

Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death
 ,
,  , ,
, ,  , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation, Culturing, and Microscopic Characterization of HUVECs
2.2. Immunocytochemical Characterization of the Proliferation Status of HUVEC Cultures
2.3. Monitoring the Senolytic Effects of Test Compounds on HUVECs with Microscopy-Based Cell Enumeration
2.4. Western Blotting
2.5. Live Cell Imaging and Real-Time Detection of Apoptosis
2.6. Statistical Analysis
3. Results
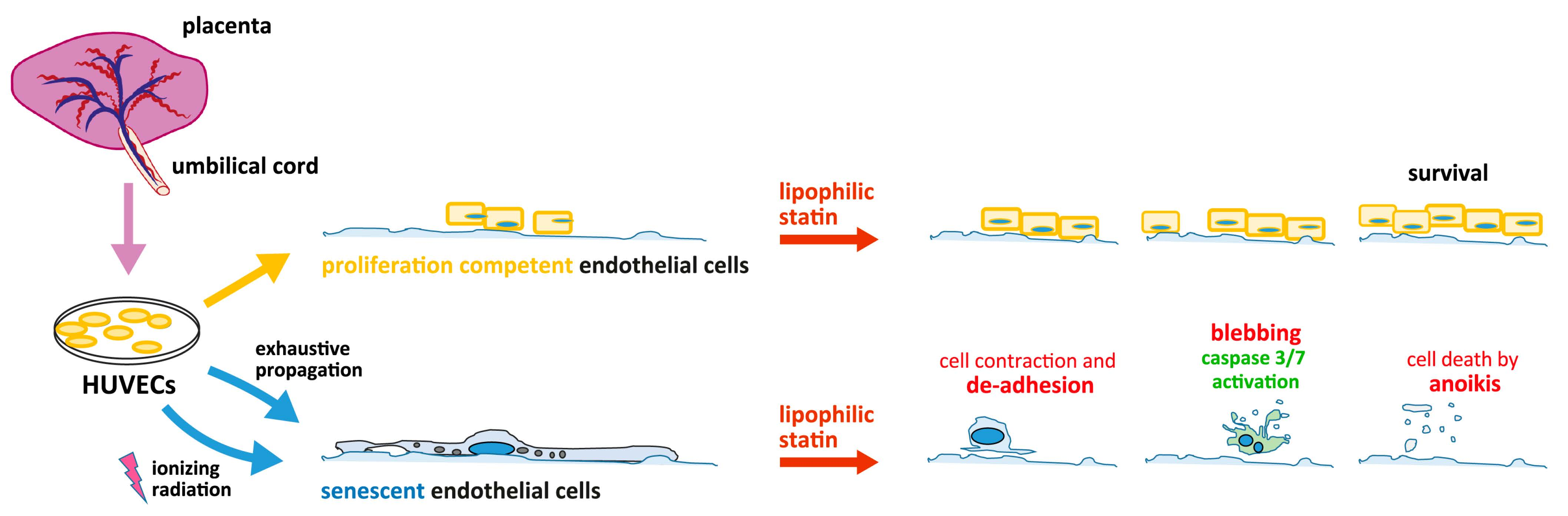
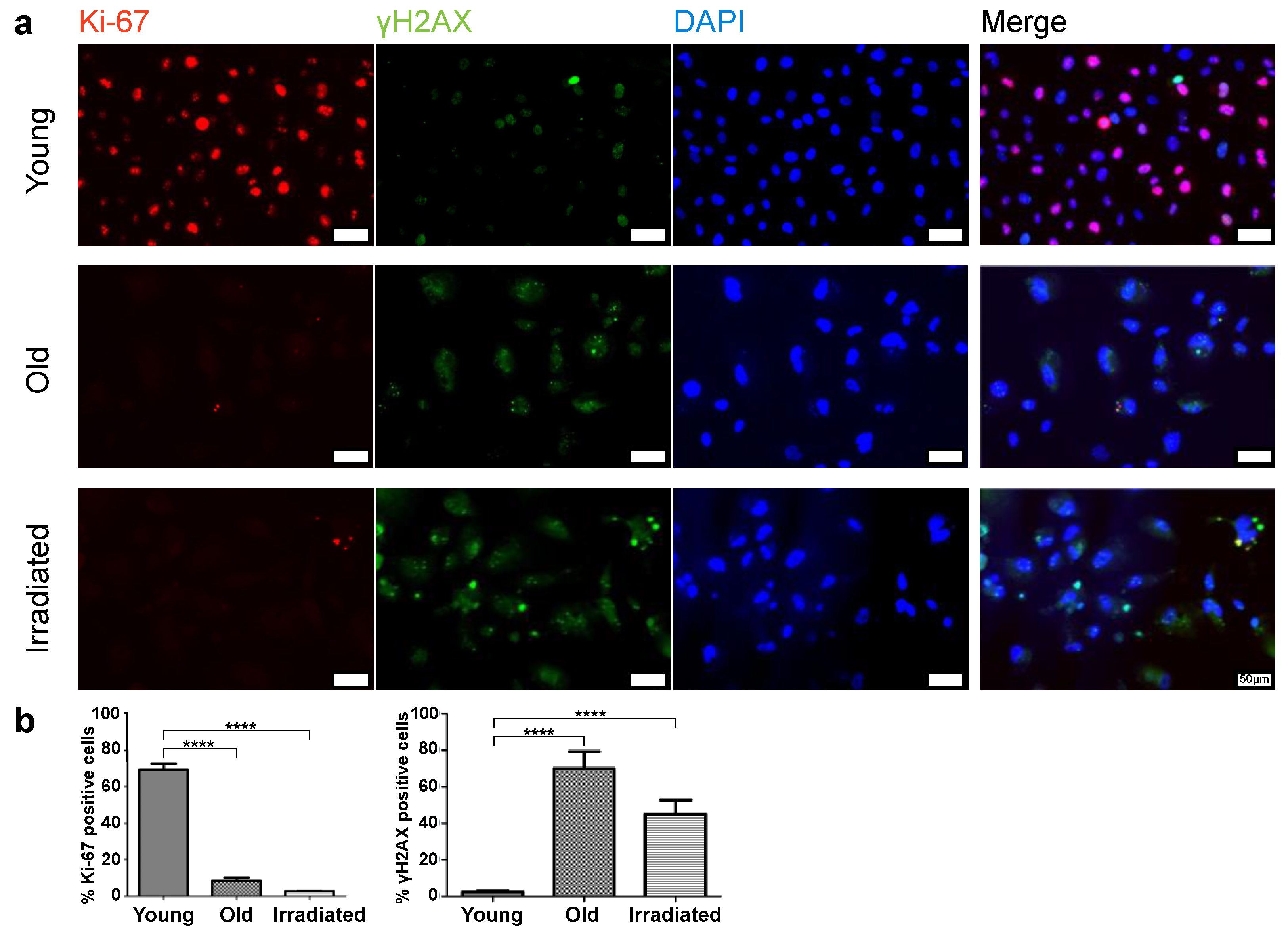
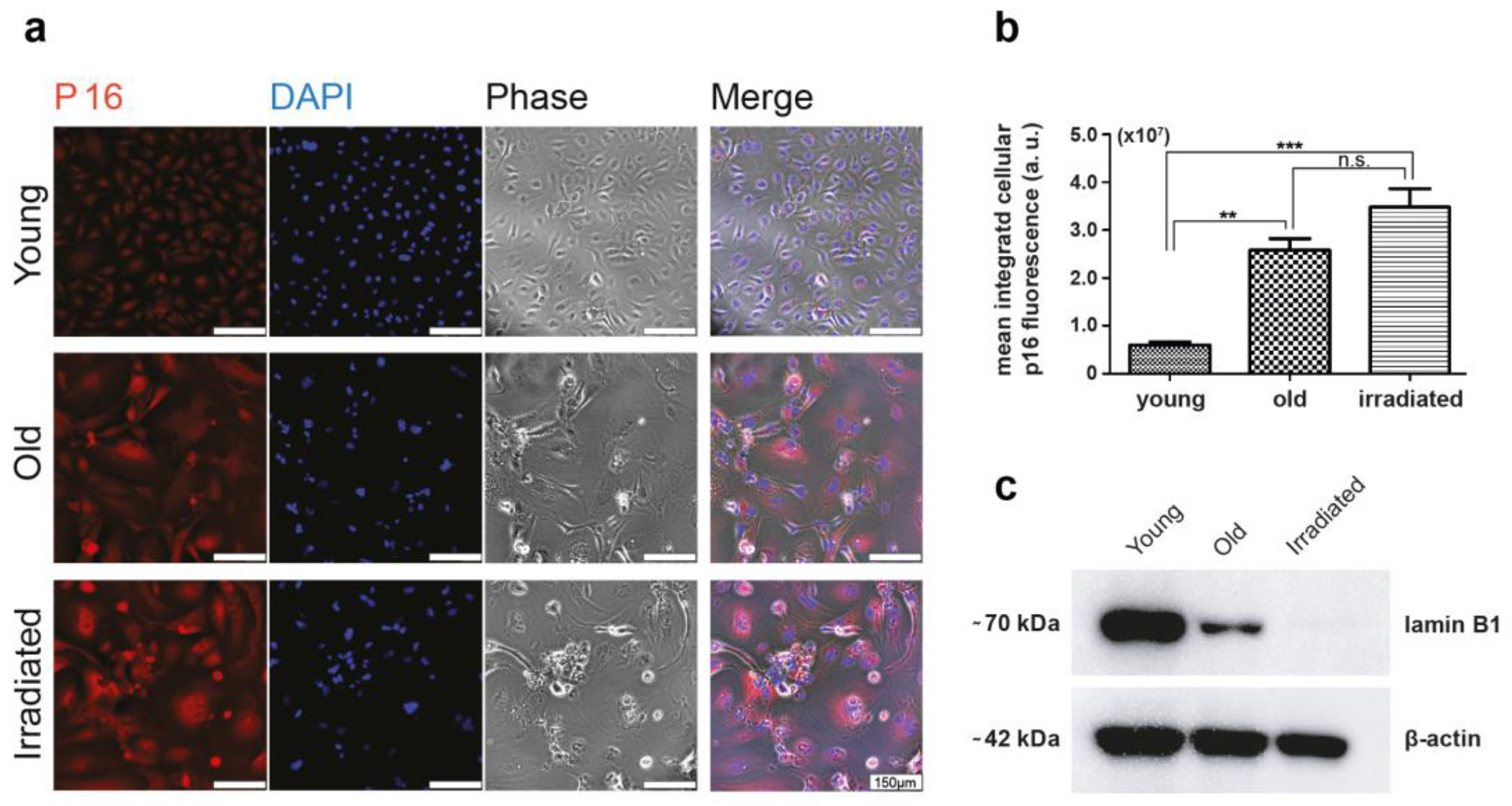
3.1. Generation of the Cells for the Two Types of Senescence Models, of Replicative Senescent HUVECs with Extensive Passaging, and of Stress-Induced Senescent HUVECs with Exposure to γ-Irradiation
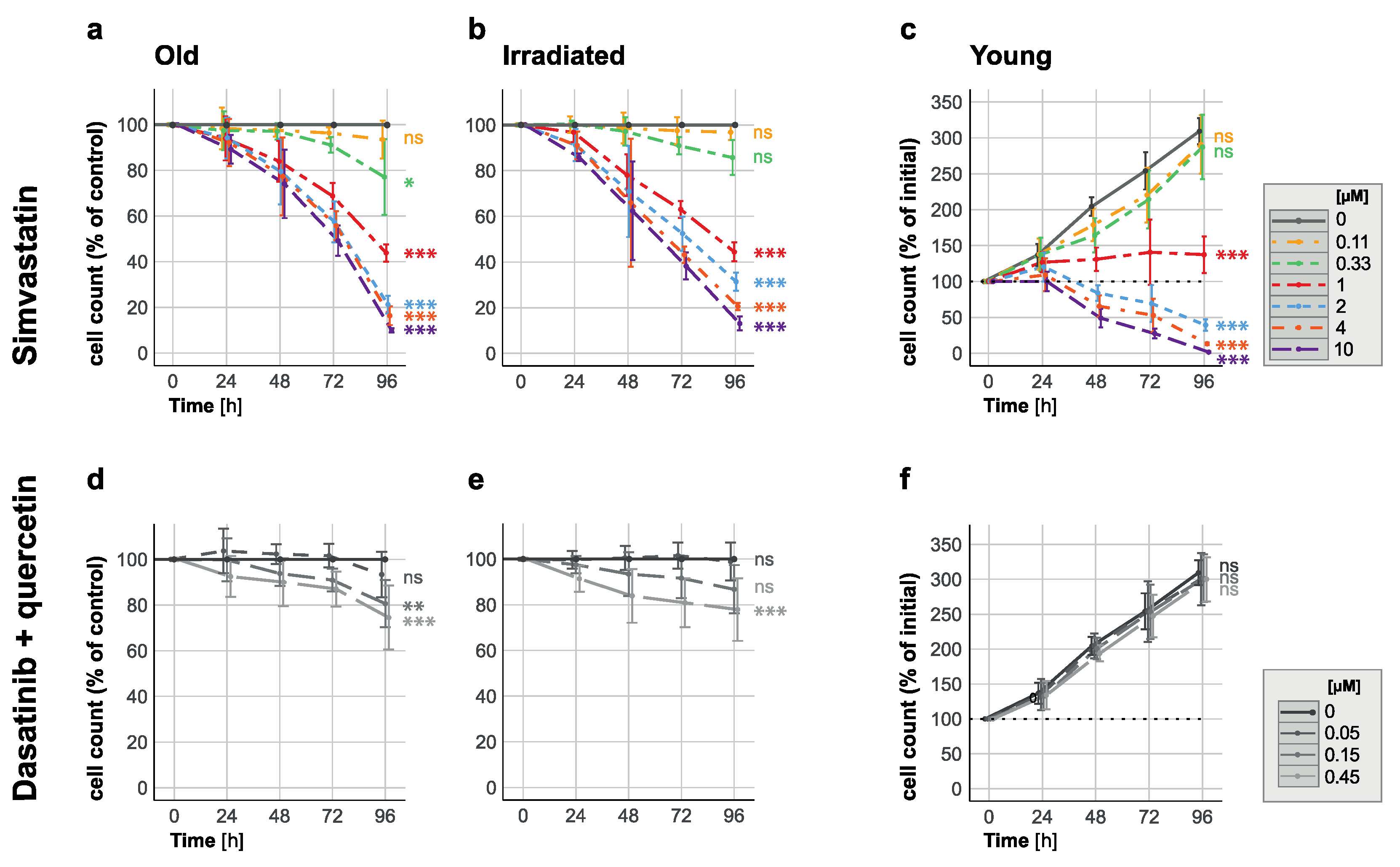
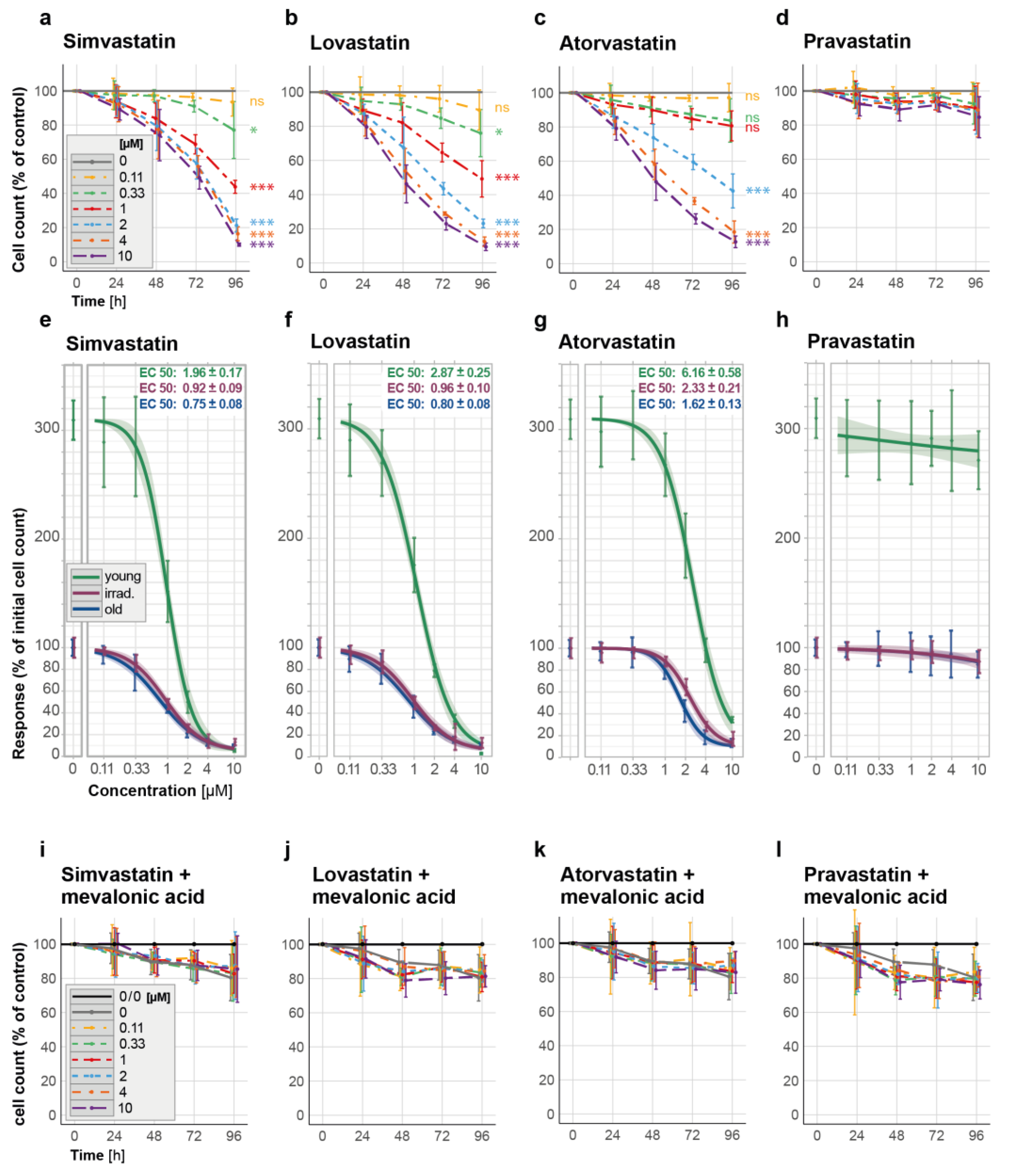
3.2. Senescent-Rendered HUVECs Are More Susceptible to Simvastatin-Induced Cell Death
3.3. All Investigated Lipophilic Statins Showed Senolytic Activity, Which Could Be Negated with the Supplementation of Mevalonic Acid
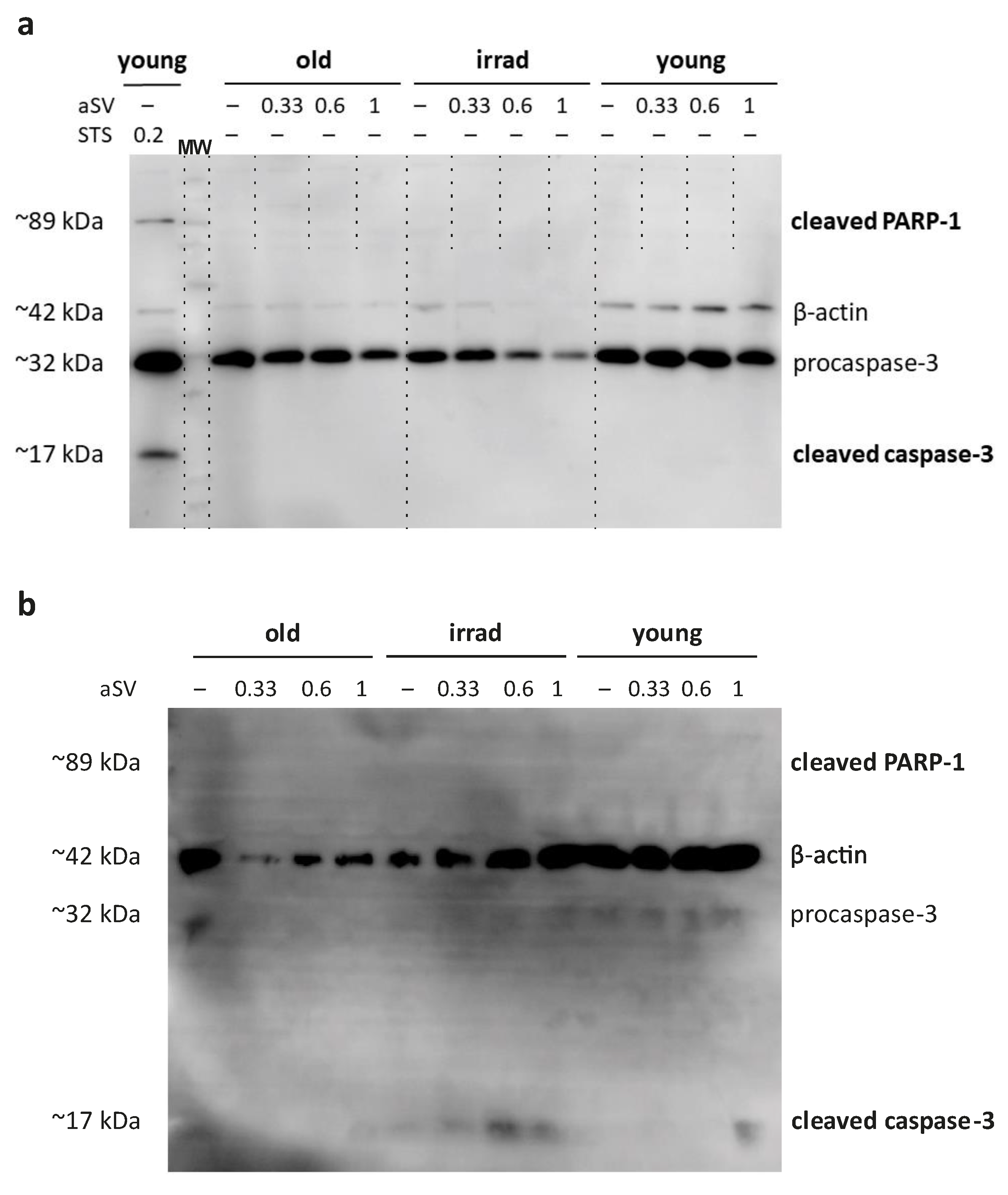
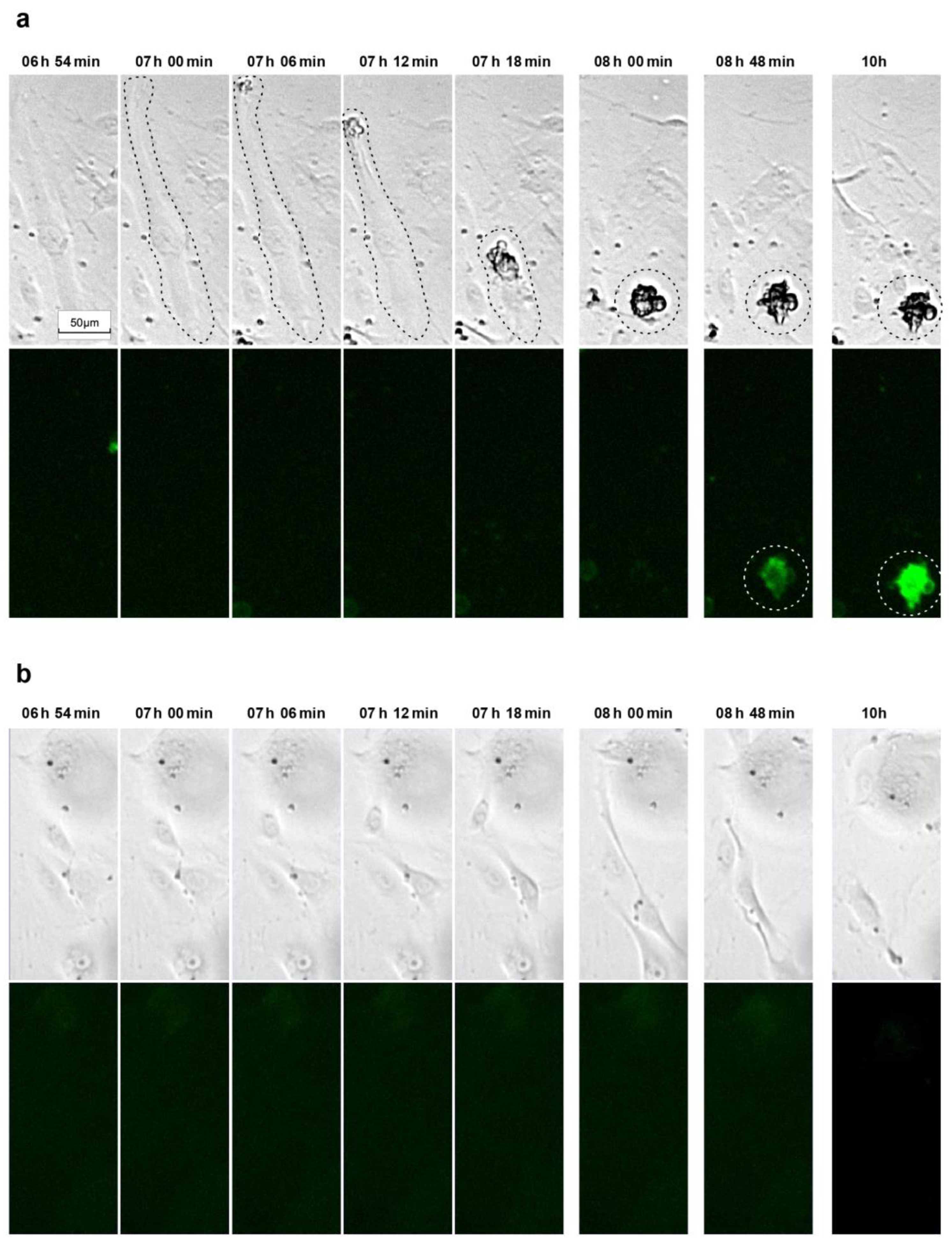
3.4. Activated Simvastatin-Induced Apoptosis/Anoikis in Senescent HUVECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and Consequences of Endothelial Cell Senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and Senolytics in Cardiovascular Disease: Promise and Potential Pitfalls. Mech. Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Cellular Senescence in Vivo: Its Relevance in Ageing and Cardiovascular Disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Warboys, C.M.; De Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed Flow Promotes Endothelial Senescence via a P53-Dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16(Ink4a)-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic Senolytic Treatment Alleviates Established Vasomotor Dysfunction in Aged or Atherosclerotic Mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Krouwer, V.J.D.; Hekking, L.H.P.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial Cell Senescence Is Associated with Disrupted Cell-Cell Junctions and Increased Monolayer Permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Mun, G.I.; Boo, Y.C. Identification of CD44 as a Senescence-Induced Cell Adhesion Gene Responsible for the Enhanced Monocyte Recruitment to Senescent Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2102–H2111. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Vallejo, S.; Peiró, C.; Sánchez-Ferrer, C.F.; Rodríguez-Mañas, L. Mechanisms Involved in the Aging-Induced Vascular Dysfunction. Front. Physiol. 2012, 3, 132. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data from 90,056 Participants in 14 Randomised Trials of Statins. Lancet Lond. Engl. 2005, 366, 1267–1278. [Google Scholar] [CrossRef]
- Chello, M.; Mastroroberto, P.; Patti, G.; D’Ambrosio, A.; Morichetti, M.C.; Di Sciascio, G.; Covino, E. Simvastatin Attenuates Leucocyte–Endothelial Interactions after Coronary Revascularisation with Cardiopulmonary Bypass. Heart 2003, 89, 538–543. [Google Scholar] [CrossRef][Green Version]
- Jain, M.K.; Ridker, P.M. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat. Rev. Drug Discov. 2005, 4, 977–987. [Google Scholar] [CrossRef]
- Bates, K.; Ruggeroli, C.E.; Goldman, S.; Gaballa, M.A. Simvastatin Restores Endothelial NO-Mediated Vasorelaxation in Large Arteries after Myocardial Infarction. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H768–H775. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Z.; Kitajima, I.; Wang, Z. Effects of Different Statins on Endothelial Nitric Oxide Synthase and AKT Phosphorylation in Endothelial Cells. Int. J. Cardiol. 2008, 127, 33–39. [Google Scholar] [CrossRef]
- Zhao, W.; Fu, H.; Chang, F.; Liu, J.; Wang, J.; Li, F.; Zhao, J. Effects of Various Doses of Atorvastatin on Vascular Endothelial Cell Apoptosis and Autophagy In vitro. Mol. Med. Rep. 2019, 19, 1919–1925. [Google Scholar] [CrossRef]
- Crampton, S.P.; Davis, J.; Hughes, C.C.W. Isolation of Human Umbilical Vein Endothelial Cells (HUVEC). J. Vis. Exp. 2007, 3, e183. [Google Scholar] [CrossRef]
- Dong, W.; Vuletic, S.; Albers, J.J. Differential Effects of Simvastatin and Pravastatin on Expression of Alzheimer’s Disease-Related Genes in Human Astrocytes and Neuronal Cells. J. Lipid Res. 2009, 50, 2095–2102. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Signorell, A.; Aho, K.; Alfons, A.; Anderegg, N.; Aragon, T.; Arppe, A.; Baddeley, A.; Barton, K.; Bolker, B.; Borchers, H.W.; et al. DescTools: Tools for Descriptive Statistics; R Package Version 0.99.27; 2019; Available online: https://cran.r-project.org/package=DescTools (accessed on 6 June 2023).
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, M.; Feng, T.; Zhan, L.; Zhou, L.; Yu, G. Use Ggbreak to Effectively Utilize Plotting Space to Deal with Large Datasets and Outliers. Front. Genet. 2021, 12, 2122. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.M.; Watts, D.G. Nonlinear Regression Analysis and Its Applications; Wiley: New York, NY, USA, 1988; ISBN 978-0-471-81643-0. [Google Scholar]
- Scholzen, T.; Gerdes, J. The Ki-67 Protein: From the Known and the Unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo Athens Greece 2008, 22, 305–309. [Google Scholar] [PubMed]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of p16INK4a and p21CIP1 Pathways to Induction of Premature Senescence of Human Endothelial Cells: Permissive Role of P53. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, H.; Zi, M.; Li, W.; Liu, J.; Yang, Y.; Zhou, D.; Kong, Q.-P.; Zhang, Y.; He, Y. Why Senescent Cells Are Resistant to Apoptosis: An Insight for Senolytic Development. Front. Cell Dev. Biol. 2022, 10, 822816. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, C.; de Martin, R.; Kumabashiri, I.; Schmid, J.A.; Binder, B.R.; Lipp, J. Nuclear Factor (NF)-kappaB-Regulated X-Chromosome-Linked Iap Gene Expression Protects Endothelial Cells from Tumor Necrosis Factor Alpha-Induced Apoptosis. J. Exp. Med. 1998, 188, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 Family: Regulators of Cell Death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.A.; Cavender, J.F. Research Article: Beta-Galactosidase Staining as a Marker of Cells Enduring Stress. Bios 2004, 75, 139–146. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Wagner, M.; Hampel, B.; Bernhard, D.; Hala, M.; Zwerschke, W.; Jansen-Dürr, P. Replicative Senescence of Human Endothelial Cells in Vitro Involves G1 Arrest, Polyploidization and Senescence-Associated Apoptosis. Exp. Gerontol. 2001, 36, 1327–1347. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The Role of Nuclear Lamin B1 in Cell Proliferation and Senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: An Update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- McFarlane, S.I.; Muniyappa, R.; Francisco, R.; Sowers, J.R. Clinical Review 145: Pleiotropic Effects of Statins: Lipid Reduction and Beyond. J. Clin. Endocrinol. Metab. 2002, 87, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Tokui, T.; Ishigami, M.; Sugiyama, Y. Tissue-Selective Uptake of Pravastatin in Rats: Contribution of a Specific Carrier-Mediated Uptake System. Biopharm. Drug Dispos. 1996, 17, 775–789. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the Mevalonate Pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulakos, J.; Ye, L.Y.; Benzaquen, M.; Moore, M.J.; Kamel-Reid, S.; Freedman, M.H.; Yeger, H.; Penn, L.Z. Differential Sensitivity of Various Pediatric Cancers and Squamous Cell Carcinomas to Lovastatin-Induced Apoptosis: Therapeutic Implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 158–167. [Google Scholar]
- Demierre, M.-F.; Higgins, P.D.R.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and Cancer Prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Tupper, J.C.; Bannerman, D.D.; Winn, R.K.; Sebti, S.M.; Hamilton, A.D.; Harlan, J.M. Inhibition of Protein Geranylgeranylation and RhoA/RhoA Kinase Pathway Induces Apoptosis in Human Endothelial Cells. J. Biol. Chem. 2002, 277, 15309–15316. [Google Scholar] [CrossRef]
- Kaneta, S.; Satoh, K.; Kano, S.; Kanda, M.; Ichihara, K. All Hydrophobic HMG-CoA Reductase Inhibitors Induce Apoptotic Death in Rat Pulmonary Vein Endothelial Cells. Atherosclerosis 2003, 170, 237–243. [Google Scholar] [CrossRef]
- Frisch, S.M.; Francis, H. Disruption of Epithelial Cell-Matrix Interactions Induces Apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef]
- Valentijn, A.J.; Zouq, N.; Gilmore, A.P. Anoikis. Biochem. Soc. Trans. 2004, 32, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics Decrease Senescent Cells in Humans: Preliminary Report from a Clinical Trial of Dasatinib plus Quercetin in Individuals with Diabetic Kidney Disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Park, H.-J.; Georgescu, S.P.; Sebti, S.M.; Hamilton, A.D.; Galper, J.B. Simvastatin Potentiates Tumor Necrosis Factor Alpha-Mediated Apoptosis of Human Vascular Endothelial Cells via the Inhibition of the Geranylgeranylation of RhoA. Life Sci. 2006, 79, 1484–1492. [Google Scholar] [CrossRef]
- Diamant, M.; Tushuizen, M.E.; Abid-Hussein, M.N.; Hau, C.M.; Böing, A.N.; Sturk, A.; Nieuwland, R. Simvastatin-Induced Endothelial Cell Detachment and Microparticle Release Are Prenylation Dependent. Thromb. Haemost. 2008, 100, 489–497. [Google Scholar] [CrossRef]
- Copaja, M.; Venegas, D.; Aranguiz, P.; Canales, J.; Vivar, R.; Avalos, Y.; Garcia, L.; Chiong, M.; Olmedo, I.; Catalán, M.; et al. Simvastatin Disrupts Cytoskeleton and Decreases Cardiac Fibroblast Adhesion, Migration and Viability. Toxicology 2012, 294, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Scerbavicius, R.; Choe, K.-H.; Cool, C.; Wood, K.; Tuder, R.M.; Burns, N.; Kasper, M.; Voelkel, N.F. Simvastatin Causes Endothelial Cell Apoptosis and Attenuates Severe Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L668–L676. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Gu, X.; Ye, Z.; Shi, J.; Zheng, X. Antiaging Effects of Simvastatin on Vascular Endothelial Cells. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2014, 20, 212–218. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, but Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J. Gerontol. A. Biol. Sci. Med. Sci. 2011, 66, 191–201. [Google Scholar] [CrossRef]
- Corsini, A.; Bellosta, S.; Baetta, R.; Fumagalli, R.; Paoletti, R.; Bernini, F. New Insights into the Pharmacodynamic and Pharmacokinetic Properties of Statins. Pharmacol. Ther. 1999, 84, 413–428. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef]
- Thibault, A.; Samid, D.; Tompkins, A.C.; Figg, W.D.; Cooper, M.R.; Hohl, R.J.; Trepel, J.; Liang, B.; Patronas, N.; Venzon, D.J.; et al. Phase I Study of Lovastatin, an Inhibitor of the Mevalonate Pathway, in Patients with Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 483–491. [Google Scholar]
- van der Spek, E.; Bloem, A.C.; van de Donk, N.W.C.J.; Bogers, L.H.; van der Griend, R.; Kramer, M.H.; de Weerdt, O.; Wittebol, S.; Lokhorst, H.M. Dose-Finding Study of High-Dose Simvastatin Combined with Standard Chemotherapy in Patients with Relapsed or Refractory Myeloma or Lymphoma. Haematologica 2006, 91, 542–545. [Google Scholar] [PubMed]
- von Kobbe, C. Targeting Senescent Cells: Approaches, Opportunities, Challenges. Aging 2019, 11, 12844–12861. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar] [CrossRef]
- Furberg, C.D. Natural Statins and Stroke Risk. Circulation 1999, 99, 185–188. [Google Scholar] [CrossRef]
- Singh, D.K.; Li, L.; Porter, T.D. Policosanol Inhibits Cholesterol Synthesis in Hepatoma Cells by Activation of AMP-Kinase. J. Pharmacol. Exp. Ther. 2006, 318, 1020–1026. [Google Scholar] [CrossRef]
- Nam, D.-E.; Yun, J.-M.; Kim, D.; Kim, O.-K. Policosanol Attenuates Cholesterol Synthesis via AMPK Activation in Hypercholesterolemic Rats. J. Med. Food 2019, 22, 1110–1117. [Google Scholar] [CrossRef]
- Francini-Pesenti, F.; Beltramolli, D.; Dall’Acqua, S.; Brocadello, F. Effect of Sugar Cane Policosanol on Lipid Profile in Primary Hypercholesterolemia. Phytother. Res. 2008, 22, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Deng, R. Therapeutic Effects of Guggul and Its Constituent Guggulsterone: Cardiovascular Benefits. Cardiovasc. Drug Rev. 2007, 25, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, C.; Nair, S.M.; Quirrin, K.-W.; Melnick, S.J. Hypolipidemic Effects of a Proprietary Commiphora Mukul Gum Resin Extract and Medium-Chain Triglyceride Preparation (GU-MCT810). J. Evid.-Based Complement. Altern. Med. 2013, 18, 248–256. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belakova, B.; Wedige, N.K.; Awad, E.M.; Hess, S.; Oszwald, A.; Fellner, M.; Khan, S.Y.; Resch, U.; Lipovac, M.; Šmejkal, K.; et al. Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death. Cells 2023, 12, 2836. https://doi.org/10.3390/cells12242836
Belakova B, Wedige NK, Awad EM, Hess S, Oszwald A, Fellner M, Khan SY, Resch U, Lipovac M, Šmejkal K, et al. Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death. Cells. 2023; 12(24):2836. https://doi.org/10.3390/cells12242836
Chicago/Turabian StyleBelakova, Barbora, Nicholas K. Wedige, Ezzat M. Awad, Simon Hess, André Oszwald, Marlene Fellner, Shafaat Y. Khan, Ulrike Resch, Markus Lipovac, Karel Šmejkal, and et al. 2023. "Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death" Cells 12, no. 24: 2836. https://doi.org/10.3390/cells12242836
APA StyleBelakova, B., Wedige, N. K., Awad, E. M., Hess, S., Oszwald, A., Fellner, M., Khan, S. Y., Resch, U., Lipovac, M., Šmejkal, K., Uhrin, P., & Breuss, J. M. (2023). Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death. Cells, 12(24), 2836. https://doi.org/10.3390/cells12242836