Kikuchi, S.; Nagasaka, T.; Nishizaki, M.; et al. PD-L1 Expression Combined with Microsatellite Instability/CD8+ Tumor Infiltrating Lymphocytes as a Useful Prognostic Biomarker in Gastric Cancer. Sci. Rep. 2019, 9, 4633. [Google Scholar] [CrossRef]
- Cho, Y.A.; Lee, H.; Kim, D.G.; Kim, H.; Ha, S.Y.; Choi, Y.-L.; Jang, K.-T.; Kim, K.-M. PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score. Cancers 2021, 13, 4659. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.F.; Bretes, L.; Furtado, I. Review of PD-1/PD-L1 Inhibitors in Metastatic DMMR/MSI-H Colorectal Cancer. Front. Oncol. 2019, 9, 396. [Google Scholar] [CrossRef]
- Xiao, Y.; Freeman, G.J. The Microsatellite Instable Subset of Colorectal Cancer Is a Particularly Good Candidate for Checkpoint Blockade Immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed]
- Seetharamu, N.; Preeshagul, I.R.; Sullivan, K.M. New PD-L1 Inhibitors in Non-Small Cell Lung Cancer—Impact of Atezolizumab. Lung Cancer 2017, 8, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo, C.-Y.; Tou, F.-F.; Wen, X.-M.; Kuang, Y.-K.; Zhu, Q.; Hu, H. Association of PD-L1 Expression Status with the Efficacy of PD-1/PD-L1 Inhibitors and Overall Survival in Solid Tumours: A Systematic Review and Meta-Analysis. Int. J. Cancer 2020, 147, 116–127. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 Kinase Destabilizes PD-L1 via Cullin 3-SPOP to Control Cancer Immune Surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Cheung, A.; Chenoweth, A.M.; Quist, J.; Sow, H.S.; Malaktou, C.; Ferro, R.; Hoffmann, R.M.; Osborn, G.; Sachouli, E.; French, E.; et al. CDK Inhibition Primes for Anti-PD-L1 Treatment in Triple-Negative Breast Cancer Models. Cancers 2022, 14, 3361. [Google Scholar] [CrossRef]
- Sun, S.-Y. Searching for the Real Function of MTOR Signaling in the Regulation of PD-L1 Expression. Transl. Oncol. 2020, 13, 100847. [Google Scholar] [CrossRef]
- Chen, B.; Hu, J.; Hu, X.; Chen, H.; Bao, R.; Zhou, Y.; Ye, Y.; Zhan, M.; Cai, W.; Li, H.; et al. DENR Controls JAK2 Translation to Induce PD-L1 Expression for Tumor Immune Evasion. Nat. Commun. 2022, 13, 2059. [Google Scholar] [CrossRef]
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 Expression Increased by IFN-γ via JAK2-STAT1 Signaling and Predicts a Poor Survival in Colorectal Cancer. Oncol. Lett. 2020, 20, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, Y.; Lv, X. IL4I1 Enhances PD-L1 Expression through JAK/STAT Signaling Pathway in Lung Adenocarcinoma. Immunogenetics 2023, 75, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Mbofung, R.M.; Malu, S.; Zhang, M.; Ashkin, E.; Devi, S.; Williams, L.; Tieu, T.; Peng, W.; Pradeep, S.; et al. The Effect of Topoisomerase I Inhibitors on the Efficacy of T-Cell-Based Cancer Immunotherapy. J. Natl. Cancer Inst. 2018, 110, 777–786. [Google Scholar] [CrossRef]
- Koyama, S.; Nagatomo, I.; Kijima, T.; Kumanogoh, A. Selecting Suitable Chemotherapies for PD-1/PD-L1 Blockade to Optimize the Tumor Immune Microenvironment. Oncotarget 2018, 9, 32552–32553. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 Years of Cancer Treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Ko, A.H. Nanomedicine Developments in the Treatment of Metastatic Pancreatic Cancer: Focus on Nanoliposomal Irinotecan. Int. J. Nanomed. 2016, 11, 1225–1235. [Google Scholar] [CrossRef]
- Xue, L.; Chen, B.; Lin, J.; Peng, J. Anti-PD-L1 Immune Checkpoint Inhibitors in Combination with Etoposide and Platinum for Extensive-Stage Small Cell Lung Cancer: A Case Report. Ann. Palliat Med. 2021, 10, 828–835. [Google Scholar] [CrossRef]
- Kataoka, N.; Kunimatsu, Y.; Tachibana, Y.; Sugimoto, T.; Sato, I.; Tani, N.; Ogura, Y.; Hirose, K.; Takeda, T. Atezolizumab in Combination with Carboplatin and Etoposide for Heavily Treated Small Cell Lung Cancer. Thorac. Cancer 2020, 11, 2740–2742. [Google Scholar] [CrossRef]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Facchinetti, F.; Di Maio, M.; Tiseo, M. Adding PD-1/PD-L1 Inhibitors to Chemotherapy for the First-Line Treatment of Extensive Stage Small Cell Lung Cancer (SCLC): A Meta-Analysis of Randomized Trials. Cancers 2020, 12, 2645. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Biamonti, G. Cellular Response to Etoposide Treatment. Cancer Lett. 2007, 252, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Théard, D.; Coisy, M.; Ducommun, B.; Concannon, P.; Darbon, J.M. Etoposide and Adriamycin but Not Genistein Can Activate the Checkpoint Kinase Chk2 Independently of ATM/ATR. Biochem. Biophys. Res. Commun. 2001, 289, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Lidonnici, M.R.; Soza, S.; Biamonti, G.; Montecucco, A. The Dispersal of Replication Proteins after Etoposide Treatment Requires the Cooperation of Nbs1 with the Ataxia Telangiectasia Rad3-Related/Chk1 Pathway. Cancer Res. 2006, 66, 1675–1683. [Google Scholar] [CrossRef]
- Weber, A.M.; Drobnitzky, N.; Devery, A.M.; Bokobza, S.M.; Adams, R.A.; Maughan, T.S.; Ryan, A.J. Phenotypic Consequences of Somatic Mutations in the Ataxia-Telangiectasia Mutated Gene in Non-Small Cell Lung Cancer. Oncotarget 2016, 7, 60807–60822. [Google Scholar] [CrossRef]
- Caporossi, D.; Porfirio, B.; Nicoletti, B.; Palitti, F.; Degrassi, F.; De Salvia, R.; Tanzarella, C. Hypersensitivity of Lymphoblastoid Lines Derived from Ataxia Telangiectasia Patients to the Induction of Chromosomal Aberrations by Etoposide (VP-16). Mutat. Res. 1993, 290, 265–272. [Google Scholar] [CrossRef]
- Kijas, A.W.; Lim, Y.C.; Bolderson, E.; Cerosaletti, K.; Gatei, M.; Jakob, B.; Tobias, F.; Taucher-Scholz, G.; Gueven, N.; Oakley, G.; et al. ATM-Dependent Phosphorylation of MRE11 Controls Extent of Resection during Homology Directed Repair by Signalling through Exonuclease 1. Nucleic Acids Res. 2015, 43, 8352–8367. [Google Scholar] [CrossRef]
- Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef]
- Maser, R.S.; Monsen, K.J.; Nelms, B.E.; Petrini, J.H. HMre11 and HRad50 Nuclear Foci Are Induced during the Normal Cellular Response to DNA Double-Strand Breaks. Mol. Cell. Biol. 1997, 17, 6087–6096. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-Coated Single-Stranded DNA as a Platform for Post-Translational Modifications in the DNA Damage Response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Loegering, D.; Arlander, S.J.H.; Hackbarth, J.; Vroman, B.T.; Roos-Mattjus, P.; Hopkins, K.M.; Lieberman, H.B.; Karnitz, L.M.; Kaufmann, S.H. Rad9 Protects Cells from Topoisomerase Poison-Induced Cell Death. J. Biol. Chem. 2004, 279, 18641–18647. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome Maintenance Mechanisms for Preventing Cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Suzuki, H.; Iiizumi, S.; Koyama, H. Hypersensitivity of Nonhomologous DNA End-Joining Mutants to VP-16 and ICRF-193: Implications for the Repair of Topoisomerase II-Mediated DNA Damage. J. Biol. Chem. 2003, 278, 35897–35902. [Google Scholar] [CrossRef]
- Malik, M.; Nitiss, K.C.; Enriquez-Rios, V.; Nitiss, J.L. Roles of Nonhomologous End-Joining Pathways in Surviving Topoisomerase II-Mediated DNA Damage. Mol. Cancer Ther. 2006, 5, 1405–1414. [Google Scholar] [CrossRef]
- Fan, Y.; Dutta, J.; Gupta, N.; Fan, G.; Gélinas, C. Regulation of Programmed Cell Death by NF-KappaB and Its Role in Tumorigenesis and Therapy. Adv. Exp. Med. Biol. 2008, 615, 223–250. [Google Scholar] [CrossRef]
- Jin, S.; Inoue, S.; Weaver, D.T. Differential Etoposide Sensitivity of Cells Deficient in the Ku and DNA-PKcs Components of the DNA-Dependent Protein Kinase. Carcinogenesis 1998, 19, 965–971. [Google Scholar] [CrossRef]
- Palmitelli, M.; de Campos-Nebel, M.; González-Cid, M. Progression of Chromosomal Damage Induced by Etoposide in G2 Phase in a DNA-PKcs-Deficient Context. Chromosome Res. 2015, 23, 719–732. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, Topoisomerase II and Cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Schonn, I.; Hennesen, J.; Dartsch, D.C. Cellular Responses to Etoposide: Cell Death despite Cell Cycle Arrest and Repair of DNA Damage. Apoptosis 2010, 15, 162–172. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.-I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging Agents That Target Signaling Pathways in Cancer Stem Cells. J. Hematol. Oncol. 2020, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Budzinska, A.; Mojzych, M.; Kontek, R. Metastasis and MAPK Pathways. Int. J. Mol. Sci. 2022, 23, 3847. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-Dependent PD-L1 Accumulation on Cancer Stem Cells Promotes Immune Evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.M.; Stupecky, M.; Sweet, P.; Osann, K.; Eklof, A.; Arquilla, E.R. Etoposide Induction of Tumor Immunity in Lewis Lung Cancer. Cancer Chemother. Pharmacol. 2001, 48, 327–332. [Google Scholar] [CrossRef]
- Slater, L.M.; Stupecky, M.; Sweet, P.; Osann, K.E. Enhancement of Leukemia Rejection by Mice Successfully Treated for L1210 Leukemia Due to Low Dose Compared to High Dose VP-16. Leuk. Res. 2002, 26, 203–206. [Google Scholar] [CrossRef]
- Slater, L.M.; Sweet, P.; Stupecky, M.; Reynolds, J.T. Cyclosporin A/VP-16 Produced Immunity to L1210 Leukemia: The Participation of Cytotoxic CD8 T-Lymphocytes. Clin. Immunol. Immunopathol. 1995, 75, 239–245. [Google Scholar] [CrossRef]
- Zhang, P.; Su, D.-M.; Liang, M.; Fu, J. Chemopreventive Agents Induce Programmed Death-1-Ligand 1 (PD-L1) Surface Expression in Breast Cancer Cells and Promote PD-L1-Mediated T Cell Apoptosis. Mol. Immunol. 2008, 45, 1470–1476. [Google Scholar] [CrossRef]
- Yang, M.; Liu, P.; Wang, K.; Glorieux, C.; Hu, Y.; Wen, S.; Jiang, W.; Huang, P. Chemotherapy Induces Tumor Immune Evasion by Upregulation of Programmed Cell Death Ligand 1 Expression in Bone Marrow Stromal Cells. Mol. Oncol. 2017, 11, 358–372. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Hu, J.; Zhang, H.; Xu, F.; He, W.; Wang, X.; Li, M.; Lu, W.; Zeng, G.; et al. CGAS/STING Axis Mediates a Topoisomerase II Inhibitor-Induced Tumor Immunogenicity. J. Clin. Investig. 2019, 129, 4850–4862. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Noll, D.M.; Mason, T.M.; Miller, P.S. Formation and Repair of Interstrand Cross-Links in DNA. Chem. Rev. 2006, 106, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of Interstrand DNA Crosslink Repair and Human Disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Kawasoe, Y.; Williams, H.; Coates, E.; Roy, U.; Shi, Y.; Beese, L.S.; Schärer, O.D.; Yan, H.; Gottesman, M.E.; et al. Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway. Cell Rep. 2017, 21, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Mizoshita, T.; Ozeki, K.; Tsukamoto, H.; Kamiya, T.; Kataoka, H.; Sakamuro, D.; Joh, T. Mechanisms of Cisplatin-Induced Apoptosis and of Cisplatin Sensitivity: Potential of BIN1 to Act as a Potent Predictor of Cisplatin Sensitivity in Gastric Cancer Treatment. Int. J. Surg. Oncol. 2012, 2012, 862879. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Sorenson, C.M.; Eastman, A. Influence of Cis-Diamminedichloroplatinum(II) on DNA Synthesis and Cell Cycle Progression in Excision Repair Proficient and Deficient Chinese Hamster Ovary Cells. Cancer Res. 1988, 48, 6703–6707. [Google Scholar]
- Osborn, M.F.; White, J.D.; Haley, M.M.; DeRose, V.J. Platinum-RNA Modifications Following Drug Treatment in S. Cerevisiae Identified by Click Chemistry and Enzymatic Mapping. ACS Chem. Biol. 2014, 9, 2404–2411. [Google Scholar] [CrossRef]
- Hostetter, A.A.; Osborn, M.F.; DeRose, V.J. RNA-Pt Adducts Following Cisplatin Treatment of Saccharomyces Cerevisiae. ACS Chem. Biol. 2012, 7, 218–225. [Google Scholar] [CrossRef]
- Karasawa, T.; Sibrian-Vazquez, M.; Strongin, R.M.; Steyger, P.S. Identification of Cisplatin-Binding Proteins Using Agarose Conjugates of Platinum Compounds. PLoS ONE 2013, 8, e66220. [Google Scholar] [CrossRef] [PubMed]
- Möltgen, S.; Piumatti, E.; Massafra, G.M.; Metzger, S.; Jaehde, U.; Kalayda, G.V. Cisplatin Protein Binding Partners and Their Relevance for Platinum Drug Sensitivity. Cells 2020, 9, 1322. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Dizon, D.S. New-Generation Platinum Agents for Solid Tumors. Future Oncol. 2009, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M. The Relationship between Cisplatin-Induced Reactive Oxygen Species, Glutathione, and BCL-2 and Resistance to Cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef]
- Poetsch, A.R. The Genomics of Oxidative DNA Damage, Repair, and Resulting Mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Shaukat, Z.; Liu, D.; Hussain, R.; Khan, M.; Gregory, S.L. The Role of JNK Signalling in Responses to Oxidative DNA Damage. Curr. Drug Targets 2016, 17, 154–163. [Google Scholar] [CrossRef]
- Jones, E.V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of P73-Mediated Apoptosis by c-Jun N-Terminal Kinase. Biochem. J. 2007, 405, 617–623. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Li, M.; Asaithamby, A. New Insights into the Roles of ATM and DNA-PKcs in the Cellular Response to Oxidative Stress. Cancer Lett. 2012, 327, 103–110. [Google Scholar] [CrossRef]
- Ditch, S.; Paull, T.T. The ATM Protein Kinase and Cellular Redox Signaling: Beyond the DNA Damage Response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Huang, S.; Mi, Q.-S.; Daniel, R.; Dong, Z. ATR-Chk2 Signaling in P53 Activation and DNA Damage Response during Cisplatin-Induced Apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Filiberti, L.; Vikhanskaya, F.; Carrassa, L.; Taya, Y.; Dincalci, M.; Broggini, M. Cisplatinum and Taxol Induce Different Patterns of P53 Phosphorylation. Neoplasia 2001, 3, 10–16. [Google Scholar] [CrossRef]
- Kondo, S.; Barna, B.P.; Kondo, Y.; Tanaka, Y.; Casey, G.; Liu, J.; Morimura, T.; Kaakaji, R.; Peterson, J.W.; Werbel, B.; et al. WAF1/CIP1 Increases the Susceptibility of P53 Non-Functional Malignant Glioma Cells to Cisplatin-Induced Apoptosis. Oncogene 1996, 13, 1279–1285. [Google Scholar]
- Yang, X.; Fraser, M.; Moll, U.M.; Basak, A.; Tsang, B.K. Akt-Mediated Cisplatin Resistance in Ovarian Cancer: Modulation of P53 Action on Caspase-Dependent Mitochondrial Death Pathway. Cancer Res. 2006, 66, 3126–3136. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, C.C.; Berberich, S.J. P53 Binds to Cisplatin-Damaged DNA. Biochim. Biophys. Acta 2001, 1517, 392–397. [Google Scholar] [CrossRef]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK Activation in Cisplatin-Induced Apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef]
- Basu, A.; Tu, H. Activation of ERK during DNA Damage-Induced Apoptosis Involves Protein Kinase Cdelta. Biochem. Biophys. Res. Commun. 2005, 334, 1068–1073. [Google Scholar] [CrossRef]
- Hayakawa, J.; Ohmichi, M.; Kurachi, H.; Kanda, Y.; Hisamoto, K.; Nishio, Y.; Adachi, K.; Tasaka, K.; Kanzaki, T.; Murata, Y. Inhibition of BAD Phosphorylation Either at Serine 112 via Extracellular Signal-Regulated Protein Kinase Cascade or at Serine 136 via Akt Cascade Sensitizes Human Ovarian Cancer Cells to Cisplatin. Cancer Res. 2000, 60, 5988–5994. [Google Scholar]
- Basu, A.; Krishnamurthy, S. Cellular Responses to Cisplatin-Induced DNA Damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- DeHaan, R.D.; Yazlovitskaya, E.M.; Persons, D.L. Regulation of P53 Target Gene Expression by Cisplatin-Induced Extracellular Signal-Regulated Kinase. Cancer Chemother. Pharmacol. 2001, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Rebillard, A.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.-T. Cisplatin Cytotoxicity: DNA and Plasma Membrane Targets. Curr. Med. Chem. 2008, 15, 2656–2663. [Google Scholar] [CrossRef]
- Abedini, M.R.; Muller, E.J.; Brun, J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Cisplatin Induces P53-Dependent FLICE-like Inhibitory Protein Ubiquitination in Ovarian Cancer Cells. Cancer Res. 2008, 68, 4511–4517. [Google Scholar] [CrossRef]
- Sun, F.; Cui, L.; Li, T.; Chen, S.; Song, J.; Li, D. Oxaliplatin Induces Immunogenic Cells Death and Enhances Therapeutic Efficacy of Checkpoint Inhibitor in a Model of Murine Lung Carcinoma. J. Recept. Signal. Transduct. Res. 2019, 39, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Golchin, S.; Alimohammadi, R.; Rostami Nejad, M.; Jalali, S.A. Synergistic Antitumor Effect of Anti-PD-L1 Combined with Oxaliplatin on a Mouse Tumor Model. J. Cell Physiol. 2019, 234, 19866–19874. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic Death of Colon Cancer Cells Treated with Oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-Induced Immune Modulation in Ovarian Cancer Mouse Models with Distinct Inflammation Profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Rojkó, L.; Reiniger, L.; Téglási, V.; Fábián, K.; Pipek, O.; Vágvölgyi, A.; Agócs, L.; Fillinger, J.; Kajdácsi, Z.; Tímár, J.; et al. Chemotherapy Treatment Is Associated with Altered PD-L1 Expression in Lung Cancer Patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1219–1226. [Google Scholar] [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.-G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in Combination with Carboplatin plus Nab-Paclitaxel Chemotherapy Compared with Chemotherapy Alone as First-Line Treatment for Metastatic Non-Squamous Non-Small-Cell Lung Cancer (IMpower130): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Qu, B.; Yang, H.; Hu, S.; Dong, X. If Small Molecules Immunotherapy Comes, Can the Prime Be Far Behind? Eur. J. Med. Chem. 2021, 218, 113356. [Google Scholar] [CrossRef]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The Pharmacology and Therapeutic Applications of Monoclonal Antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takaoka, A. Comparing Antibody and Small-Molecule Therapies for Cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Baxi, S.; Yang, A.; Gennarelli, R.L.; Khan, N.; Wang, Z.; Boyce, L.; Korenstein, D. Immune-Related Adverse Events for Anti-PD-1 and Anti-PD-L1 Drugs: Systematic Review and Meta-Analysis. BMJ 2018, 360, k793. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Dömling, A. Inhibitors of Programmed Cell Death 1 (PD-1): A Patent Review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 973–977. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-Molecule Inhibitors of PD-1/PD-L1 Immune Checkpoint Alleviate the PD-L1-Induced Exhaustion of T-Cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yao, Z.; Wang, S.; Xie, T.; Wu, G.; Zhang, H.; Zhang, P.; Wu, Y.; Yuan, H.; Sun, H. Syntheses, Biological Evaluations, and Mechanistic Studies of Benzo[c][1,2,5]Oxadiazole Derivatives as Potent PD-L1 Inhibitors with In Vivo Antitumor Activity. J. Med. Chem. 2021, 64, 8391–8409. [Google Scholar] [CrossRef]
- Qin, M.; Cao, Q.; Zheng, S.; Tian, Y.; Zhang, H.; Xie, J.; Xie, H.; Liu, Y.; Zhao, Y.; Gong, P. Discovery of [1,2,4]Triazolo [4,3- a]Pyridines as Potent Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2019, 62, 4703–4715. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, Y.; Yu, C.; Du, H.; Liu, J.; Li, H.; Huang, S.; Zhu, Q.; Xu, Y.; Zou, Y. Discovery of Novel Small-Molecule Inhibitors of PD-1/PD-L1 Interaction via Structural Simplification Strategy. Molecules 2021, 26, 3347. [Google Scholar] [CrossRef]
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik-Krol, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Weglarczyk, K.; et al. Terphenyl-Based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 11614–11636. [Google Scholar] [CrossRef]
- Chen, H.; Wang, K.; Yang, Y.; Huang, X.; Dai, X.; Feng, Z. Design, Synthesis, and Structure-Activity Relationship of Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction Inhibitors Bearing a Benzo[d]Isothiazole Scaffold. Eur. J. Med. Chem. 2021, 217, 113377. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, K.; Chen, H.; Feng, Z. Design, Synthesis, Evaluation, and SAR of 4-Phenylindoline Derivatives, a Novel Class of Small-Molecule Inhibitors of the Programmed Cell Death-1/ Programmed Cell Death-Ligand 1 (PD-1/PD-L1) Interaction. Eur. J. Med. Chem. 2021, 211, 113001. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S.; et al. Checkpoint Inhibition through Small Molecule-Induced Internalization of Programmed Death-Ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Guo, Y.; Pan, Z.; Zhong, S.; Jin, X.; Zhuang, W.; Chen, S.; Gao, J.; Huang, W.; et al. Discovery of Phenyl-Linked Symmetric Small Molecules as Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. Eur. J. Med. Chem. 2021, 223, 113637. [Google Scholar] [CrossRef] [PubMed]
- Surmiak, E.; Magiera-Mularz, K.; Musielak, B.; Muszak, D.; Kocik-Krol, J.; Kitel, R.; Plewka, J.; Holak, T.A.; Skalniak, L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 11797. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic Effects of Chemotherapy-Induced Tumor Cell Death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA Damage Response in Cancer Therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in Synthetic Lethality for Cancer Therapy: Cellular Mechanism and Clinical Translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Myers, S.H.; Ortega, J.A.; Cavalli, A. Synthetic Lethality through the Lens of Medicinal Chemistry. J. Med. Chem. 2020, 63, 14151–14183. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP Inhibitors: Review of Mechanisms of Action and BRCA1/2 Mutation Targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent Advances in DDR (DNA Damage Response) Inhibitors for Cancer Therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 8199. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953. [Google Scholar] [CrossRef]
- Baxter, J.S.; Zatreanu, D.; Pettitt, S.J.; Lord, C.J. Resistance to DNA Repair Inhibitors in Cancer. Mol. Oncol. 2022, 16, 3811–3827. [Google Scholar] [CrossRef]
- Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23, 1701. [Google Scholar] [CrossRef]
- Huang, J.-L.; Chang, Y.-T.; Hong, Z.-Y.; Lin, C.-S. Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 3238. [Google Scholar] [CrossRef]
- Lee, E.C.Y.; Kok, J.S.T.; Teh, B.T.; Lim, K.S. Interplay between the DNA Damage Response and Immunotherapy Response in Cancer. Int. J. Mol. Sci. 2022, 23, 13356. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, M.; Peng, Z.; Fan, R.; He, Y.; Zhang, H.; Xiong, W.; Jiang, W. Advances of DNA Damage Repair-Related Drugs and Combination With Immunotherapy in Tumor Treatment. Front. Immunol. 2022, 13, 854730. [Google Scholar] [CrossRef]
- Joshi, M.; Grivas, P.; Mortazavi, A.; Monk, P.; Clinton, S.K.; Sue-Ann Woo, M.; Holder, S.L.; Drabick, J.J.; Yin, M. Alterations of DNA Damage Response Genes Correlate with Response and Overall Survival in Anti-PD-1/PD-L1-Treated Advanced Urothelial Cancer. Cancer Med 2020, 9, 9365–9372. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Felipe Fernandez, M.; Khabibov, M.; Garifullin, A.; Dressler, D.; Topchu, I.; Patel, J.D.; Weinberg, F.; Boumber, Y. The Prognostic and Therapeutic Potential of DNA Damage Repair Pathway Alterations and Homologous Recombination Deficiency in Lung Cancer. Cancers 2022, 14, 5305. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Figueroa, A.; Sotelo, M.J. Immunotherapy in Microsatellite Instability Metastatic Colorectal Cancer: Current Status and Future Perspectives. J. Clin. Transl. Res. 2021, 7, 511–522. [Google Scholar]
- Chang, L.; Chang, M.; Chang, H.M.; Chang, F. Microsatellite Instability: A Predictive Biomarker for Cancer Immunotherapy. Appl. Immunohistochem. Mol. Morphol. 2018, 26, e15–e21. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Kuang, C.; Hao, S.; Yu, J.; Huang, Y.; Zhang, L. Immunotherapy Efficacy on Mismatch Repair-Deficient Colorectal Cancer: From Bench to Bedside. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188447. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch Repair Deficiency/Microsatellite Instability-High as a Predictor for Anti-PD-1/PD-L1 Immunotherapy Efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Viale, G.; Trapani, D.; Curigliano, G. Mismatch Repair Deficiency as a Predictive Biomarker for Immunotherapy Efficacy. Biomed. Res. Int. 2017, 2017, 4719194. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Wagner, M.; Jasek, M.; Karabon, L. Immune Checkpoint Molecules-Inherited Variations as Markers for Cancer Risk. Front. Immunol. 2020, 11, 606721. [Google Scholar] [CrossRef]
- Sato, H.; Jeggo, P.A.; Shibata, A. Regulation of Programmed Death-ligand 1 Expression in Response to DNA Damage in Cancer Cells: Implications for Precision Medicine. Cancer Sci. 2019, 110, 3415–3423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, J.; Chen, J.; Zhou, Q. The Developing Landscape of Combinatorial Therapies of Immune Checkpoint Blockade with DNA Damage Repair Inhibitors for the Treatment of Breast and Ovarian Cancers. J. Hematol. Oncol. 2021, 14, 206. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-Induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Sun, L.-L.; Yang, R.-Y.; Li, C.-W.; Chen, M.-K.; Shao, B.; Hsu, J.-M.; Chan, L.-C.; Yang, Y.; Hsu, J.L.; Lai, Y.-J.; et al. Inhibition of ATR Downregulates PD-L1 and Sensitizes Tumor Cells to T Cell-Mediated Killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/Caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
- Li, R.-Y.; Zheng, Z.-Y.; Li, Z.-M.; Heng, J.-H.; Zheng, Y.-Q.; Deng, D.-X.; Xu, X.-E.; Liao, L.-D.; Lin, W.; Xu, H.-Y.; et al. Cisplatin-Induced Pyroptosis Is Mediated via the CAPN1/CAPN2-BAK/BAX-Caspase-9-Caspase-3-GSDME Axis in Esophageal Cancer. Chem. Biol. Interact. 2022, 361, 109967. [Google Scholar] [CrossRef]
- Wu, M.; Wang, Y.; Yang, D.; Gong, Y.; Rao, F.; Liu, R.; Danna, Y.; Li, J.; Fan, J.; Chen, J.; et al. A PLK1 Kinase Inhibitor Enhances the Chemosensitivity of Cisplatin by Inducing Pyroptosis in Oesophageal Squamous Cell Carcinoma. EBioMedicine 2019, 41, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-C.; Li, C.-G.; Wang, Y.-F.; Xu, L.-H.; He, X.-H.; Zeng, Q.-Z.; Zeng, C.-Y.; Mai, F.-Y.; Hu, B.; Ouyang, D.-Y. Chemotherapeutic Paclitaxel and Cisplatin Differentially Induce Pyroptosis in A549 Lung Cancer Cells via Caspase-3/GSDME Activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Li, Y.; Xia, W.; Wu, M.; Yin, J.; Wang, Q.; Li, S.; Zhang, A.; Huang, S.; Zhang, Y.; Jia, Z. Activation of GSDMD Contributes to Acute Kidney Injury Induced by Cisplatin. Am. J. Physiol. Ren. Physiol. 2020, 318, F96–F106. [Google Scholar] [CrossRef]
- Shen, X.; Wang, H.; Weng, C.; Jiang, H. Caspase 3/GSDME-Dependent Pyroptosis Contributes to Chemotherapy Drug-Induced Nephrotoxicity. Cell Death Dis. 2021, 12, 186. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, G.; Wu, D.; Wu, S.; Gong, L.; Li, Z.; Luo, G.; Hu, J.; Chen, J.; Huang, X.; et al. Lobaplatin Induces Pyroptosis through Regulating CIAP1/2, Ripoptosome and ROS in Nasopharyngeal Carcinoma. Biochem. Pharmacol. 2020, 177, 114023. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ge, L.; Shi, X.; Liu, J.; Ruan, H.; Heng, D.; Ye, C. Lobaplatin Induces Pyroptosis in Cervical Cancer Cells via the Caspase-3/GSDME Pathway. Anticancer Agents Med. Chem. 2022, 22, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by Caspase-3 Determines Lobaplatin-Induced Pyroptosis in Colon Cancer Cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME Mediates Caspase-3-Dependent Pyroptosis in Gastric Cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation during Apoptosis and Inflammasome Activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Dessouki, F.B.A.; Kukreja, R.C.; Singla, D.K. Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Muscle Toxicity through Counteracting Pyroptosis. Pharmaceuticals 2020, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, H.; Li, D.; Zhou, X.; Qin, Q.; Zhang, Q. Caspase-3-Mediated GSDME Induced Pyroptosis in Breast Cancer Cells through the ROS/JNK Signalling Pathway. J. Cell Mol. Med. 2021, 25, 8159–8168. [Google Scholar] [CrossRef]
- Yu, P.; Wang, H.-Y.; Tian, M.; Li, A.-X.; Chen, X.-S.; Wang, X.-L.; Zhang, Y.; Cheng, Y. Eukaryotic Elongation Factor-2 Kinase Regulates the Cross-Talk between Autophagy and Pyroptosis in Doxorubicin-Treated Human Melanoma Cells in Vitro. Acta Pharmacol. Sin. 2019, 40, 1237–1244. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Tsuchiya, K. Switching from Apoptosis to Pyroptosis: Gasdermin-Elicited Inflammation and Antitumor Immunity. Int. J. Mol. Sci. 2021, 22, 426. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Ding, J.; Wang, C.; Zhou, X.; Gao, W.; Huang, H.; Shao, F.; Liu, Z. A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis. Nature 2020, 579, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A New Paradigm of Cell Death for Fighting against Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Sun, Z. Eliciting Pyroptosis to Fuel Cancer Immunotherapy: Mechanisms and Strategies. Cancer Biol. Med. 2022, 19, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.; Zhu, Y.; Yang, P.; Chen, Z.; Xia, Y.; Qiao, C.; Liu, W.; Deng, H.; Li, J.; Ning, P.; et al. Pyroptosis in Inflammatory Diseases and Cancer. Theranostics 2022, 12, 4310–4329. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, X.; Deng, Y.; Li, S.; Xu, X.; Qin, Y.; Peng, L. Pyroptosis Provides New Strategies for the Treatment of Cancer. J. Cancer 2023, 14, 140–151. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Kołat, D.; Kałuzińska-Kołat, Ż.; Gawrysiak, M.; Drozda, R.; Celik, I.; Kontek, R. PD-1/PD-L1 and DNA Damage Response in Cancer. Cells 2023, 12, 530. https://doi.org/10.3390/cells12040530
Kciuk M, Kołat D, Kałuzińska-Kołat Ż, Gawrysiak M, Drozda R, Celik I, Kontek R. PD-1/PD-L1 and DNA Damage Response in Cancer. Cells. 2023; 12(4):530. https://doi.org/10.3390/cells12040530
Chicago/Turabian StyleKciuk, Mateusz, Damian Kołat, Żaneta Kałuzińska-Kołat, Mateusz Gawrysiak, Rafał Drozda, Ismail Celik, and Renata Kontek. 2023. "PD-1/PD-L1 and DNA Damage Response in Cancer" Cells 12, no. 4: 530. https://doi.org/10.3390/cells12040530
APA StyleKciuk, M., Kołat, D., Kałuzińska-Kołat, Ż., Gawrysiak, M., Drozda, R., Celik, I., & Kontek, R. (2023). PD-1/PD-L1 and DNA Damage Response in Cancer. Cells, 12(4), 530. https://doi.org/10.3390/cells12040530